Week 5 HW: Protein Design Part II
SOD1 Binder Peptide Design (From Pranam)
Part 1: Generate Binders with PepMLM
Colab link: https://colab.research.google.com/drive/1_l-gF1EFDOHIyetFlJT4wAGmYJr-raXB

Sequence (non-mutated): MATKAVCVLKGDGPVQGIINFEQKESNGPVKVWGSIKGLTEGLHGFHVHEFGDNTAGCTSAGPHFNPLSRKHGGPKDEERHVGDLGNVTADKDGVADVSIEDSVISLSGDHCIIGRTLVVHEKADDLGKGGNEESTKTGNAGSRLACGVIGIAQ
Sequence (A4V): MATKVVCVLKGDGPVQGIINFEQKESNGPVKVWGSIKGLTEGLHGFHVHEFGDNTAGCTSAGPHFNPLSRKHGGPKDEERHVGDLGNVTADKDGVADVSIEDSVISLSGDHCIIGRTLVVHEKADDLGKGGNEESTKTGNAGSRLACGVIGIAQ
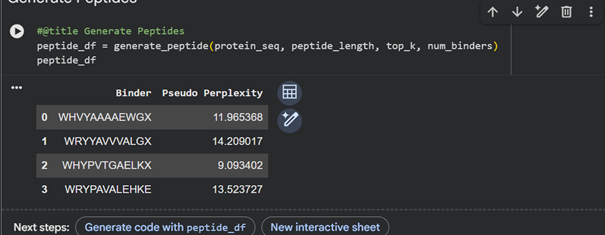
| Peptide | Sequence | Perplexity |
|---|---|---|
| Peptide 1 | WHHVYAAAAEWG | 11.965 |
| Peptide 2 | WRYYAVVVALGA | 14.209 |
| Peptide 3 | WHYPVTGAELKA | 9.093 |
| Peptide 4 | WRPYAVALEHKE | 13.523 |
| Known peptide | FLYRWLPSRRGG | - |
Part 2: Evaluate Binders with AlphaFold3
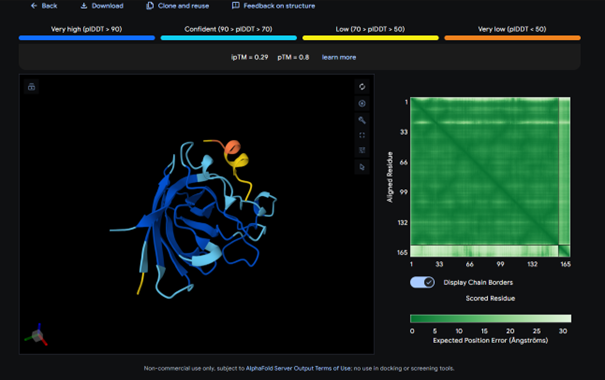
AlphaFold results for peptide 1:
The complex has a low ipTM score (0.29), indicating weak binding. The peptide does not localize near the N-terminus where A4V sits. It appears to bind to a surface loop region and is surface bound rather than partially buried. It does not engage the β-barrel region or approach the dimer interface.
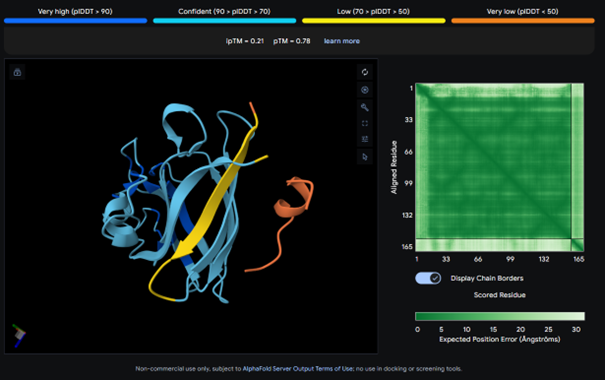
AlphaFold results for peptide 2:
This complex also has a low ipTM score (ipTM 0.21). It is surface bound and is not binded near terminus. It seems to bind on the outside of the β-barrel.
