Living Modified Organism(LMO) Cotton Textiles That Maintain Hygiene and Emit Pleasant Scents
This application envisions a cotton-based textile integrated with genetically engineered microorganisms that are designed to function stably on the fabric surface. These microorganisms would metabolize organic compounds derived from human sweat and skin debris—such as fatty acids, ammonia, and keratin degradation byproducts—thereby maintaining hygiene and reducing odor formation.
Unlike conventional approaches that rely on antimicrobial chemicals or synthetic fragrances, this system would operate as an engineered living material, in which biological activity is deliberately programmed to occur only within a non-natural environment (the textile itself). Conventional perfumes and detergents repeatedly introduce chemically stable, exogenous compounds that may unintentionally interfere with human endocrine systems or disrupt the skin microbiome through persistent and non-selective exposure.
Part 0: Basics of Gel Electrophoresis #S.S: 부트캠프와 Wk2 수업을 들은 후, 과제를 확인했지만 정확한 개념 정리가 안되어 있어서 뭘 진행하는지 이해하기 어려웠다. 따라서 ChatGPT를 활용, it became clearer once I understood the concept of restriction enzyme(molecular scissors) and gel electrophoresis(DNA size filter).
Prompt: "restriction enzyme biology 와 gel electrophoresis에 대해서 아주 간단히 설명해줘" 를 통해 다음과 같은 답변을 얻었다. 1. Restriction enzyme (제한효소) biology 제한효소는 특정한 DNA 염기서열을 인식해서 그 지점을 자르는 효소입니다. 원래는 박테리아의 방어 시스템으로, 침입한 바이러스 DNA를 잘라 파괴합니다. 각 효소는 고유한 인식 서열(예: GAATTC)을 가지며, 그 위치에서 DNA를 정확히 절단합니다. 이 특성 덕분에 분자생물학에서는 DNA를 원하는 조각으로 자르는 도구로 사용됩니다. 2. Gel electrophoresis (겔 전기영동) 겔 전기영동은 DNA 조각을 크기별로 분리하는 실험 방법입니다. DNA는 음전하를 띠므로 전기를 걸면 양극 방향으로 이동합니다. 작은 DNA 조각일수록 겔 속을 더 빨리, 더 멀리 이동합니다. 결과적으로 DNA 조각들이 밴드 형태로 나타나며, 크기를 비교·확인할 수 있습니다. One-line summary -Restriction enzymes: molecular scissors that cut DNA at specific sequences -Gel electrophoresis: a method to separate DNA fragments by size Part 1: Benchling & In-silico Gel Art #S.S: Benchling is such a cool platform that we can edit DNA in-silico. I uploaded ‘Lambda DNA.txt’ and managed digest by choosing each enzyme that cuts DNA strand. My initial thought was to see the ladder that each enzyme makes to decide which figure/shape I’d like on my artwork, but the shown result were not a single bar but of multiple layers. — at this stage, I have no idea how to compose an gel art.
A group HW: Design cell-free RNA biosensors RNA toehold switch가 작동하는 원리를 recitation에서 Ana가 설명해 주었는데, 나는 우선 mRNA가 단백질로 translation되는 기전을 이해하고나서 이런 biosensor의 기능을 이해할 수 있었다.
mRNA에 ribosome이 와서 결합한다. 찾아다닌다기 보다는 cell내에서 자유롭게 떠다니던 ribosome이 충돌하여 결합하는 것에 가깝다고. Eukaryotes의 경우는 rough endoplasmic reticulum(RER, 세포소기관organelle)에 거칠어 보이도록 ribosome들이 붙어있어서 단백질을 생산, 반면에 Prokaryotes, 즉, bacteria의 경우 ribosome은 free-floating 상태로만 cytoplasm(세포질)에 존재.
Part A. Conceptual Questions
Answer any NINE of the following questions from Shuguang Zhang: (i.e. you can select two to skip)
How many molecules of amino acids do you take with a piece of 500 grams of meat? (on average an amino acid is ~100 Daltons) Why do humans eat beef but do not become a cow, eat fish but do not become fish? Why are there only 20 natural amino acids? Can you make other non-natural amino acids? Design some new amino acids. Where did amino acids come from before enzymes that make them, and before life started? If you make an α-helix using D-amino acids, what handedness (right or left) would you expect? Can you discover additional helices in proteins? Why are most molecular helices right-handed? Why do β-sheets tend to aggregate? What is the driving force for β-sheet aggregation? Why do many amyloid diseases form β-sheets? Can you use amyloid β-sheets as materials? Design a β-sheet motif that forms a well-ordered structure. Part B: Protein Analysis and Visualization
Part A: SOD1 Binder Peptide Design (From Pranam) Superoxide dismutase 1 (SOD1) is a cytosolic antioxidant enzyme that converts superoxide radicals into hydrogen peroxide and oxygen. In its native state, it forms a stable homodimer and binds copper and zinc.
Mutations in SOD1 cause familial Amyotrophic Lateral Sclerosis (ALS). Among them, the A4V mutation (Alanine → Valine at residue 4) leads to one of the most aggressive forms of the disease. The mutation subtly destabilizes the N-terminus, perturbs folding energetics, and promotes toxic aggregation.
DNA Assembly Answer these questions about the protocol in this week’s lab:
What are some components in the Phusion High-Fidelity PCR Master Mix and what is their purpose? What are some factors that determine primer annealing temperature during PCR? There are two methods from this class that create linear fragments of DNA: PCR, and restriction enzyme digests. Compare and contrast these two methods, both in terms of protocol as well as when one may be preferable to use over the other. How can you ensure that the DNA sequences that you have digested and PCR-ed will be appropriate for Gibson cloning? How does the plasmid DNA enter the E. coli cells during transformation? Describe another assembly method in detail (such as Golden Gate Assembly) Explain the other method in 5 - 7 sentences plus diagrams (either handmade or online). Model this assembly method with Benchling or Asimov Kernel! Asimov Kernel
Living Modified Organism(LMO) Cotton Textiles That Maintain Hygiene and Emit Pleasant Scents
This application envisions a cotton-based textile integrated with genetically engineered microorganisms that are designed to function stably on the fabric surface. These microorganisms would metabolize organic compounds derived from human sweat and skin debris—such as fatty acids, ammonia, and keratin degradation byproducts—thereby maintaining hygiene and reducing odor formation.
Unlike conventional approaches that rely on antimicrobial chemicals or synthetic fragrances, this system would operate as an engineered living material, in which biological activity is deliberately programmed to occur only within a non-natural environment (the textile itself). Conventional perfumes and detergents repeatedly introduce chemically stable, exogenous compounds that may unintentionally interfere with human endocrine systems or disrupt the skin microbiome through persistent and non-selective exposure.
In contrast, genetically engineered microorganisms can be designed to activate only in specific physiological contexts and to simultaneously regulate both the production and degradation of fragrance-related compounds, thereby reducing the structural risks associated with continuous, indiscriminate chemical exposure.
