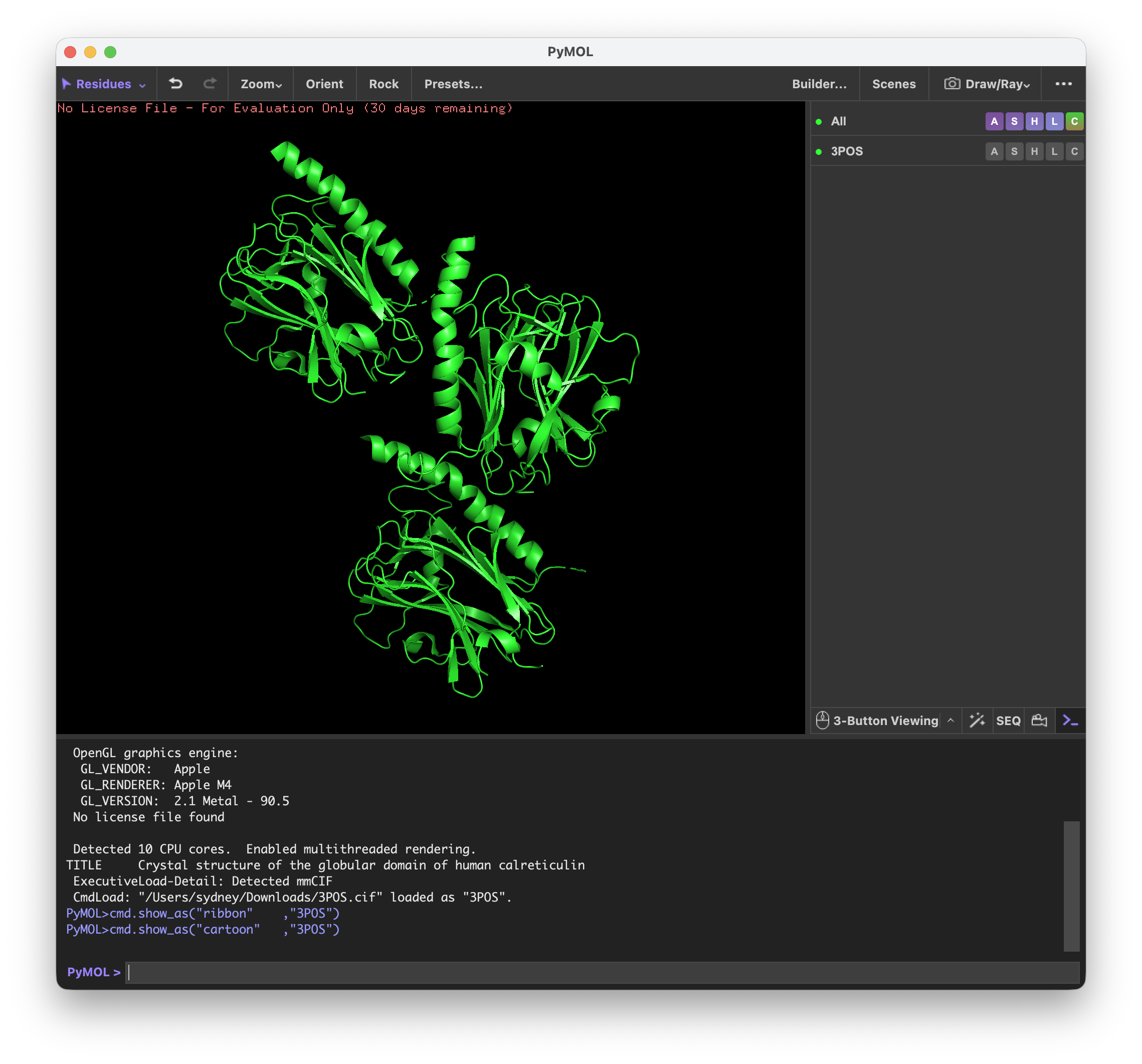
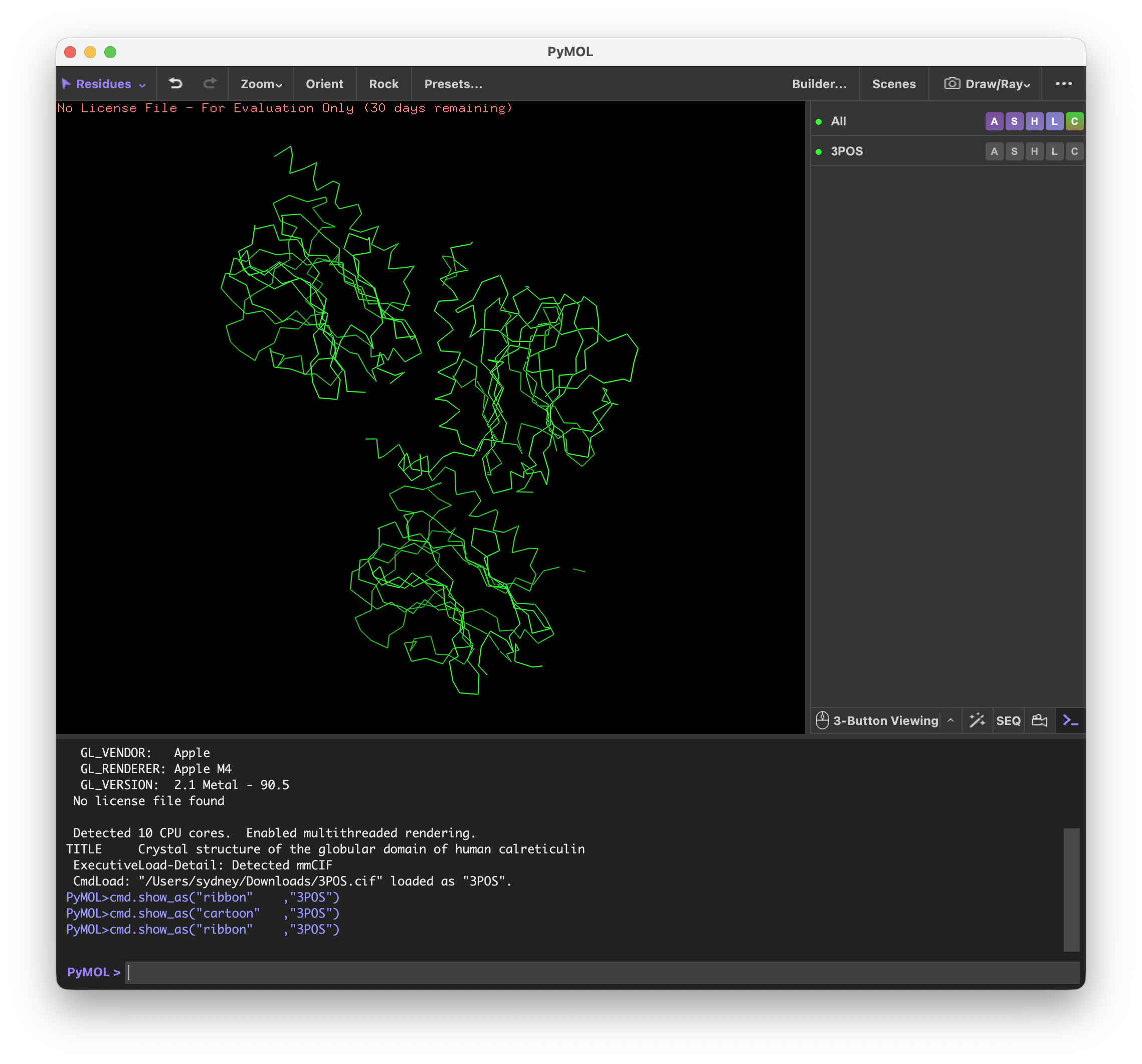
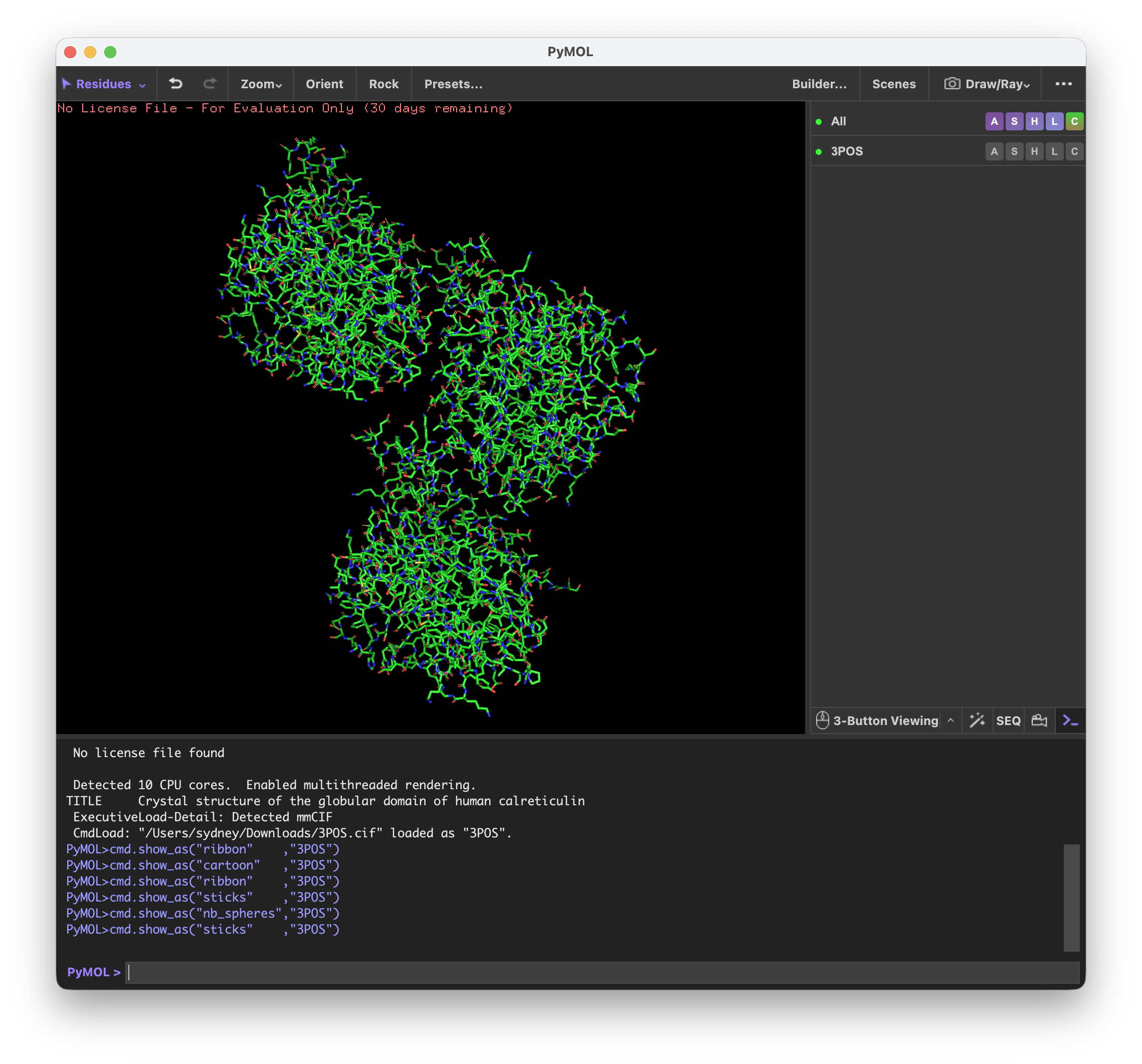
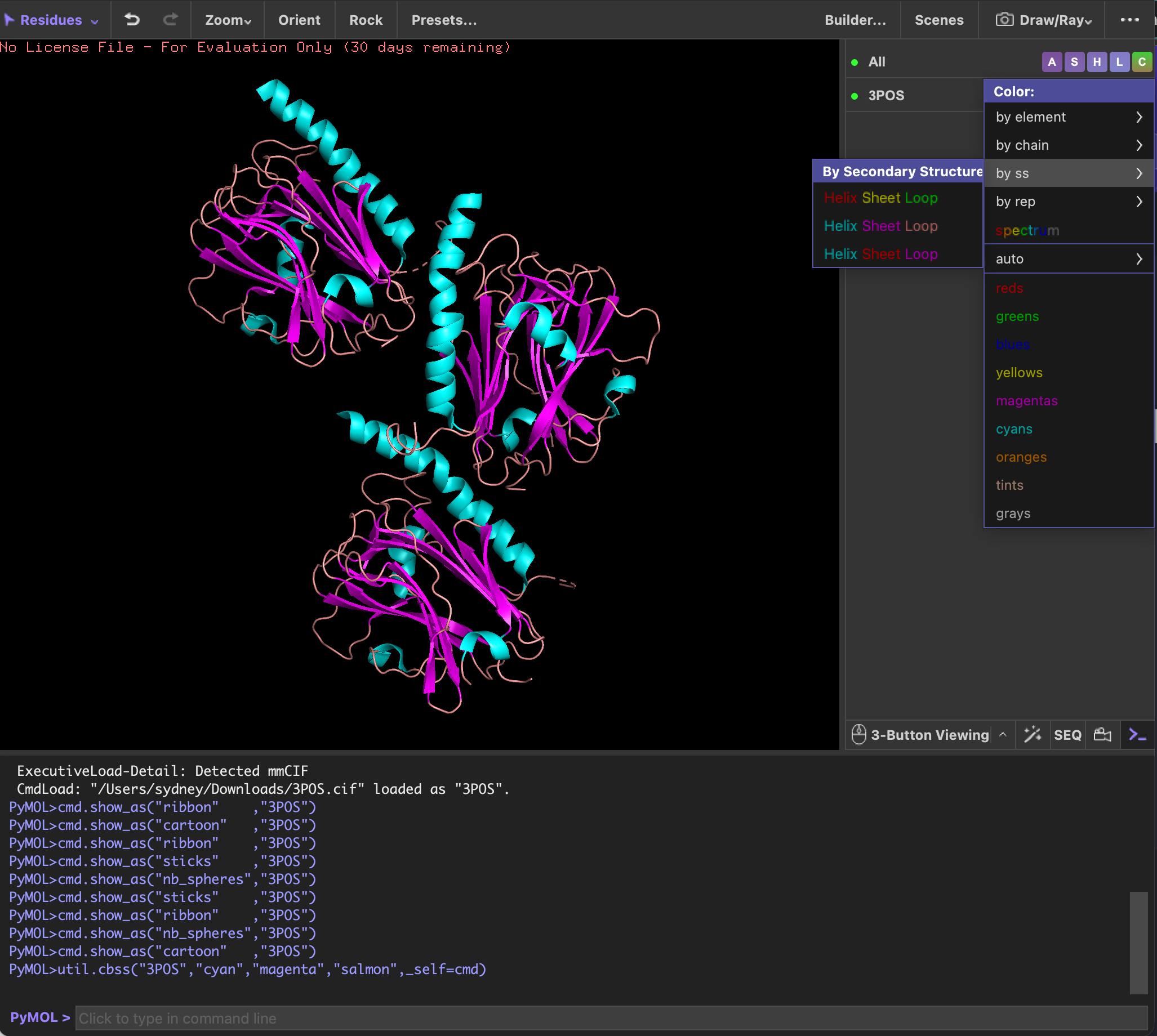
Homework
Weekly homework submissions:
Week 1: Principles and Practices
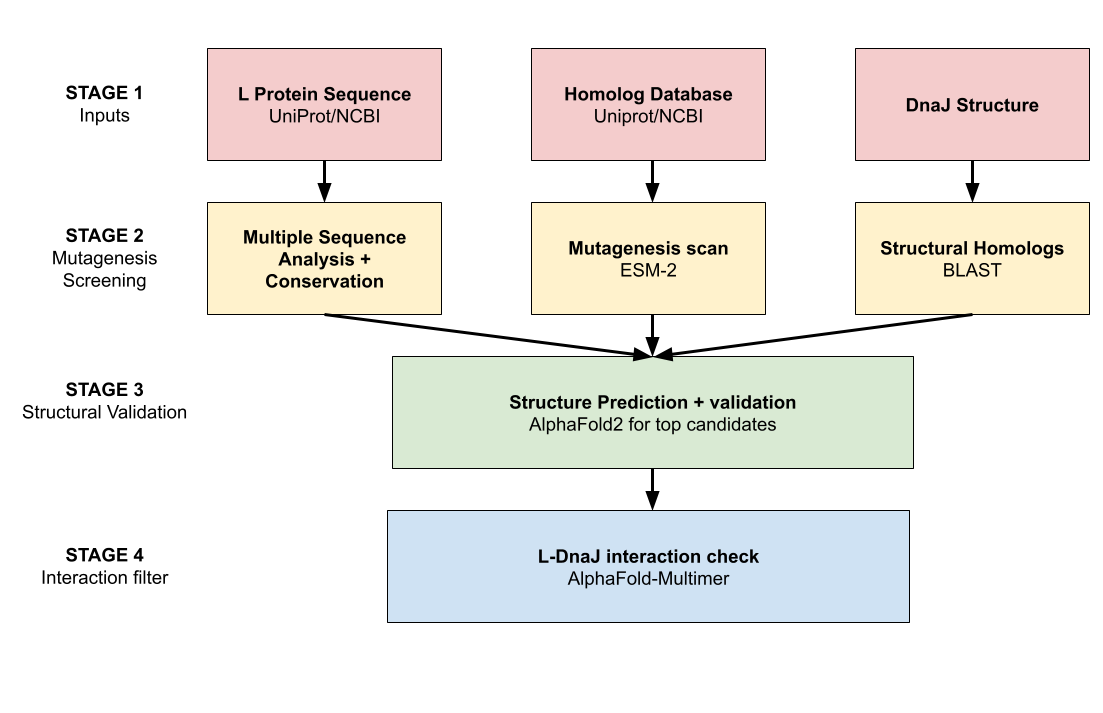
Class Assignment Project Idea Chronic wounds and surgical site infections affect millions of patients and cost heathcare systems tens of billions of dollars annually, yet closure devices often remain as passive stitches that do not actively orchestrate local immunity or regeneration [1][2]. Drug-eluting sutures have shown that suture material can safely deliver local therapeutics, but current designs provide only finite, non-adaptive release of single agents such as antibiotics or growth factors [3][4]. Cell-filled sutures packed with mesenchymal stem cells already demonstrate that viable cells can be integrated into suture structures and enhance healing, but these cells are unmodified and lack controllable, multi-functional outputs [5]. Separately, engineered combinatorial cell devices in fiber-like formats can secrete optimized cocktails of growth factors to accelerate wound and bone repair, but they are not load-bearing sutures and do not address infection or scar modulation at the incision line [6].