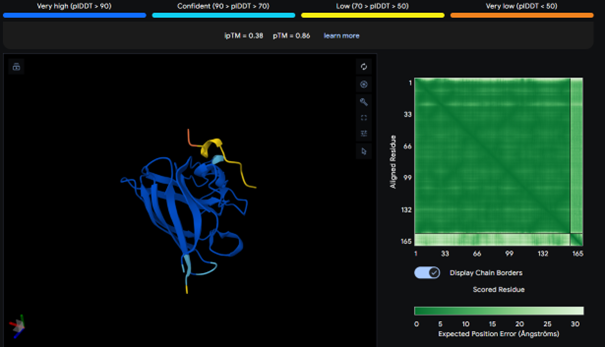
AlphaFold results for peptide 3:
It has the highest binding score (ipTM = 0.38) among the tested models, but this still indicates relatively weak binding. The peptide does not bind to the N-terminus and appears surface-bound. There is no strong interaction with the β-barrel or the dimer interface.
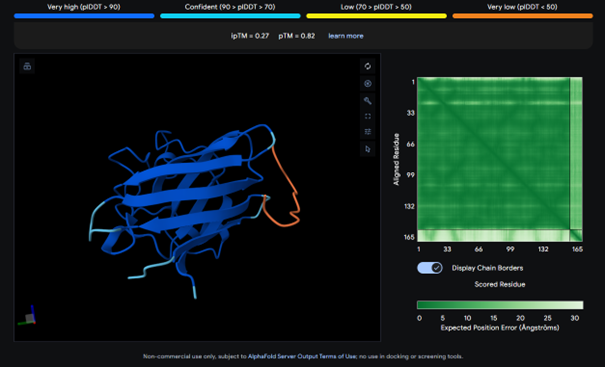
AlphaFold results for peptide 4:
This complex has a low binding score (ipTM = 0.24), meaning weak interaction. The peptide does not bind to the N-terminus and bounds to surface. There is no strong interaction with the β-barrel or the dimer interface.
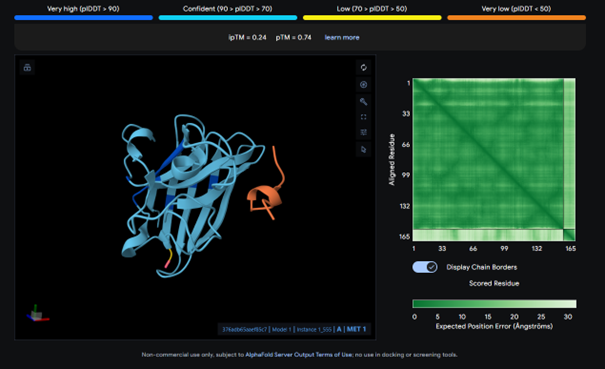
AlphaFold results for the known binder:
I expected the known binder to have a higher score, but it also shows a low binding score (ipTM = 0.27). Considering this, the result for peptide 3 is actually higher than the known binder, even though it is still relatively low.
All ipTM values indicate weak binding confidence. The known binder has an ipTM of about 0.27, which is also relatively low. Among the PepMLM-generated peptides, peptide 3 shows the highest ipTM (0.38) and therefore exceeds the score of the known binder. In addition, none of the peptides appear to bind near the N-terminus and they all appear surface-bound.
Part 3: Evaluate Properties of Generated Peptides in the PeptiVerse
Then I moved to Peptiverse and evaluated the predicted binding affinity, solubility, hemolysis probability, net charge (pH 7), and molecular weight for all peptides.