However, this approach also introduces ethical, safety, and governance challenges related to long-term human contact, environmental release, and biological containment.
Governance and Policy Goals for Living Cotton Textiles in KR
:: “To Ensure Biosafety and Biosecurity”
a) Prevent disruption of the human skin microbiome
b) Minimize risks of unintended environmental dissemination
c) Ensure users are fully informed that the product contains living, engineered organisms
option 1) Technical Containment via Textile-Dependent Microbial Design
Purpose:
Biosafety guidance tends to be general (prevent acute harm, prevent release), without microbiome-specific containment requirements for long-duration skin contact consumer products. Thus, require microbiome-protective technical containment for living textiles. Engineered microbes must be textile-restricted in survival, localization, and activity, and demonstrably non-colonizing to skin.
Design:
Self-limiting activity circuit
Fail-safe kill switch triggered off-textile
Localization constraint (stay on fiber, not on skin)
In addition to general biosafety guidelines(합성생물학 육성 방안, 2025. 3. 11.), require application-specific assessment of impacts on the realease of living cotton textiles products intended for prolonged skin contact.
Introduce a mandatory, application-specific Environmental Impact Assessment (EIA) for LMOs, to proactively evaluate environmental risks unique to engineered biological systems, in addition to compliance with existing chemical regulations.
Require an Environmental Impact Assessment prior to deployment for synthetic biology applications with potential environmental exposure
Environmental persistence and degradation pathways
Potential spread beyond intended containment
Interaction with natural ecosystems and microbial communities
Actors:
Developers in Academia/Industry (conduct and submit EIA)
Korea Disease Control and Prevention Agency (KDCA) / Ministry of Science and ICT (MSIT)
Environmental and chemical safety authorities
Risks of Failure and “Success”:
EIA may become a procedural formality without capturing dynamic biological risks
Increased compliance burden may disadvantage smaller research groups or startups
Insufficient data to assess long-term ecological effects
option 3) User-Centered Governance Through Labeling and Disclosure
Purpose:
Consumers are often unaware of the biological mechanisms underlying novel products (esp. GMO/LMO). This option prioritizes transparency and informed consent.
Design:
Clear labeling would indicate the presence of engineered microorganisms, describe their function, and provide guidance for use and disposal.
Actors:
Manufacturers
Consumer protection authorities
Assumptions:
Users will engage with and understand disclosed information
Risks of Failure and “Success”:
Information overload may lead to disregard
Transparency may unintentionally provoke public anxiety or resistance
Does the option:
Option 1
Option 2
Option 3
Enhance Biosecurity
• By preventing incidents
1
2
3
• By helping respond
3
2
1
Foster Lab Safety
• By preventing incident
1
2
n/a
• By helping respond
3
3
n/a
Protect the environment
• By preventing incidents
1
1
3
• By helping respond
3
2
2
Other considerations
• Minimizing costs and burdens to stakeholders
1
3
2
• Feasibility?
3
2
1
• Not impede research
3
3
2
• Promote constructive applications
1
2
2
Drawing on the scoring results, Option 1—technical containment through precisely engineered microbial circuits—emerges as the most feasible and effective governance priority.
It consistently scores highest in preventing biosecurity, laboratory, and environmental incidents while minimizing costs and avoiding unnecessary constraints on research. By embedding safety directly into genetic and circuit-level design, this approach addresses risks at their source rather than relying on external oversight or post hoc responses.
Although it offers limited capacity for incident response, this reflects a deliberate and ethically preferable trade-off; systems designed to fail safely reduce the need for downstream intervention. While this strategy assumes long-term stability of engineered circuits and involves uncertainty around evolutionary dynamics, it provides a practical foundation that can later be complemented by regulatory review and transparency measures, making it the most realistic starting point for governing synthetic biology applications with close human and environmental interfaces.
…ethical concerns that arose:
GMO라는 용어나 개념은 음식과 관련이 있어서인지 매우 친숙하게 느껴졌는데,
미생물을 유전적으로 변형한 LMO라는 개념은 처음 알게 되었다.
또한, 유전자변형미생물의 환경에 대한 영향은 더욱 판단하기 어려울 것으로 보여져서,
앞으로 합성생물학 제품들이 개발됨에 따라 많은 규제와 이슈들이 있을 것으로 생각된다.
Week 2 HW: DNA Read, Write and Edit
Part 0: Basics of Gel Electrophoresis
#S.S: 부트캠프와 Wk2 수업을 들은 후, 과제를 확인했지만 정확한 개념 정리가 안되어 있어서 뭘 진행하는지 이해하기 어려웠다.
따라서 ChatGPT를 활용,
it became clearer once I understood the concept of restriction enzyme(molecular scissors) and gel electrophoresis(DNA size filter).
Prompt: "restriction enzyme biology 와 gel electrophoresis에 대해서 아주 간단히 설명해줘"
를 통해 다음과 같은 답변을 얻었다.
1. Restriction enzyme (제한효소) biology
제한효소는 특정한 DNA 염기서열을 인식해서 그 지점을 자르는 효소입니다.
원래는 박테리아의 방어 시스템으로, 침입한 바이러스 DNA를 잘라 파괴합니다.
각 효소는 고유한 인식 서열(예: GAATTC)을 가지며, 그 위치에서 DNA를 정확히 절단합니다.
이 특성 덕분에 분자생물학에서는 DNA를 원하는 조각으로 자르는 도구로 사용됩니다.
2. Gel electrophoresis (겔 전기영동)
겔 전기영동은 DNA 조각을 크기별로 분리하는 실험 방법입니다.
DNA는 음전하를 띠므로 전기를 걸면 양극 방향으로 이동합니다.
작은 DNA 조각일수록 겔 속을 더 빨리, 더 멀리 이동합니다.
결과적으로 DNA 조각들이 밴드 형태로 나타나며, 크기를 비교·확인할 수 있습니다.
One-line summary
-Restriction enzymes: molecular scissors that cut DNA at specific sequences
-Gel electrophoresis: a method to separate DNA fragments by size
Part 1: Benchling & In-silico Gel Art
#S.S: Benchling is such a cool platform that we can edit DNA in-silico.
I uploaded ‘Lambda DNA.txt’ and managed digest by choosing each enzyme that cuts DNA strand.
My initial thought was to see the ladder that each enzyme makes to decide which figure/shape I’d like on my artwork,
but the shown result were not a single bar but of multiple layers. — at this stage, I have no idea how to compose an gel art.