Week 2: DNA Read, Write, and Edit
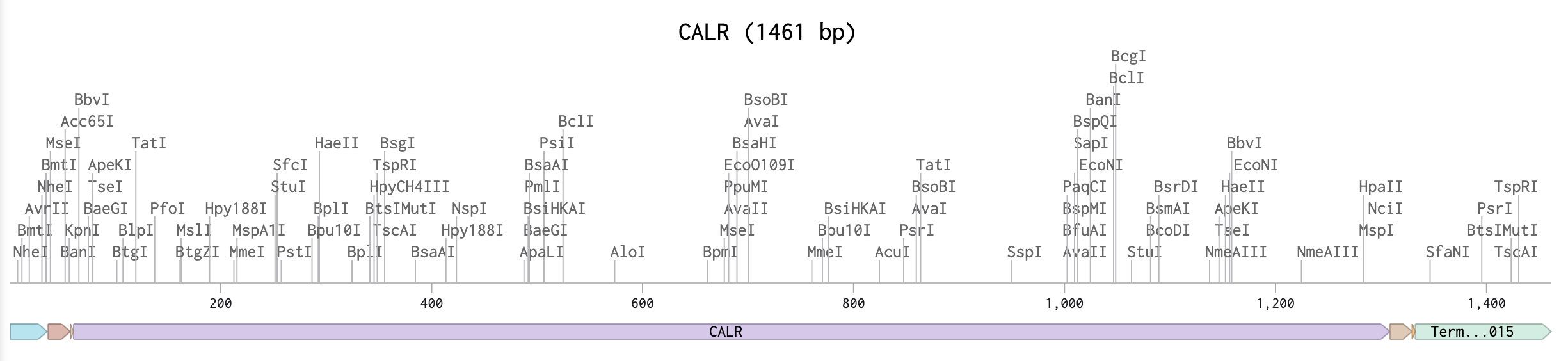
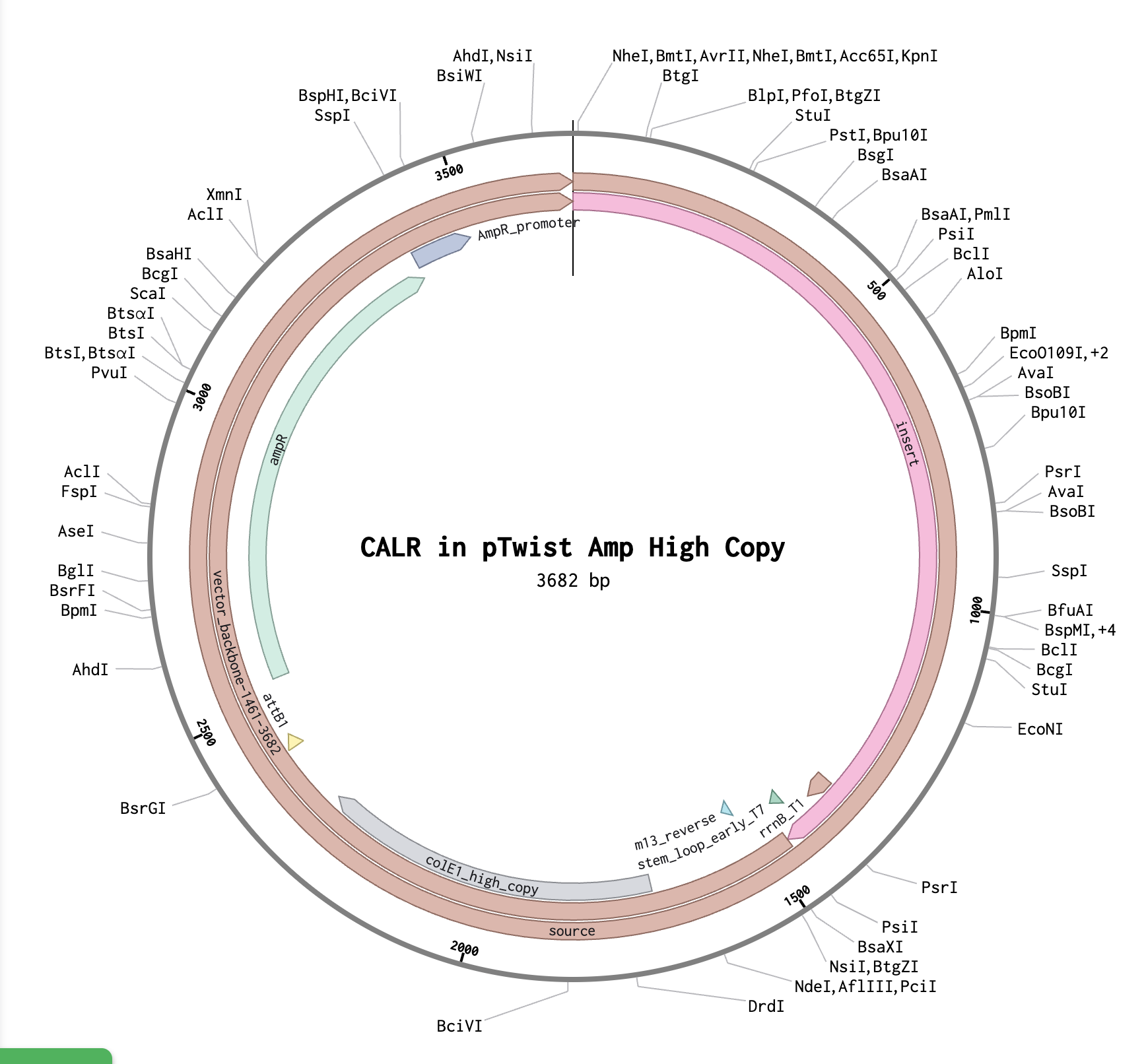
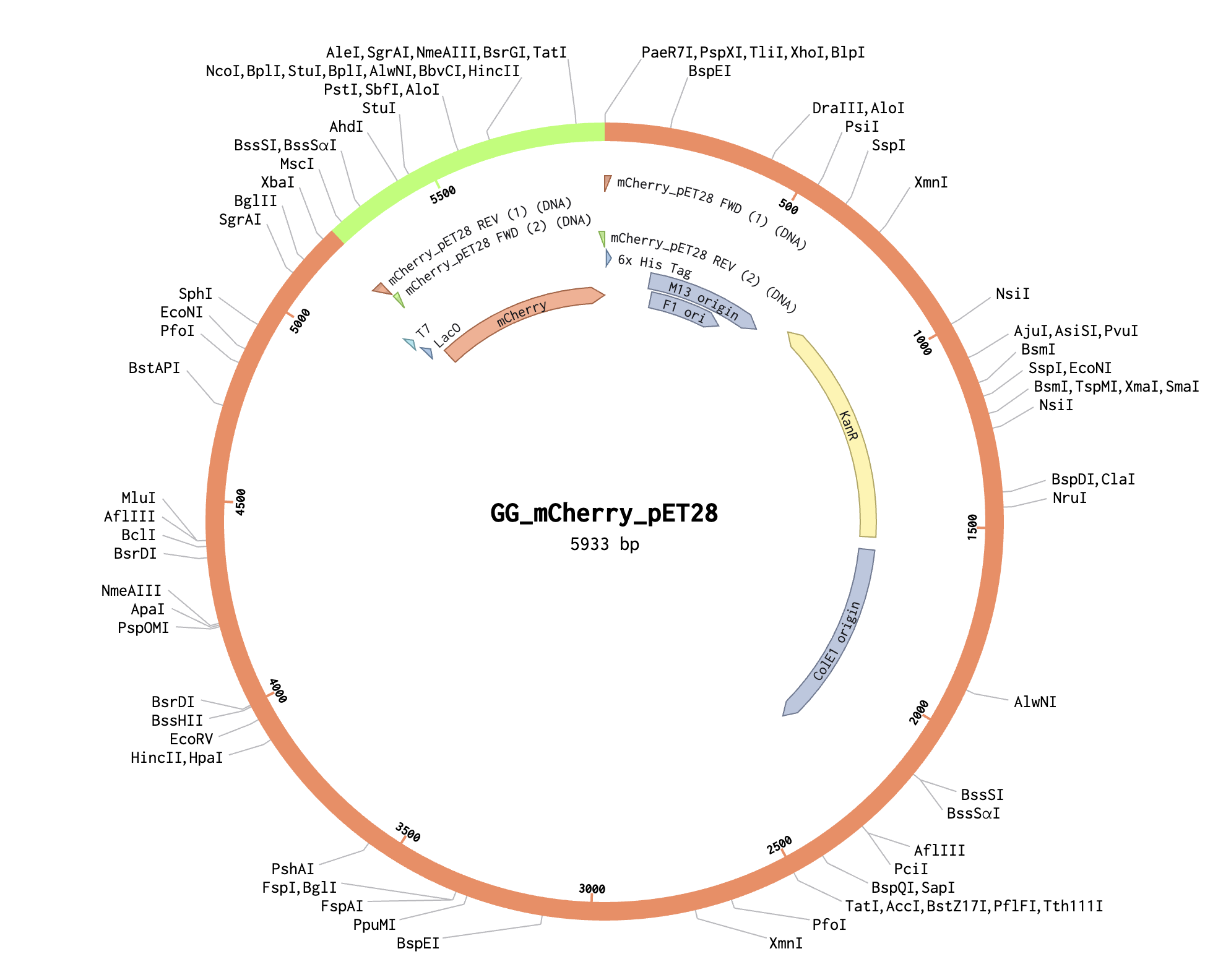
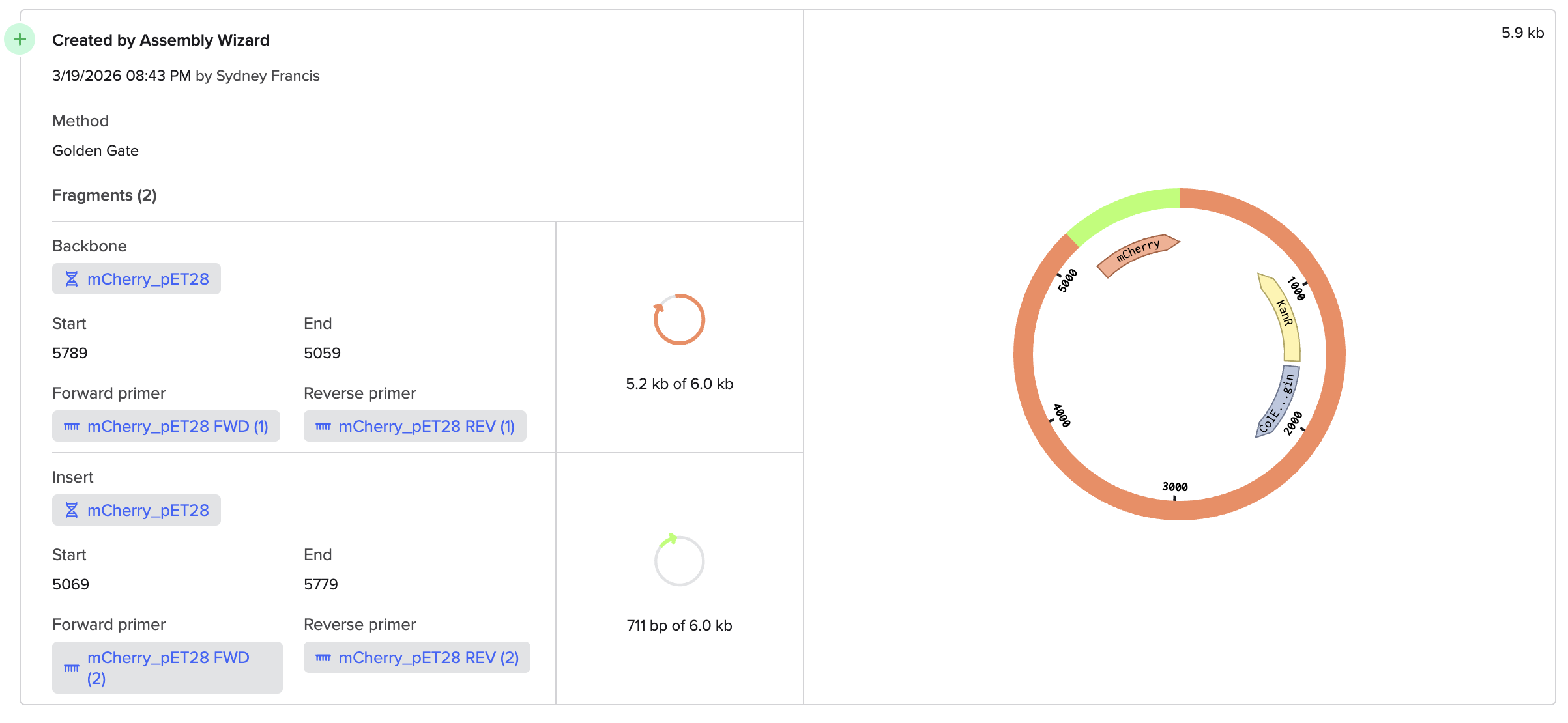
Benchling and In-Silico Gel Art Simulate Restriction Enzyme Digest I found this process quite intuitive, as I’ve done similar simulations with the application SnapGene, but it was interesting to notice the small interface differences between the two!