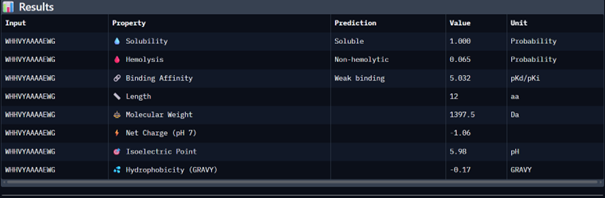
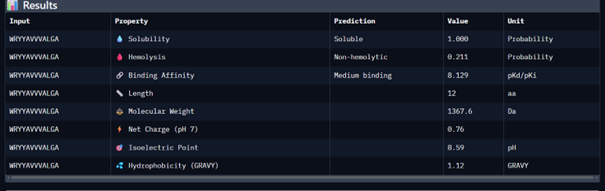
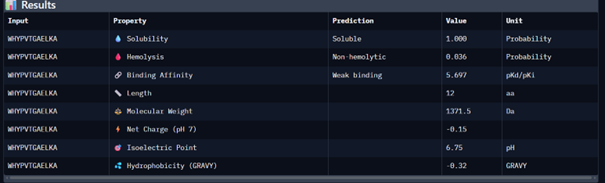
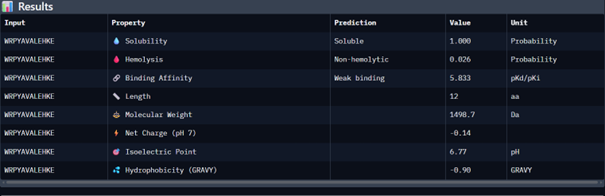
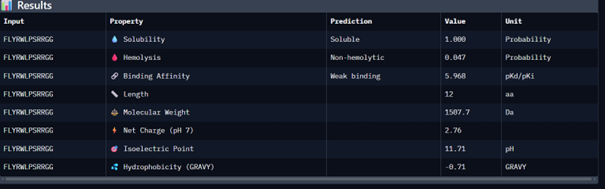
Peptide 1 appeared slightly hydrophilic, peptide 2 more hydrophobic, and peptides 3 and 4 strongly hydrophilic. All peptides were predicted to be soluble. None showed a significant hemolytic effect, although peptide 2 had the highest hemolysis probability, while peptides 3 and 4 had the lowest. Predicted binding affinity was generally low for all peptides, including the known binder, but unlike the AlphaFold results, Peptiverse predicted the highest binding potential for peptide 2.
Part 4: Generate Optimized Peptides with moPPIt
Colab link: https://colab.research.google.com/drive/1YxRRQkXeqlYSoXEwLAPAu4BLaHGLGW0b
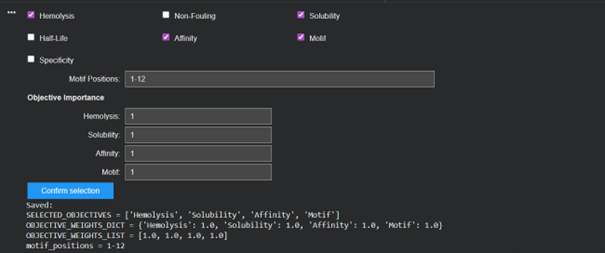
| Peptide | Sequence | Hemolysis | Solubility | Affinity | Motif |
|---|---|---|---|---|---|
| Peptide 1 | KEDYGYGCQGAL | 0.937 | 0.833 | 6.62 | 0.452 |
| Peptide 2 | EQKTYGYGCTHK | 0.977 | 1.000 | 5.51 | 0.603 |
| Peptide 3 | TEKSQCGTTGEK | 0.971 | 1.000 | 5.84 | 0.513 |
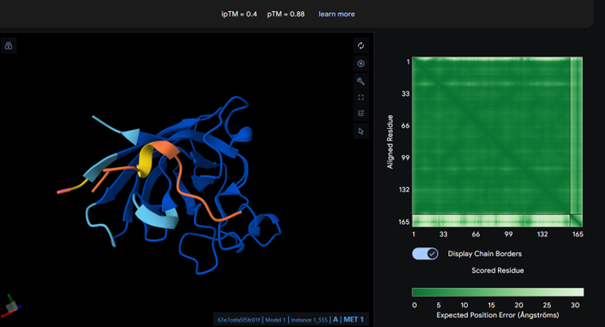
The peptide binds near the N-terminal region, which is where the A4V mutation site is located, as intended in the design. The peptide still appears mostly surface-bound rather than deeply buried, but it is positioned closer to the targeted N-terminal region compared to the other peptides, which was the motif I chose for the generated peptides to bind.
The main reason for the difference between the peptides suggested by the two models is likely that PepMLM generates peptides that could bind anywhere on the protein, whereas moPPIt works by specifying the region we want to target and then designing sequences optimized according to multiple criteria. Because of this, moPPIt provides peptides that are not only target-guided but also optimized during the design process, while PepMLM searches more broadly for generally compatible binders.
Before moving to clinical studies, I would first evaluate whether the peptide is safe, starting by checking that it does not cause hemolysis. Then I would examine whether the peptide is soluble. Among the peptides that meet these criteria, I would select those with the highest predicted binding affinity. From these candidates, I would choose the peptide that is biochemically easier to produce and has suitable biological properties (such as pH-related charge and molecular weight) before advancing it toward clinical studies.