===
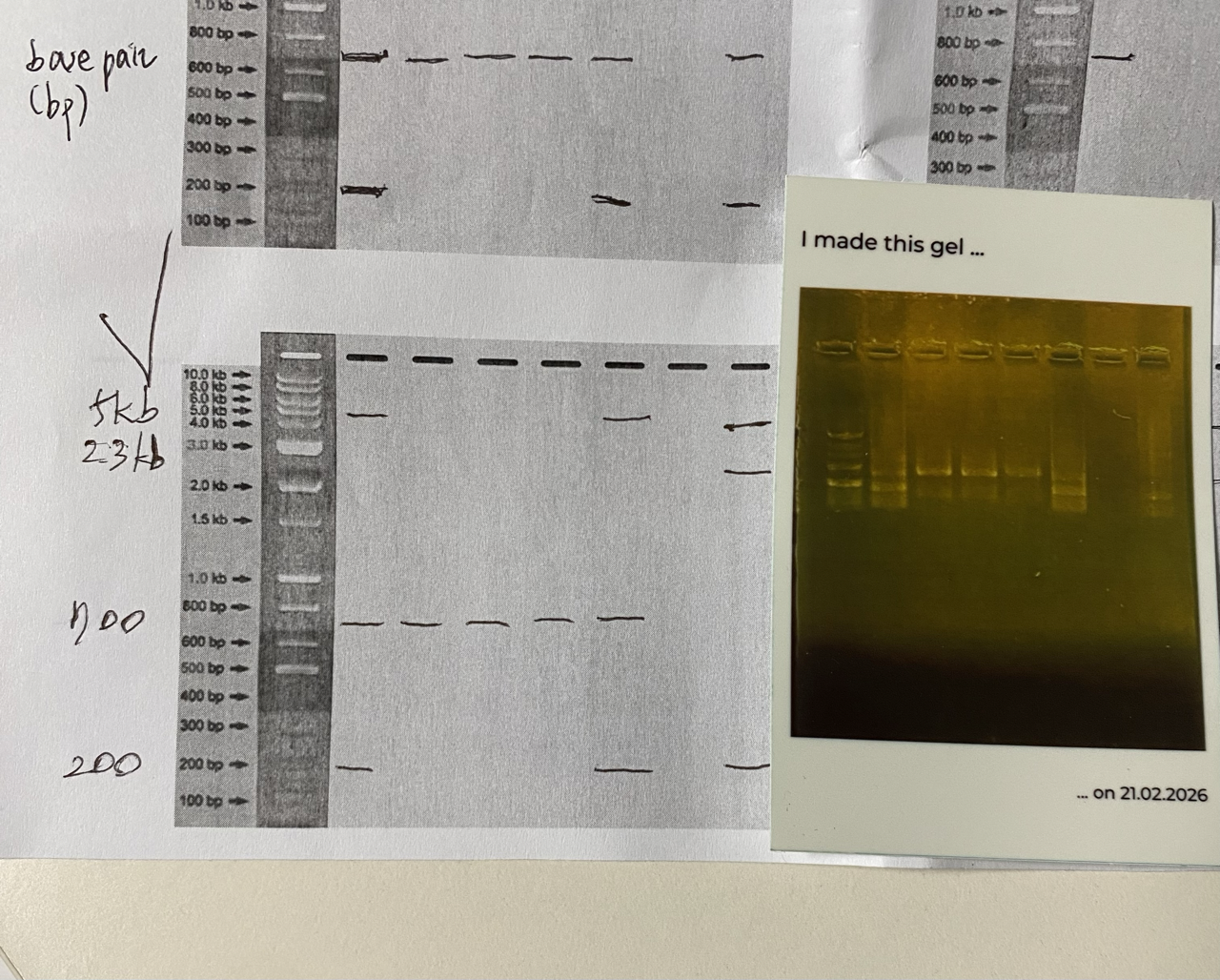
#S.S: Little update on my Gel Art, after attending a session from Designer Cells lab in Songdo, S. Korea.
Ana, one of our TAs, printed out the Gel Electrophoresis result for us, using an instant photo printer. The photo on the right bottom. It also is a sticker !!!
So, my first ever wetlab experience started with how to use pipettes. Next, we moved on to the main dish, how to perform gel electrophoresis.
mix TAE and agarose(powder) and the solution would be microwaved until it is clear, which means it is completely dissolved.
I missed the reasons- to add 1uL of SYBR Safe DNA stain, and ChatGPT explains as below:
Place the comb(or we can put them later-it depends your way of doing it-just avoid forming bubbles) and pour the agarose gel.
When the gel is solidified enough, remove the comb(which generated wells) and start loading the wells with your DNA stands.
There were 4 different sized DNA strands (200bp, 700bp, 2.3kb, 5kb) ready for us, thanks to one of our TAs, Hyunseo.
I designed my gel art saying “Hi”, but the result is shown rather like “HI”, which still, luckily conveys the intended meaning ;D
Hyunseo guessed 5kb and 2.3kb might gone bad? and failed to show on our Gel.
I am presenting my artwork again below just to show off.
The most challenging part of wet lab was actually doing the math and figuring out volumn units. I would refer the summary from ChatGPT for my future references.
Basic Units and Calculations in Biology
1️⃣ Volume Units (Metric System)
All volume units are based on powers of 10.
1 L (liter) = base unit
1 mL = 10⁻³ L
1 µL = 10⁻⁶ L
1 nL = 10⁻⁹ L
Key rule:
Each step (milli → micro → nano) differs by 1000×.
It is simply a very large counting unit for atoms or molecules.
3️⃣ Molarity (M)
Molarity is concentration.
[
M = \frac{\text{moles}}{\text{liters}}
]
Example:
1 M solution = 1 mole dissolved in 1 liter.
Important:
mol = amount
M = concentration
4️⃣ Dilution Formula
[
C_1 V_1 = C_2 V_2
]
Used to prepare diluted solutions from a concentrated stock.
Example:
To make 100 mL of 1 M solution from 10 M stock:
[
(10M)V_1 = (1M)(100mL)
]
V₁ = 10 mL
5️⃣ Key Takeaways
milli, micro, nano differ by 1000× each step
M means mol/L
Most lab work uses µL volumes
DNA amounts are often measured in ng (10⁻⁹ g)
Part 3: DNA Design Challenge
#S.S: now, this is going to be a fun exercise!