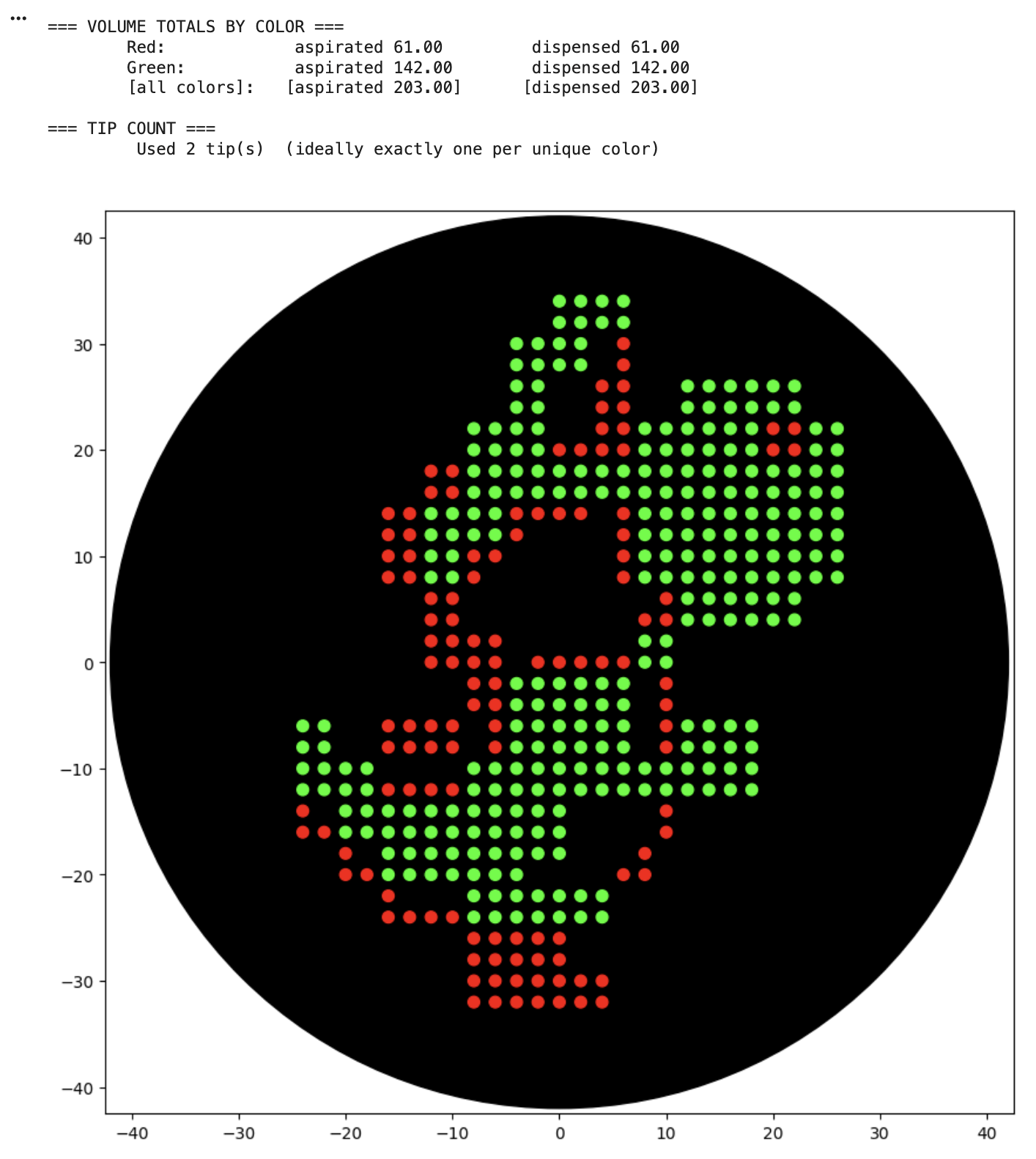
Python script for Opentrons artwork For the art portion of this week’s assignment, I decided to code Yoshi from Super Mario Brothers since the Designer Cells node only had the red and green colors. I used this photo as reference. From there, I started to code for the Opentron automation.
Conceptual Questions (Question 1) A 500g piece of meat would weight about 3.011x1026 Daltons, and since each amino acid is equal to about 100 Daltons, that would mean that by consuming this piece of meat, you are consuming 3.011x1024 amino acids. (Questions 2) When we eat sources of meat, we physically and enzymatically break down the proteins into amino acids, fatty acids, and sugars, which in turn are used to provide energy to our bodies.