3.1. Choose your protein : Fibronectin type III domain-containing protein 10
3.2. Reverse Translate: Protein (amino acid) sequence to DNA (nucleotide) sequence.
'>sp|F2Z333|FND10_HUMAN Fibronectin type III domain-containing protein 10 OS=Homo sapiens OX=9606 GN=FNDC10 PE=1 SV=1 MRAPPLLLLLAACAPPPCAAAAPTPPGWEPTPDAPWCPYKVLPEGPEAGGGRLCFRSPARGFRCQAPGCVLHAPAGRSLRASVLRNRSVLLQWRLAPAAARRVRAFALNCSWRGAYTRFPCERVLLGASCRDYLLPDVHDSVLYRLCLQPLPLRAGPAAAAPETPEPAECVEFTAEPAGMQDIVVAMTAVGGSICVMLVVICLLVAYITENLMRPALARPGLRRHP'
'>reverse translation of sp|F2Z333|FND10_HUMAN Fibronectin type III domain-containing protein 10 OS=Homo sapiens OX=9606 GN=FNDC10 PE=1 SV=1 to a 678 base sequence of consensus codons.
atgmgngcnccnccnytnytnytnytnytngcngcntgygcnccnccnccntgygcngcn
gcngcnccnacnccnccnggntgggarccnacnccngaygcnccntggtgyccntayaar
gtnytnccngarggnccngargcnggnggnggnmgnytntgyttymgnwsnccngcnmgn
ggnttymgntgycargcnccnggntgygtnytncaygcnccngcnggnmgnwsnytnmgn
gcnwsngtnytnmgnaaymgnwsngtnytnytncartggmgnytngcnccngcngcngcn
mgnmgngtnmgngcnttygcnytnaaytgywsntggmgnggngcntayacnmgnttyccn
tgygarmgngtnytnytnggngcnwsntgymgngaytayytnytnccngaygtncaygay
wsngtnytntaymgnytntgyytncarccnytnccnytnmgngcnggnccngcngcngcn
gcnccngaracnccngarccngcngartgygtngarttyacngcngarccngcnggnatg
cargayathgtngtngcnatgacngcngtnggnggnwsnathtgygtnatgytngtngtn
athtgyytnytngtngcntayathacngaraayytnatgmgnccngcnytngcnmgnccn
ggnytnmgnmgncayccn'
3.3. Codon optimization.
The purpose of codon optimisation is to improve expression efficiency. It would not alter the encoded protein but it will redesign the DNA sequence to match a host organism.
'>Improved DNA:
ATGGGCCCCCTGTTCGCCCTGGCCCCCCCCGGCGCCCGCCCCCCCCCCGT
GGGCCCCCCCGAGCCCTGGTGCCTGAAGTTCCGCGGCCGCGCCGGCGGCC
TGCTGCCCCGCGTGGTGCAGCCCGGCGGCAGCGCCCGCGGCTGGCGCTAA
CGCTTCCACGGCTGCCCCCGCGCCGGCGGCCTGCTGATGTGGGGCGCCAA
CGTGCCCGGCGGCCTGGGCTGGGACATCCCCAGCCAGAGCCTGGTGGTGA
CCAGCTGGCGCCCCCGCGCCCCCGAGCCCGACCGCGACGGCGACTACGCC
ACCGCCGACGCCGAGTGCGTGCACGACGCCTGGGGCCTGGTGTGCGTGTA
CTGCTGCGCCAACACCAAGTACGGCCGCTGCGCCGGCGGCACC'
{ref.} https://www.jcat.de/ --- codon optimisation tool, free online access
3.4. You have a sequence! Now what?
Using plasmid? The DNA could be used to produce protein.
3.5. How does it work in nature/biological systems?
#S.S(3.1.): Firstly, I had to recognise the differences between ‘gene vs. protein’.
From my understanding, the central dogma goes like this : DNA(gene) -> RNA(transcription) -> Protein(translation)
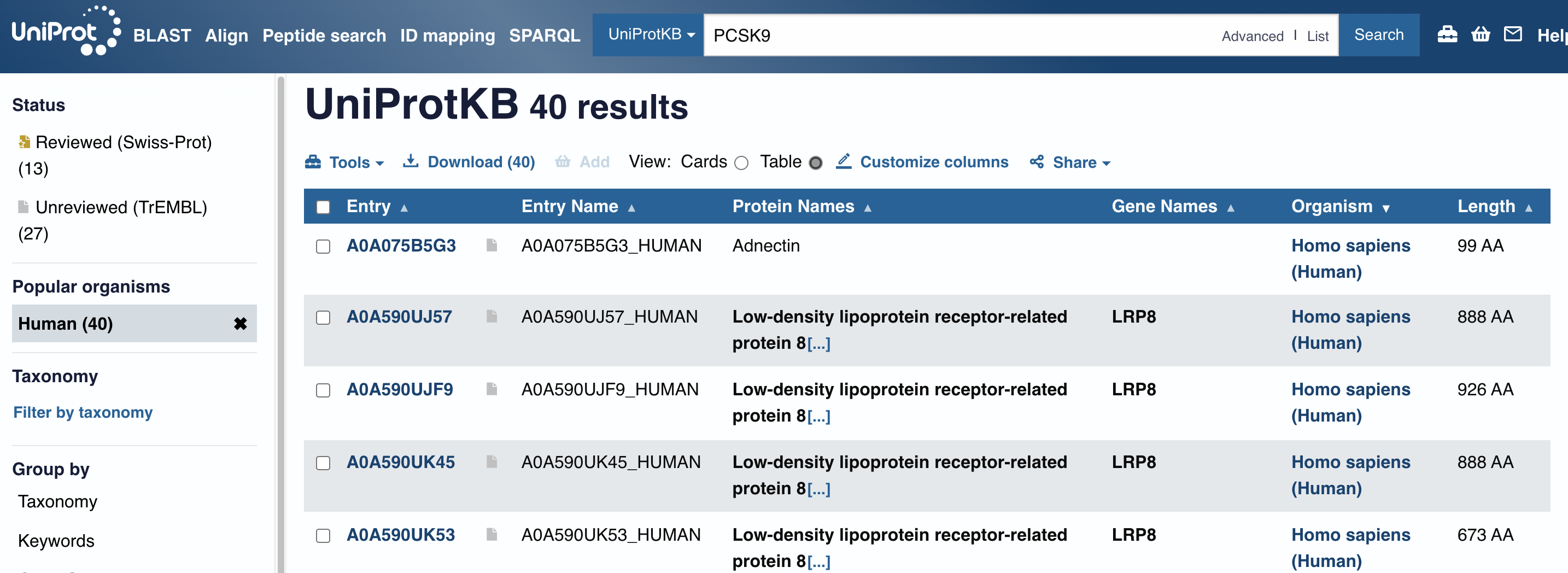
The problem is I am not familiar with names of proteins, thus come up with a gene that I’d encountered in the past! – such as, PCSK9
There are PCSK9 inhibitors in the market that are precsribed to regulated high LDL-C level of Dyslipidemia or FH(Familial Hyperlipidemia).
But, I wasn’t happy with the NCBI lists of protein accession (NP_777596.2, etc.) since I don’t understand what they mean.
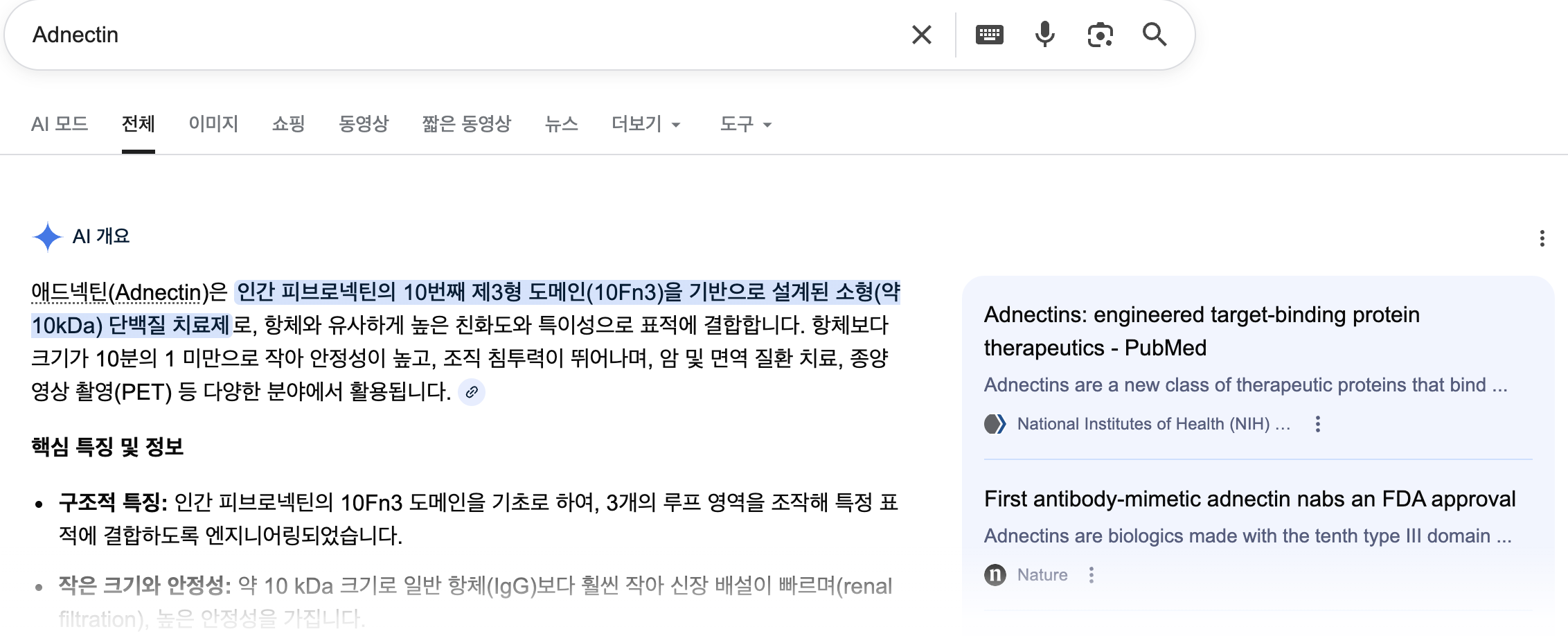
In UniProtKB, however, I came across an interesting name, ‘Adnectin’ - I was curious, why does it have a special name on it?
It is a protein therapeutics, according to below resources, and naturally I decided to look into Human Fibronectin deeper.
{UniProt}
{google}
{PubMed. NCBI}
Part 5: DNA Read/Write/Edit
5.1 DNA Read
(i) What DNA would you want to sequence (e.g., read) and why? This could be DNA related to human health (e.g. genes related to disease research), environmental monitoring (e.g., sewage waste water, biodiversity analysis), and beyond (e.g. DNA data storage, biobank).
I would like to sequence my own DNA, and especially check on the currently available cancer gene test.
(ii) In lecture, a variety of sequencing technologies were mentioned. What technology or technologies would you use to perform sequencing on your DNA and why? Also answer the following questions:
Is your method first-, second- or third-generation or other? How so?
NGS(second) method would fit for my own DNA read, since it's fast enough and relatively cheaper that the third-gen method.
5.2 DNA Write
(i) What DNA would you want to synthesize (e.g., write) and why? These could be individual genes, clusters of genes or genetic circuits, whole genomes, and beyond. As described in class thus far, applications could range from therapeutics and drug discovery (e.g., mRNA vaccines and therapies) to novel biomaterials (e.g. structural proteins), to sensors (e.g., genetic circuits for sensing and responding to inflammation, environmental stimuli, etc.), to art (DNA origamis). If possible, include the specific genetic sequence(s) of what you would like to synthesize! You will have the opportunity to actually have Twist synthesize these DNA constructs! :)
I remember writing DNA of a bateriophage to used it as a therapeutic.
(ii) What technology or technologies would you use to perform this DNA synthesis and why? Also answer the following questions:
What are the essential steps of your chosen sequencing methods?
What are the limitations of your sequencing method (if any) in terms of speed, accuracy, scalability?
5.3 DNA Edit
(i) What DNA would you want to edit and why? In class, George shared a variety of ways to edit the genes and genomes of humans and other organisms. Such DNA editing technologies have profound implications for human health, development, and even human longevity and human augmentation. DNA editing is also already commonly leveraged for flora and fauna, for example in nature conservation efforts, (animal/plant restoration, de-extinction), or in agriculture (e.g. plant breeding, nitrogen fixation). What kinds of edits might you want to make to DNA (e.g., human genomes and beyond) and why?
I'm more interested in utilizing RNA than editing DNA form.
(ii) What technology or technologies would you use to perform these DNA edits and why? Also answer the following questions:
How does your technology of choice edit DNA? What are the essential steps?
What preparation do you need to do (e.g. design steps) and what is the input (e.g. DNA template, enzymes, plasmids, primers, guides, cells) for the editing?
What are the limitations of your editing methods (if any) in terms of efficiency or precision?