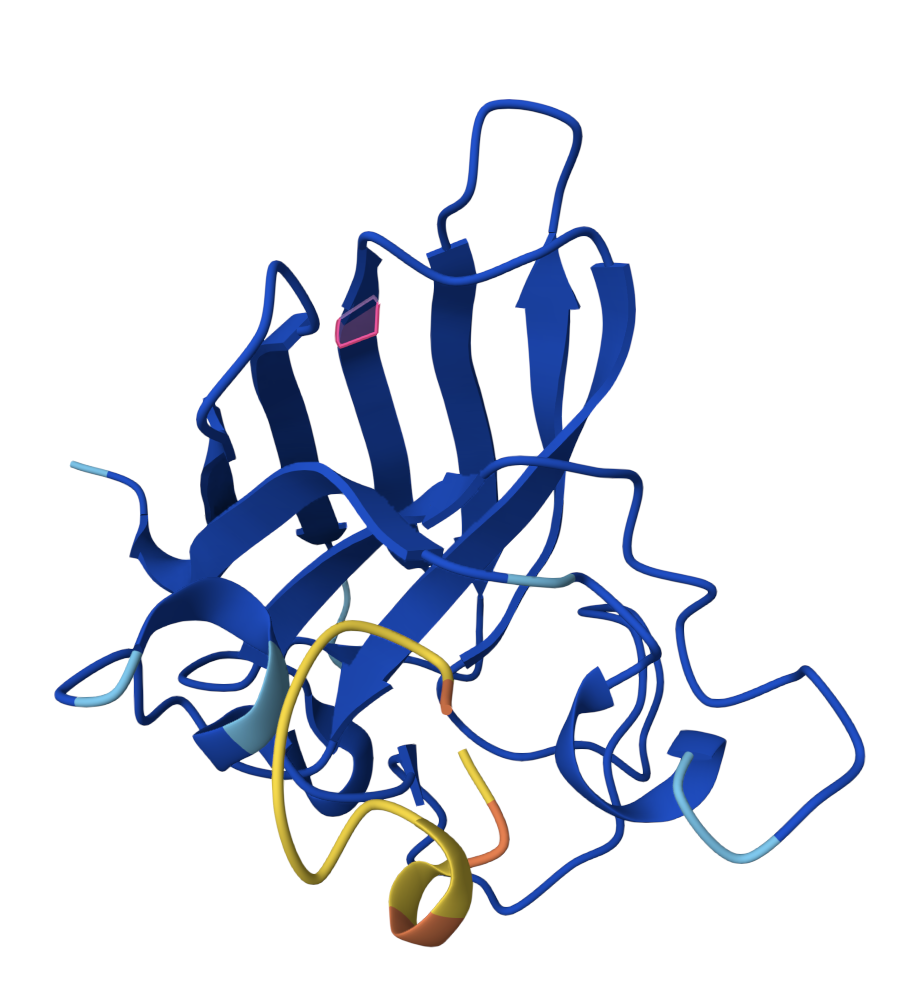
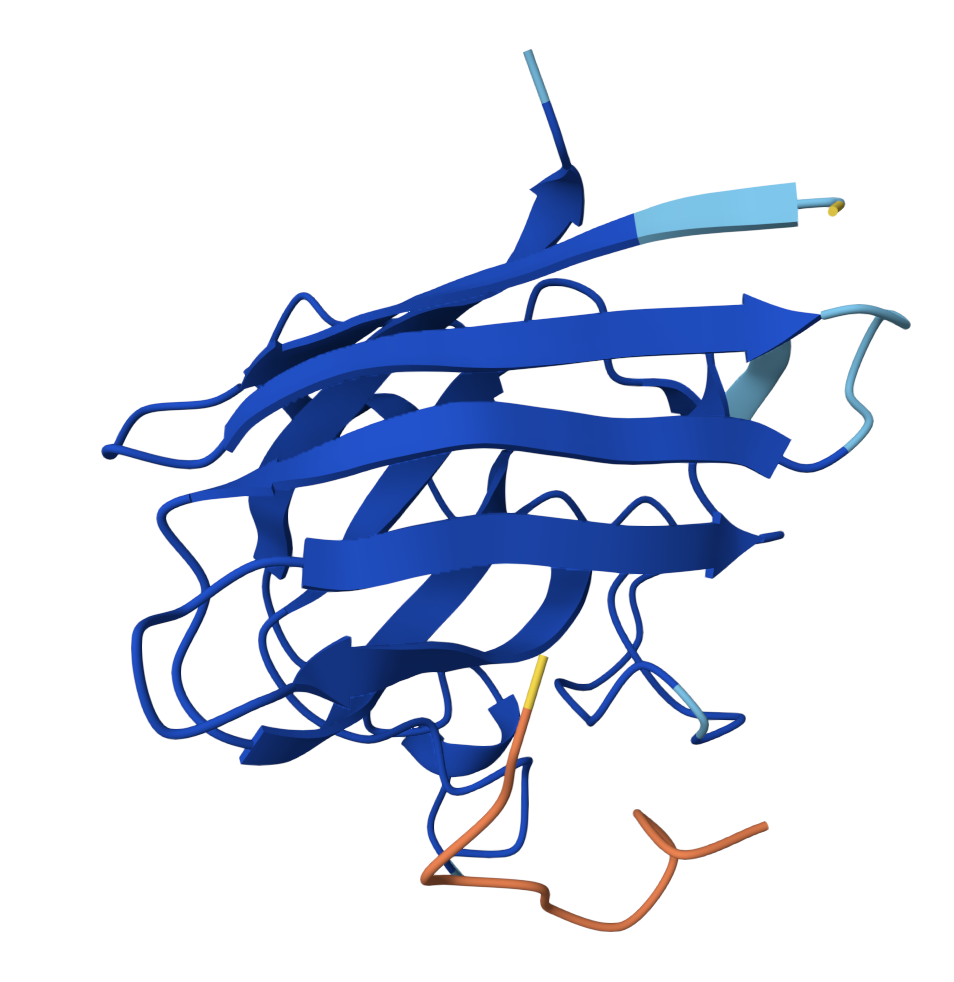
SOD1 binder peptide design Generate binders with PepMLM The original SOD1 sequence[1] is as follows: >sp|P00441|SODC_HUMAN Superoxide dismutase [Cu-Zn] OS=Homo sapiens OX=9606 GN=SOD1 PE=1 SV=2 MATKAVCVLKGDGPVQGIINFEQKESNGPVKVWGSIKGLTEGLHGFHVHEFGDNTAGCTS AGPHFNPLSRKHGGPKDEERHVGDLGNVTADKDGVADVSIEDSVISLSGDHCIIGRTLVV HEKADDLGKGGNEESTKTGNAGSRLACGVIGIAQ The A4V mutation changes the alanine to valine at codon 4, which results in
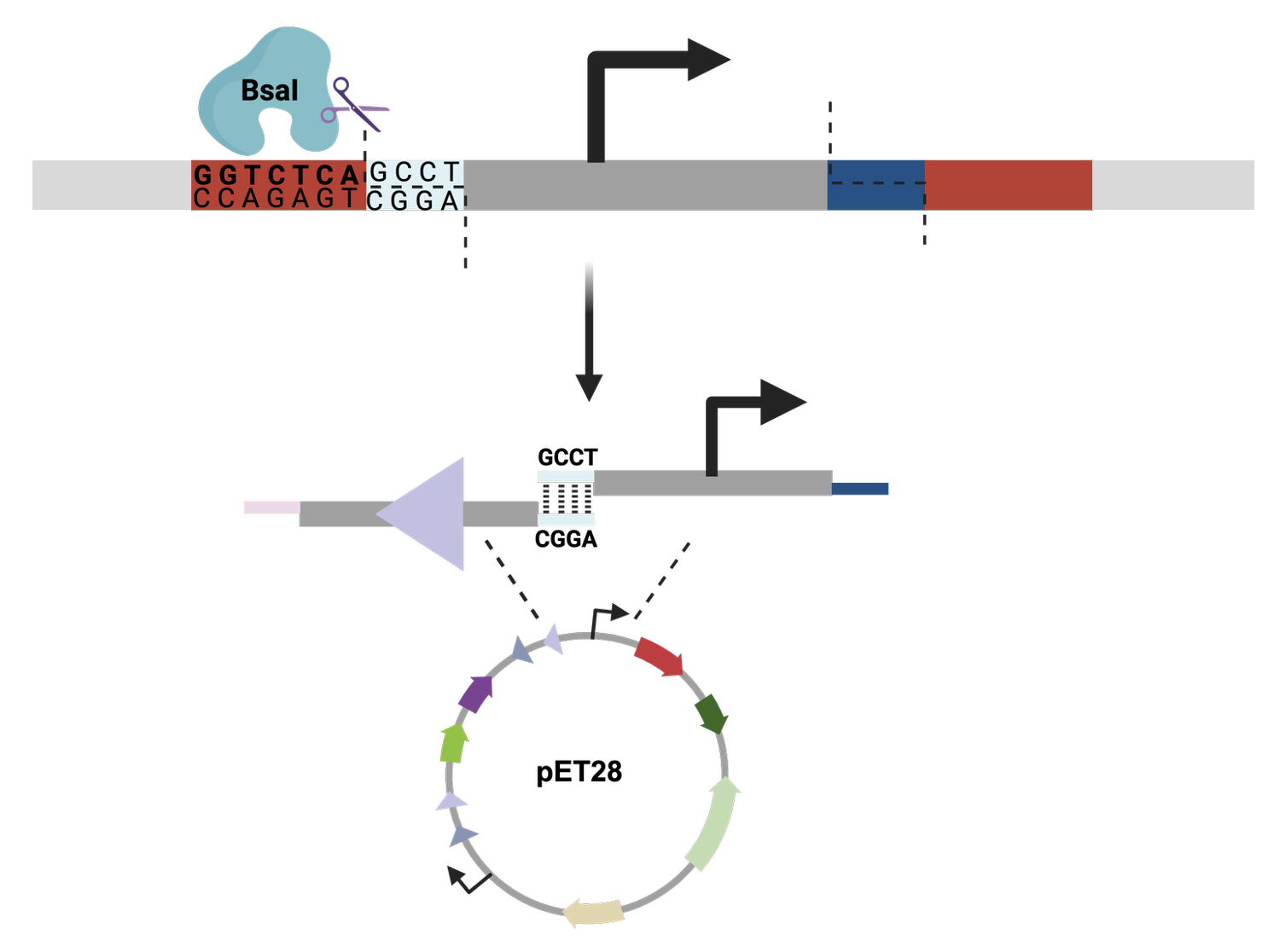
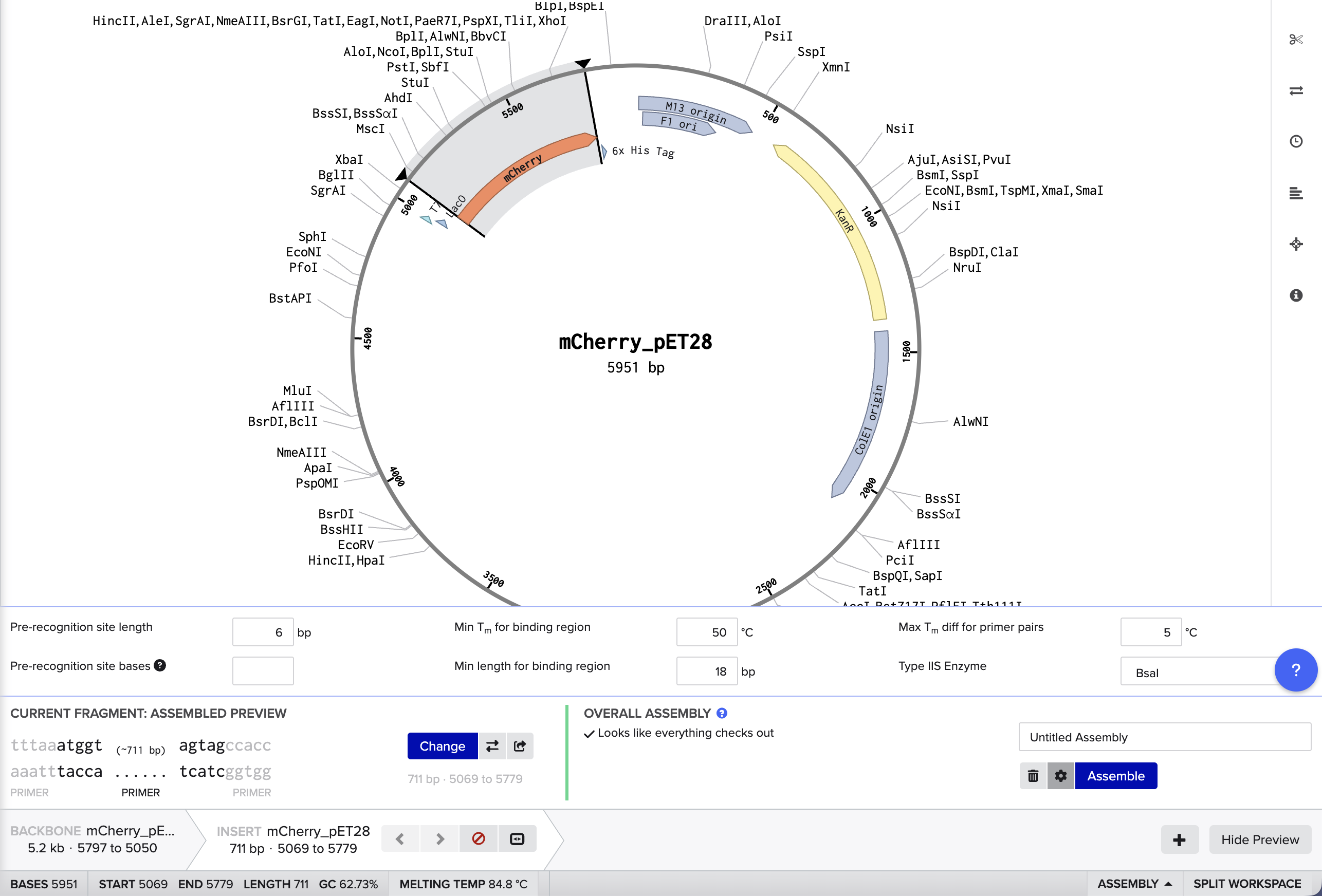
DNA Assembly (Question 1) Within Phusion High-Fidelity Master Mix [1], there is a Phusion DNA Polymerase (which enzymatically synthesizes the DNA in the 5’ to 3’ direction), nucleotides (the building blocks of the synthesized DNA), and an optimized reaction buffer (maintains optimal conditions for the polymerase). (Question 2) Some factors that determine primer annealing temperature are the primer length, the GC content, and the salt concentration.