Week 3 HW: Lab Automation
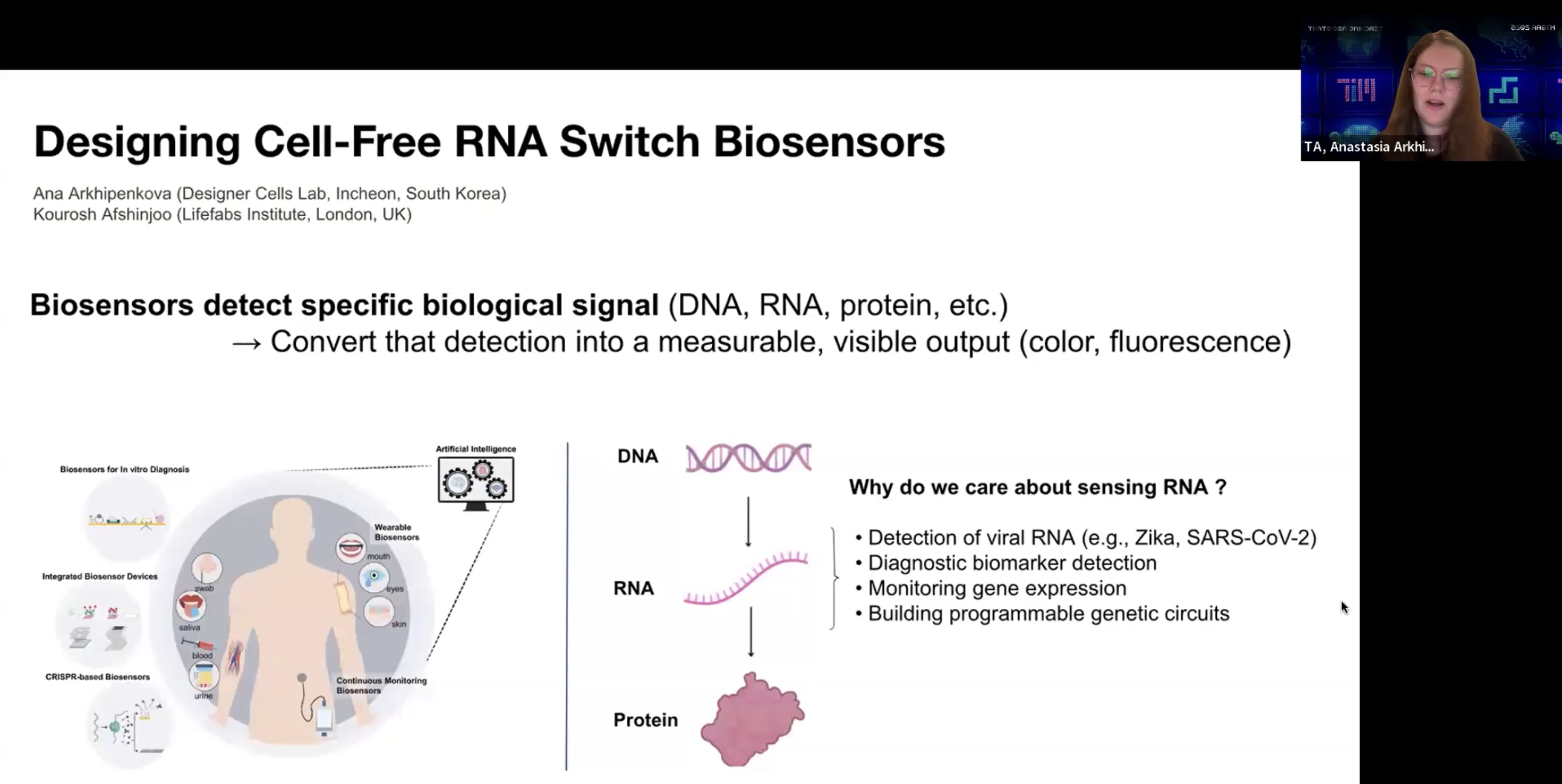
A group HW: Design cell-free RNA biosensors
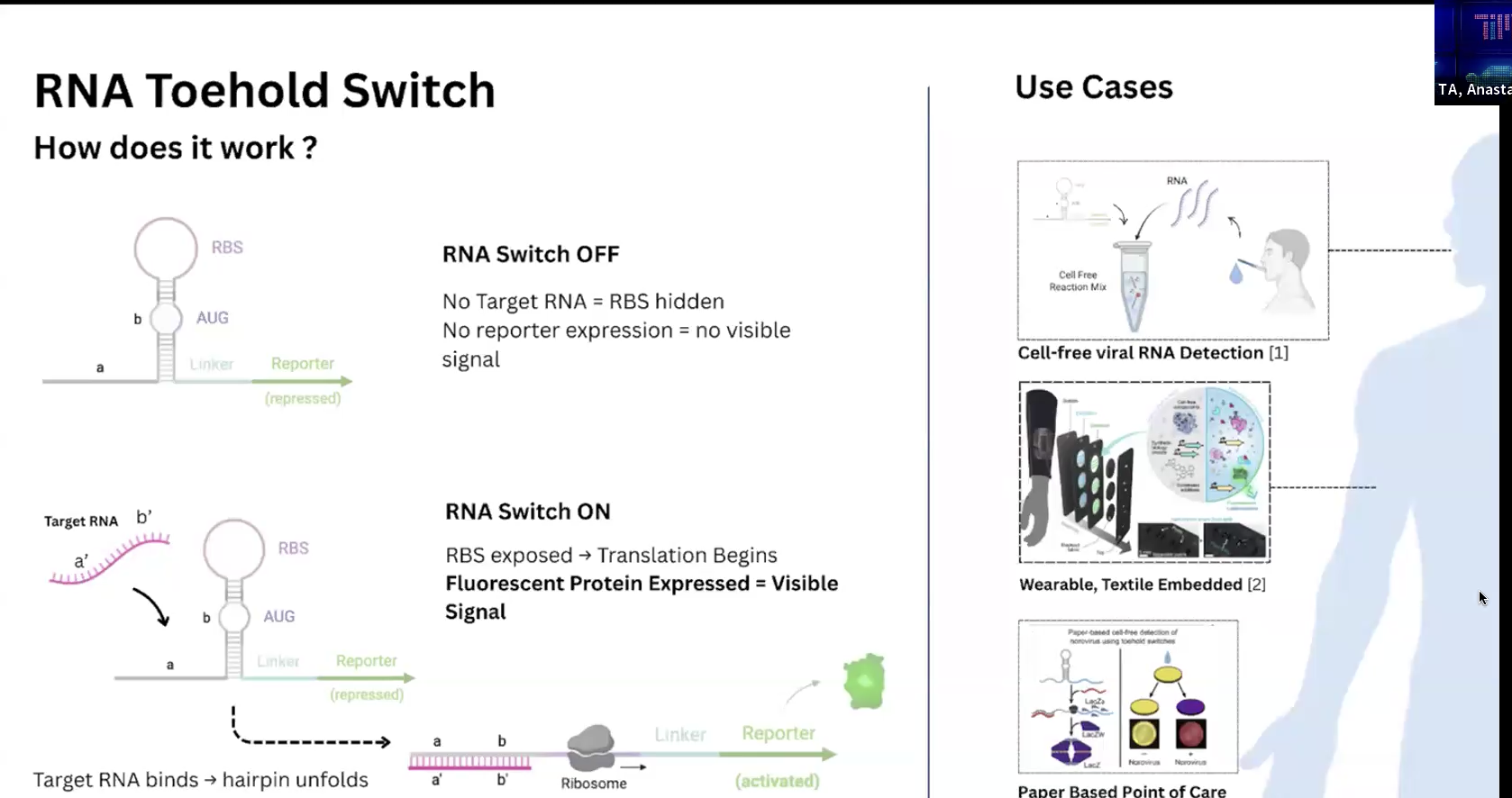
RNA toehold switch가 작동하는 원리를 recitation에서 Ana가 설명해 주었는데,
나는 우선 mRNA가 단백질로 translation되는 기전을 이해하고나서 이런 biosensor의 기능을 이해할 수 있었다.
mRNA에 ribosome이 와서 결합한다. 찾아다닌다기 보다는 cell내에서 자유롭게 떠다니던 ribosome이 충돌하여 결합하는 것에 가깝다고.