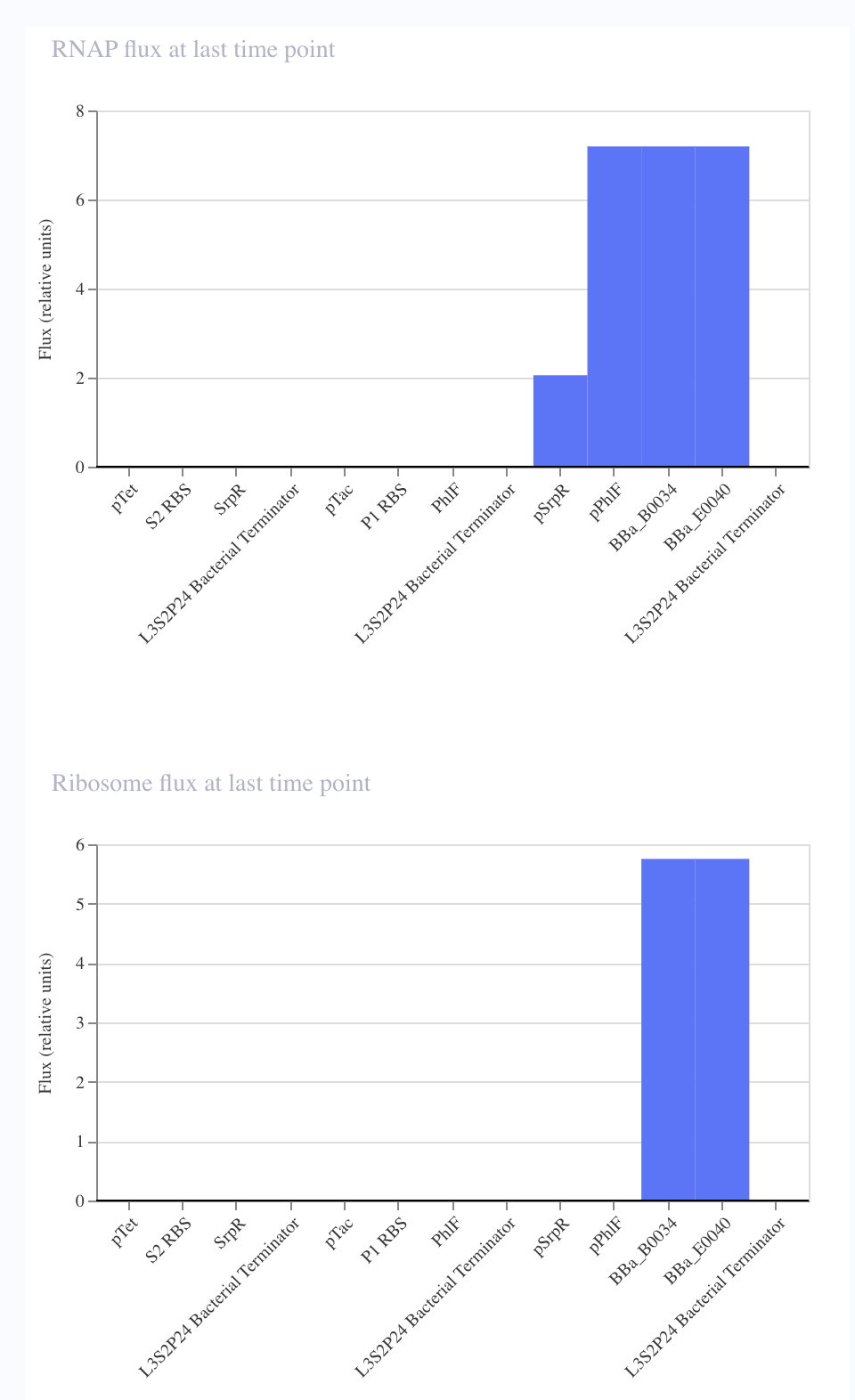
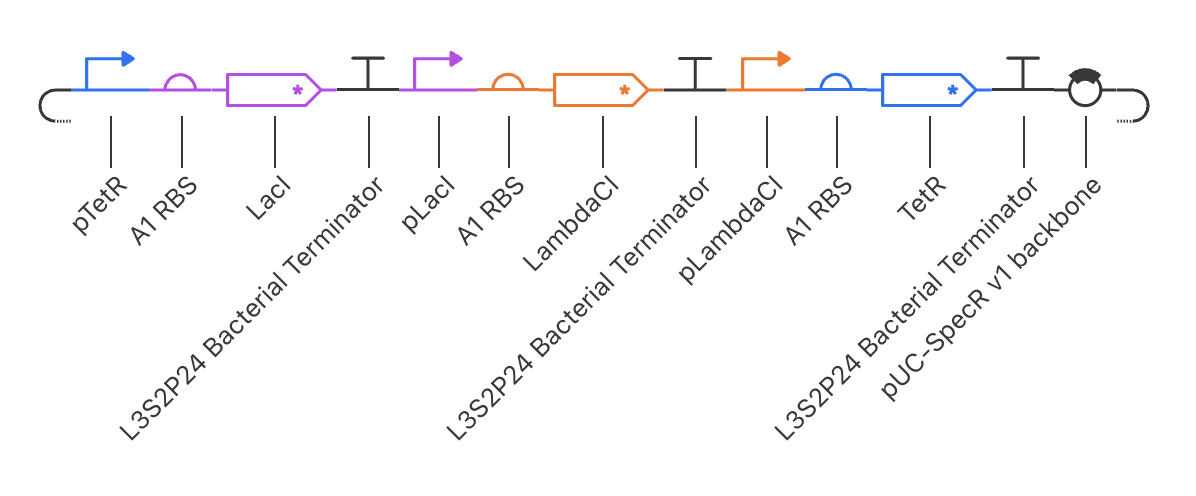
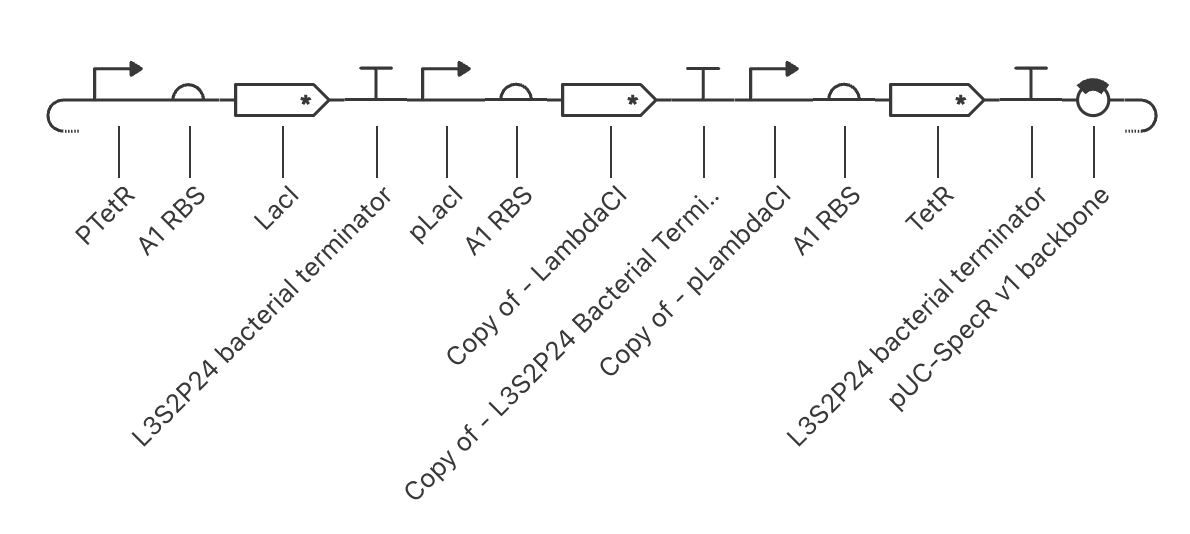
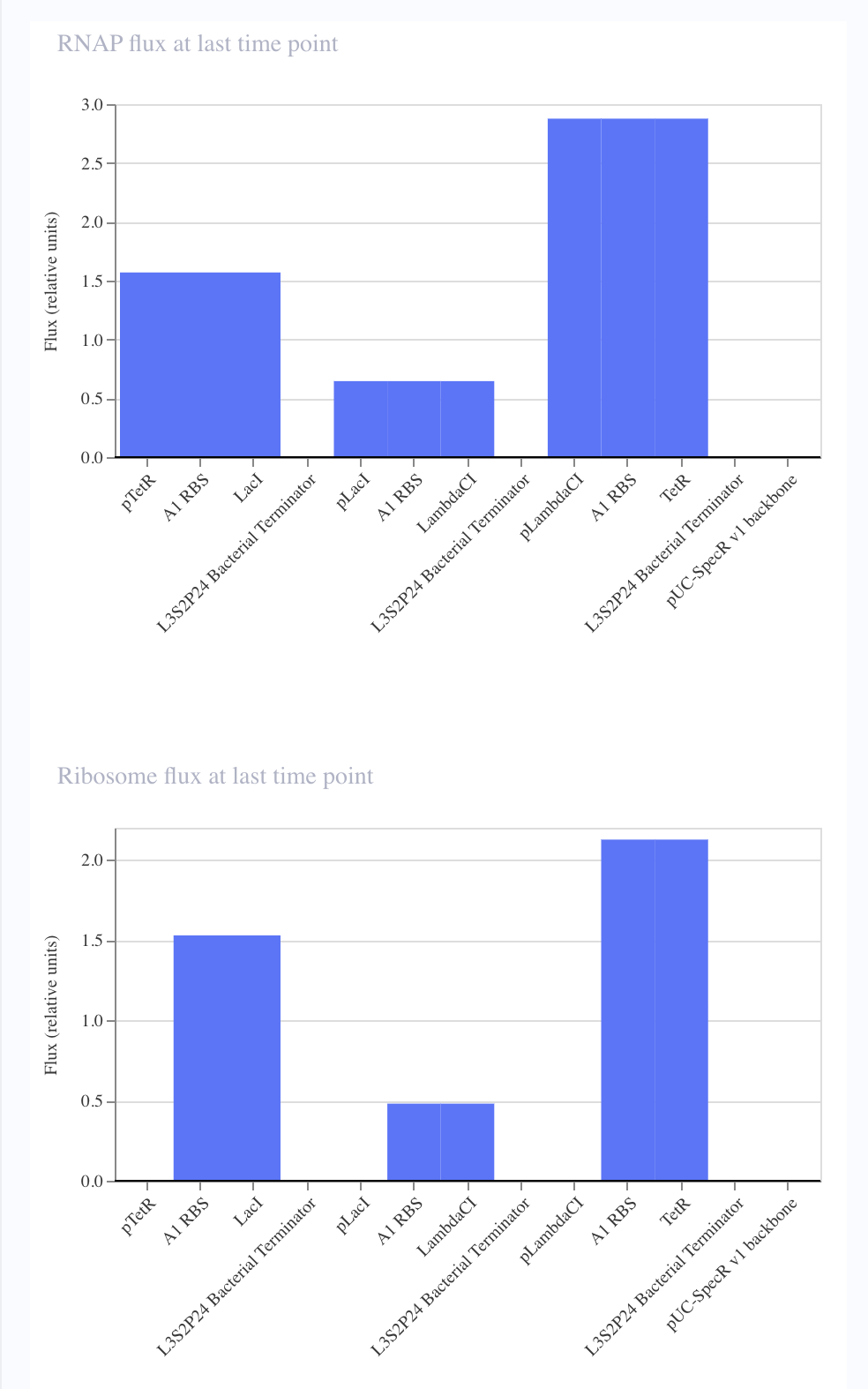
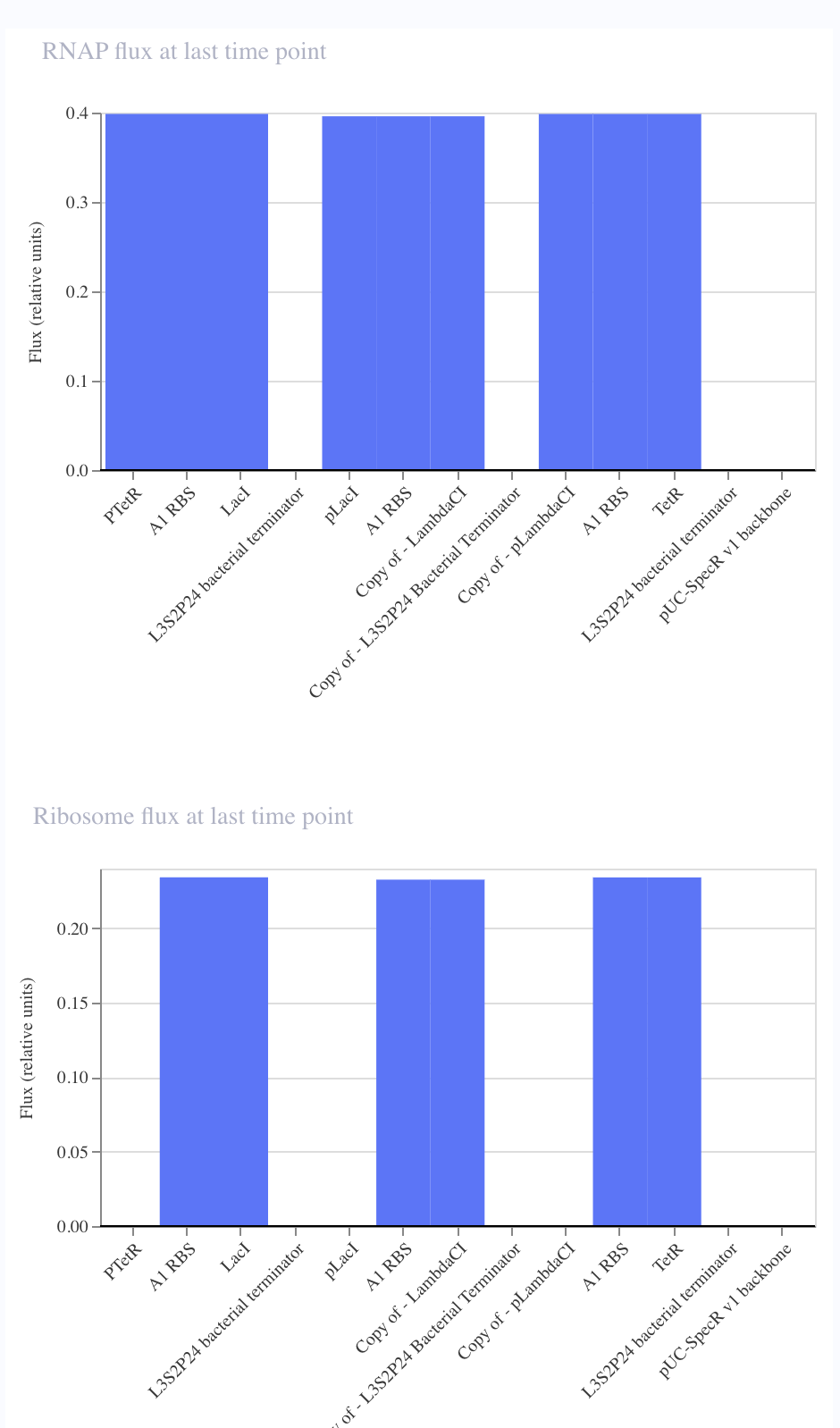
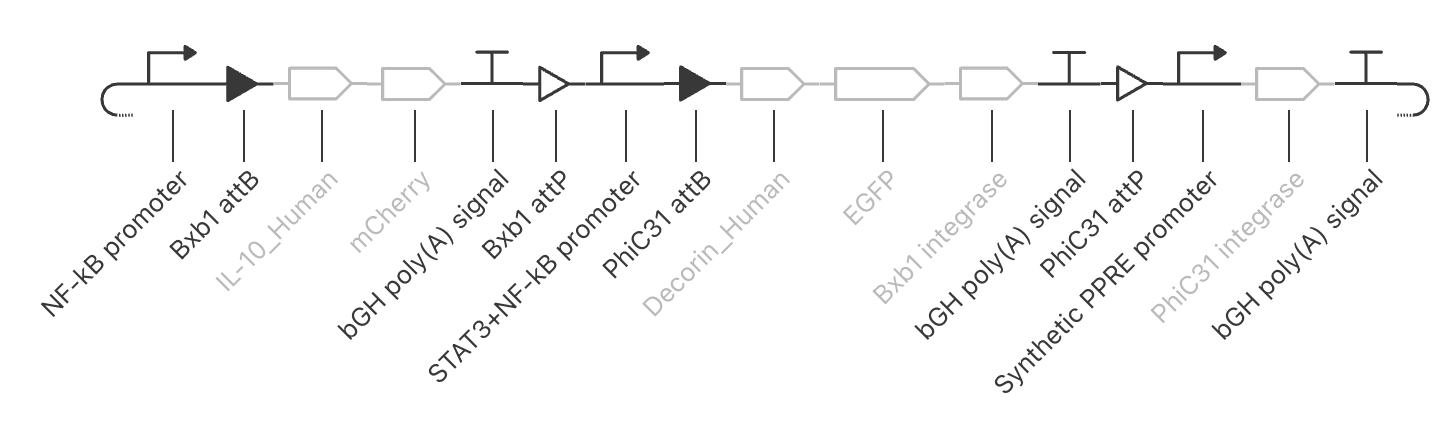
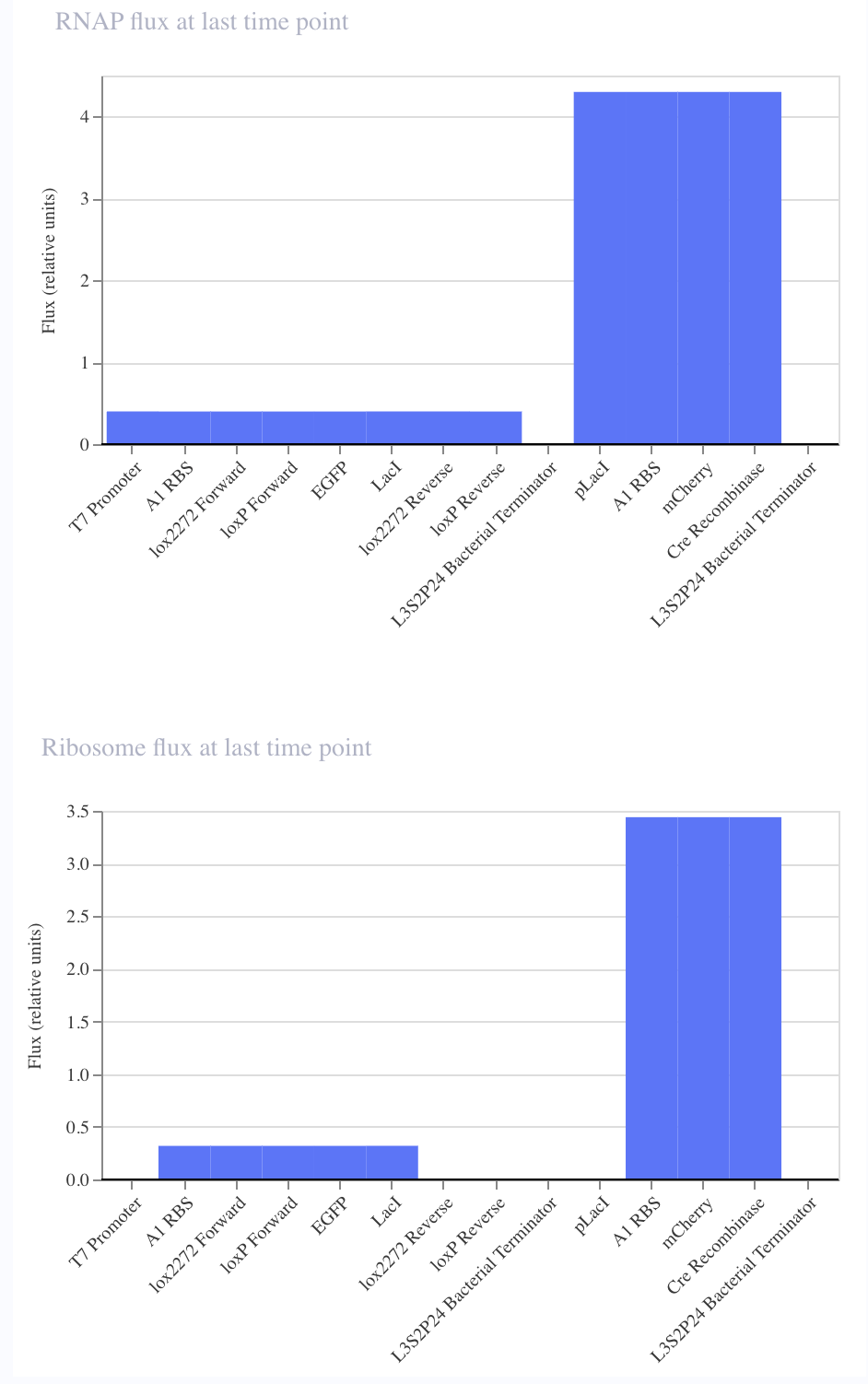
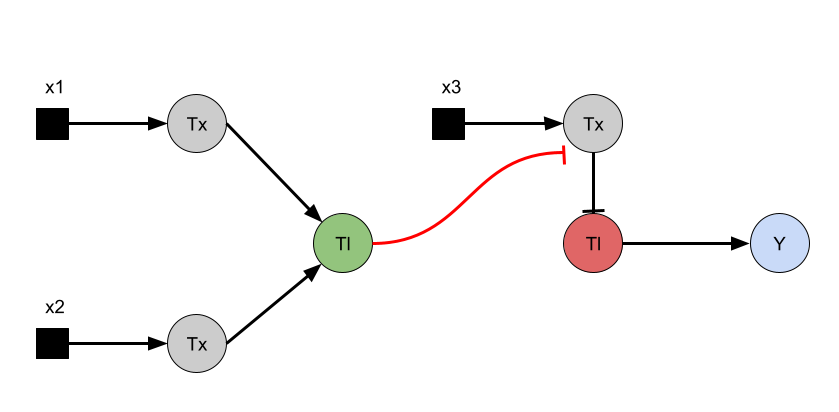
Intrancellular Artificial Neural Networks (IANNs) (Question 1) IANNs have the advantagae of providing a more nuanced approach to using genetic circuits by allowing continuous input and output response, where as