Eukaryotes의 경우는 rough endoplasmic reticulum(RER, 세포소기관organelle)에 거칠어 보이도록 ribosome들이 붙어있어서 단백질을 생산,
반면에 Prokaryotes, 즉, bacteria의 경우 ribosome은 free-floating 상태로만 cytoplasm(세포질)에 존재.
요새 기본개념 학습에 도움을 받고 있는 biology podcast(https://podcasts.apple.com/kr/podcast/teach-me-biology/id1525760514?l=en-GB&i=1000486622621)의 기억을 떠올리면,
Eukaryotes/animal cell에서 RER은 protein을 생산하고, smooth ER(w/o ribosome)은 lipid를 생산한다.
free ribosome은 세포 내부에서 사용할 단백질을 생산한다고 한다.
추가로, mitochondria는 release energy & produce ATP. 그리고 risosome은 pathogen이나 dead cell, etc를 없애는 역할을 하는 것 같다.
mRNA에는 RBS(ribosome binding site)가 있어서 ribosome이 부착되고 번역을 시작할 수 있다.
이 RBS는 bacteria에서는 Shine–Dalgarno sequence라고 불린다고. 시작코돈 AUG 앞에 위치한다.
하나 더 몰랐던 개념, RNA는 folding landscape, 동적으로 접혔다 풀렸다 하는 구조집합을 보이는데,
toehold switch에 나온 hairpin 구조는 rna 염기 가닥이 서로 연결되지 않은 loop, 그리고 일부가 상보적으로 연결된 stem으로 구성된 loop-stem 구조이다.
모든 RNA가 이렇게 모양이 생긴건 아니고 염기서열에 따라서 동적인듯 하다. 그렇지만 박테리아는 비교적 mRNA 구조가 단순하므로 의도적으로 hairpin구조로 만들어서 활용하는 듯 하다.
Finally, toehold switch에서 hairpin구조의 RNA switch는 평소(off status) RBS(is hidden within stem)와 AUG를 가림으로써 ribosome의 접근을 막아서 protein translation을 막는다. When switched-on(target RNA가 접근 시, it will bind and zip-open the stem followed by strand displacement(헤어핀 붕괴), in the end, ribosome can start translation process.
약간 이해가 안되었던 부분은 RNA가 지금 2종이 나온다는 거였는데,
chatGPT에게 질문하려고 개념을 써보다 보니 얼추 아래와 같이 이해가 되었다.
toehold switch는 bacteria의 RNA가 표현되기를 hairpin 구조를 취하도록 re-engineered it.
hairpin 상태에 있다가, target RNA(외부의 phage같은 것들의 input 등)가 들어오면 protein을 만들도록 설계.
ChapGPT의 도움을 받아 재정리해보면,
Bacterial mRNA의 5’UTR (AUG가 붙은 앞쪽 부분으로 예상.)을 hairpin 구조로 합성한 것!
(자연적으로 형성된 switch로 Riboswitch라는게 존재함)
#s.s: At this stage, I wondered what it means to be ‘cell-free’ and why it matters.
Below is an examplary table from ChatGPT to help my understanding.
It seems cell-free system could partially avoid GMO-regulatory risks or make things easier!
(Which helps me narrow down the desired setting for my final project, i guess)
Category (항목)
Cell-based (세포 기반)
Cell-free (세포 비의존)
Viability (생존)
Required (필요)
Not required (불필요)
Proliferation (증식)
Present (있음)
Absent (없음)
Regulation (규제)
GMO-regulated (GMO 규제)
Relatively relaxed (상대적 완화)
Response Speed (반응 속도)
Slow (느림)
Fast (빠름)
Circuit Complexity (회로 복잡성)
High (높음)
Low (낮음)
Field Deployability (현장 적용성)
Low (낮음)
High (높음)
Brainstorming on RNA Biosensor, assisted by ChatGPT
Target AMR(Antimicrobial Resistance) gene
Development of resistance for antibiotics seemed like a major issue in the medical field. It is routinely tested in hospital already with AST (antimicrobial susceptibility testing). Also, molecular testing for AMR gene is performed as an early guidance. Thus, RNA biosensor have potential to be the fastest detection tool replacing the current molecular test in urgent scenarios, although it might lack precision compared to AST.
Microneedle patch for Vaccination
Could a biosensor detect humoral immunity after vaccine innoculation? It is possible to check interstitial fluid (ISF) on one’s skin, but immunity is proved mostly by protein(as an antibody) and RNA sensor is not the best option to detect a vaccination result… so, I would dump this idea.
Virus RNA biosensor
Back to the familiar idea that I was introduced from the class, which is the detection of RNA viruses.
RNA is THE genetic material of a pathogene and the sensor have potential to complement or partially replace PCR.
In addition, it would have significant societal value - high impact on public health.
So, I’m thinking “broad coronavirus biosensor” as a front-line detector.
#GPT: “Detecting viral RNA directly is important because
The presence of viral RNA typically indicates an active infection, rather than past exposure or residual immunity.
In regarding Coronaviruses, related viruses such as SARS-CoV (2003) and MERS-CoV (2012) had already caused outbreaks and the coronavirus family was already known. However, SARS-CoV-2(COVID-19) was a newly emerged zoonotic virus that humans had not previously encountered at population scale. So COVID-19 was not created “from nothing,” but it represented a new spillover event of a genetically distinct virus within a known viral family.
— A biosensor targeting conserved coronavirus regions could potentially detect certain future zoonotic spillovers within the CoV family.
But:
It would not detect unrelated viral families.
It would not replace sequencing.
It must be part of an integrated surveillance system.
Week 4 HW: Protein Design Part I
Part A. Conceptual Questions
Answer any NINE of the following questions from Shuguang Zhang: (i.e. you can select two to skip)
How many molecules of amino acids do you take with a piece of 500 grams of meat? (on average an amino acid is ~100 Daltons)
Why do humans eat beef but do not become a cow, eat fish but do not become fish?
Why are there only 20 natural amino acids?
Can you make other non-natural amino acids? Design some new amino acids.
Where did amino acids come from before enzymes that make them, and before life started?
If you make an α-helix using D-amino acids, what handedness (right or left) would you expect?
Can you discover additional helices in proteins?
Why are most molecular helices right-handed?
Why do β-sheets tend to aggregate?
What is the driving force for β-sheet aggregation?
Why do many amyloid diseases form β-sheets?
Can you use amyloid β-sheets as materials?
Design a β-sheet motif that forms a well-ordered structure.
Part B: Protein Analysis and Visualization
In this part of the homework, you will be using online resources and 3D visualization software to answer questions about proteins. Pick any protein (from any organism) of your interest that has a 3D structure and answer the following questions:
Briefly describe the protein you selected and why you selected it.
Identify the amino acid sequence of your protein.
How long is it? What is the most frequent amino acid? You can use this Colab notebook to count the frequency of amino acids.
How many protein sequence homologs are there for your protein? Hint: Use Uniprot’s BLAST tool to search for homologs.
Does your protein belong to any protein family?
Identify the structure page of your protein in RCSB
When was the structure solved? Is it a good quality structure? Good quality structure is the one with good resolution. Smaller the better (Resolution: 2.70 Å)
Are there any other molecules in the solved structure apart from protein?
Does your protein belong to any structure classification family?
Open the structure of your protein in any 3D molecule visualization software:
PyMol Tutorial Here (hint: ChatGPT is good at PyMol commands)
Visualize the protein as “cartoon”, “ribbon” and “ball and stick”.
Color the protein by secondary structure. Does it have more helices or sheets?
Color the protein by residue type. What can you tell about the distribution of hydrophobic vs hydrophilic residues?
Visualize the surface of the protein. Does it have any “holes” (aka binding pockets)?
Part C. Using ML-Based Protein Design Tools
Week 5 HW: Protein Design Part II
Part A: SOD1 Binder Peptide Design (From Pranam)
Superoxide dismutase 1 (SOD1) is a cytosolic antioxidant enzyme that converts superoxide radicals into hydrogen peroxide and oxygen. In its native state, it forms a stable homodimer and binds copper and zinc.
Mutations in SOD1 cause familial Amyotrophic Lateral Sclerosis (ALS). Among them, the A4V mutation (Alanine → Valine at residue 4) leads to one of the most aggressive forms of the disease. The mutation subtly destabilizes the N-terminus, perturbs folding energetics, and promotes toxic aggregation.
Your challenge:
Design short peptides that bind mutant SOD1.
Then decide which ones are worth advancing toward therapy.
You will use three models developed in our lab:
PepMLM: target sequence-conditioned peptide generation via masked language modeling
PeptiVerse: therapeutic property prediction
moPPIt: motif-specific multi-objective peptide design using Multi-Objective Guided Discrete Flow Matching (MOG-DFM)
Part 1: Generate Binders with PepMLM
Begin by retrieving the human SOD1 sequence from UniProt (P00441) and introducing the A4V mutation.
Using the PepMLM Colab linked from the HuggingFace PepMLM-650M model card:
Generate four peptides of length 12 amino acids conditioned on the mutant SOD1 sequence.
To your generated list, add the known SOD1-binding peptide FLYRWLPSRRGG for comparison.
Record the perplexity scores that indicate PepMLM’s confidence in the binders.
Part 2: Evaluate Binders with AlphaFold3
Navigate to the AlphaFold Server: alphafoldserver.com
For each peptide, submit the mutant SOD1 sequence followed by the peptide sequence as separate chains to model the protein-peptide complex.
Record the ipTM score and briefly describe where the peptide appears to bind. Does it localize near the N-terminus where A4V sits? Does it engage the β-barrel region or approach the dimer interface? Does it appear surface-bound or partially buried?
In a short paragraph, describe the ipTM values you observe and whether any PepMLM-generated peptide matches or exceeds the known binder.
Part 3: Evaluate Properties of Generated Peptides in the PeptiVerse
Structural confidence alone is insufficient for therapeutic development. Using PeptiVerse, let’s evaluate the therapeutic properties of your peptide! For each PepMLM-generated peptide:
Paste the peptide sequence.
Paste the A4V mutant SOD1 sequence in the target field.
Check the boxes
Predicted binding affinity
Solubility
Hemolysis probability
Net charge (pH 7)
Molecular weight
Compare these predictions to what you observed structurally with AlphaFold3. In a short paragraph, describe what you see. Do peptides with higher ipTM also show stronger predicted affinity? Are any strong binders predicted to be hemolytic or poorly soluble? Which peptide best balances predicted binding and therapeutic properties?
Choose one peptide you would advance and justify your decision briefly.
Part 4: Generate Optimized Peptides with moPPIt
Now, move from sampling to controlled design. moPPIt uses Multi-Objective Guided Discrete Flow Matching (MOG-DFM) to steer peptide generation toward specific residues and optimize binding and therapeutic properties simultaneously. Unlike PepMLM, which samples plausible binders conditioned on just the target sequence, moPPIt lets you choose where you want to bind and optimize multiple objectives at once.
Open the moPPit Colab linked from the HuggingFace moPPIt model card
Make a copy and switch to a GPU runtime.
In the notebook:
Paste your A4V mutant SOD1 sequence.
Choose specific residue indices on SOD1 that you want your peptide to bind (for example, residues near position 4, the dimer interface, or another surface patch).
Set peptide length to 12 amino acids.
Enable motif and affinity guidance (and solubility/hemolysis guidance if available). Generate peptides.
After generation, briefly describe how these moPPit peptides differ from your PepMLM peptides. How would you evaluate these peptides before advancing them to clinical studies?
Part C: Final Project: L-Protein Mutants
High level summary: The objective of this assignment is to improve the stability and auto-folding of the lysis protein of a MS2-phage. This mechanism is key to the understanding of how phages can potentially solve antibiotic-resistance.
Week 6 HW: Genetic Circuits - Part I
DNA Assembly
Answer these questions about the protocol in this week’s lab:
What are some components in the Phusion High-Fidelity PCR Master Mix and what is their purpose?
What are some factors that determine primer annealing temperature during PCR?
There are two methods from this class that create linear fragments of DNA: PCR, and restriction enzyme digests. Compare and contrast these two methods, both in terms of protocol as well as when one may be preferable to use over the other.
How can you ensure that the DNA sequences that you have digested and PCR-ed will be appropriate for Gibson cloning?
How does the plasmid DNA enter the E. coli cells during transformation?
Describe another assembly method in detail (such as Golden Gate Assembly)
Explain the other method in 5 - 7 sentences plus diagrams (either handmade or online).
Model this assembly method with Benchling or Asimov Kernel!
Asimov Kernel
Create a Repository for your work
Create a blank Notebook entry to document the homework and save it to that Repository
Explore the devices in the Bacterial Demos Repo to understand how the parts work together by running the Simulator on various examples, following the instructions for the simulator found in the “Info” panel (click the “i” icon on the right to open the Info panel)
Create a blank Construct and save it to your Repository
Recreate the Repressilator in that empty Construct by using parts from the Characterized Bacterial Parts repository
Search the parts using the Search function in the right menu
Drag and drop the parts into the Construct
Confirm it works as expected by running the Simulator (“play” button) and compare your results with the Repressilator Construct found in the Bacterial Demos repository
Document all of this work in your Notebook entry - you can copy the glyph image and the simulator graphs, and paste them into your Notebook
Build three of your own Constructs using the parts in the Characterized Bacterials Parts Repo
Explain in the Notebook Entry how you think each of the Constructs should function
Run the simulator and share your results in the Notebook Entry
If the results don’t match your expectations, speculate on why and see if you can adjust the simulator settings to get the expected outcome