genetic circuits, which use Boolean logic, often respond in a more binary manner. (Question 2) The introduction of IANNs raised an interesting question in my individual project idea. Since my final project involves the design of a genetic circuit that can sense and then respond to the formation of fibrotic scarring, IANNs could be used as a more sophisticated approach to this problem by increasing the specificity of the circuit to only activate in a truly fibrotic wound microenvironment. In my original circuit, I had aimed to have part of my circuit sense both STAT3 and NF-kB as a trigger to secrete the anti-fibrotic factor, decorin. However, by incorporating IANN instead, I could further decrease the noise from transient inflammatory spikes through encoding three synthetic transcription factors whose expression is driven by STAT3, NF-kB, TGF-B, and HIF-1a promoters respectively. The second section of my genetic circuit would then be placed under a promoter that would require the binding of all three synthetic transcription factors.
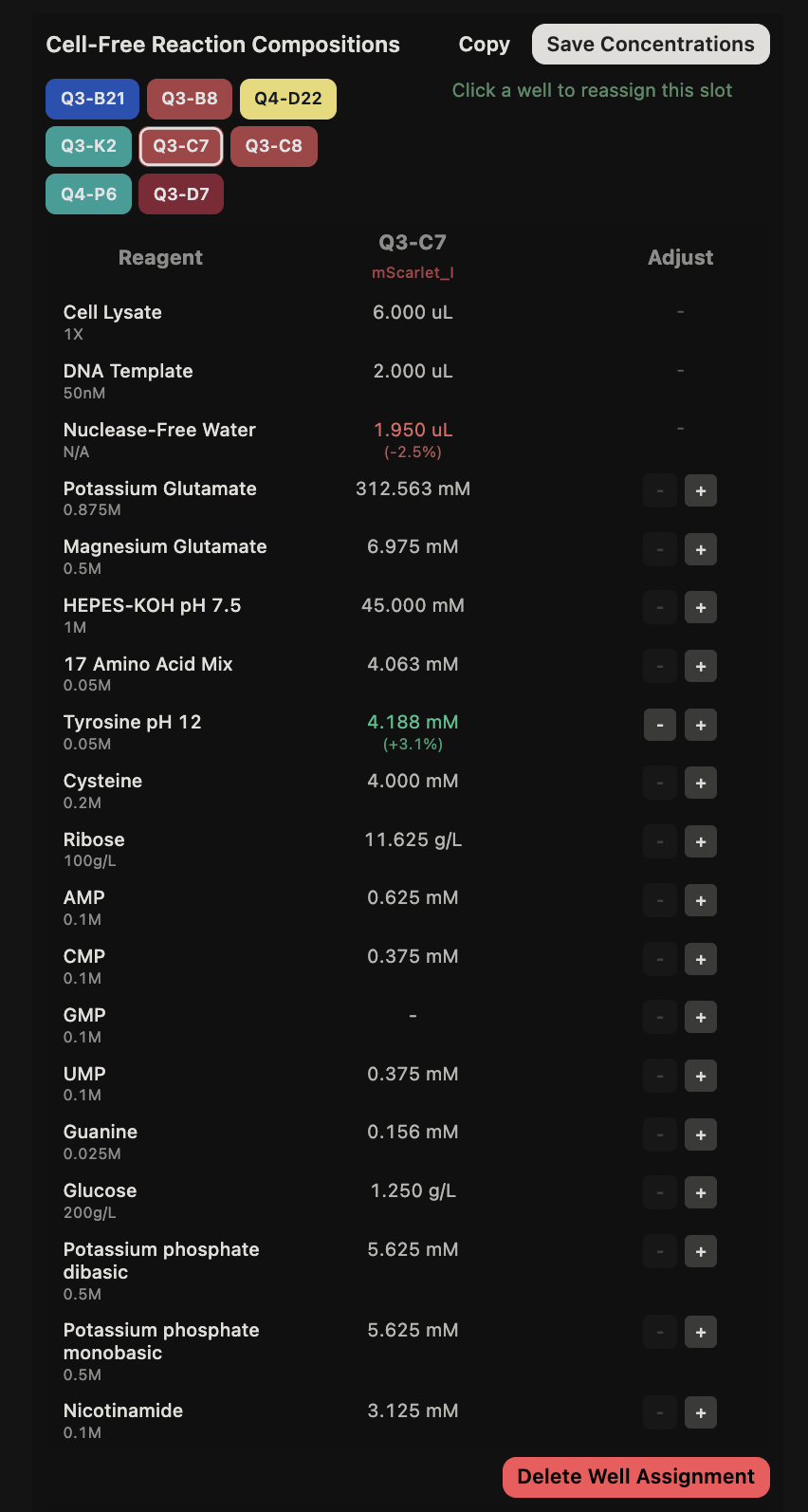
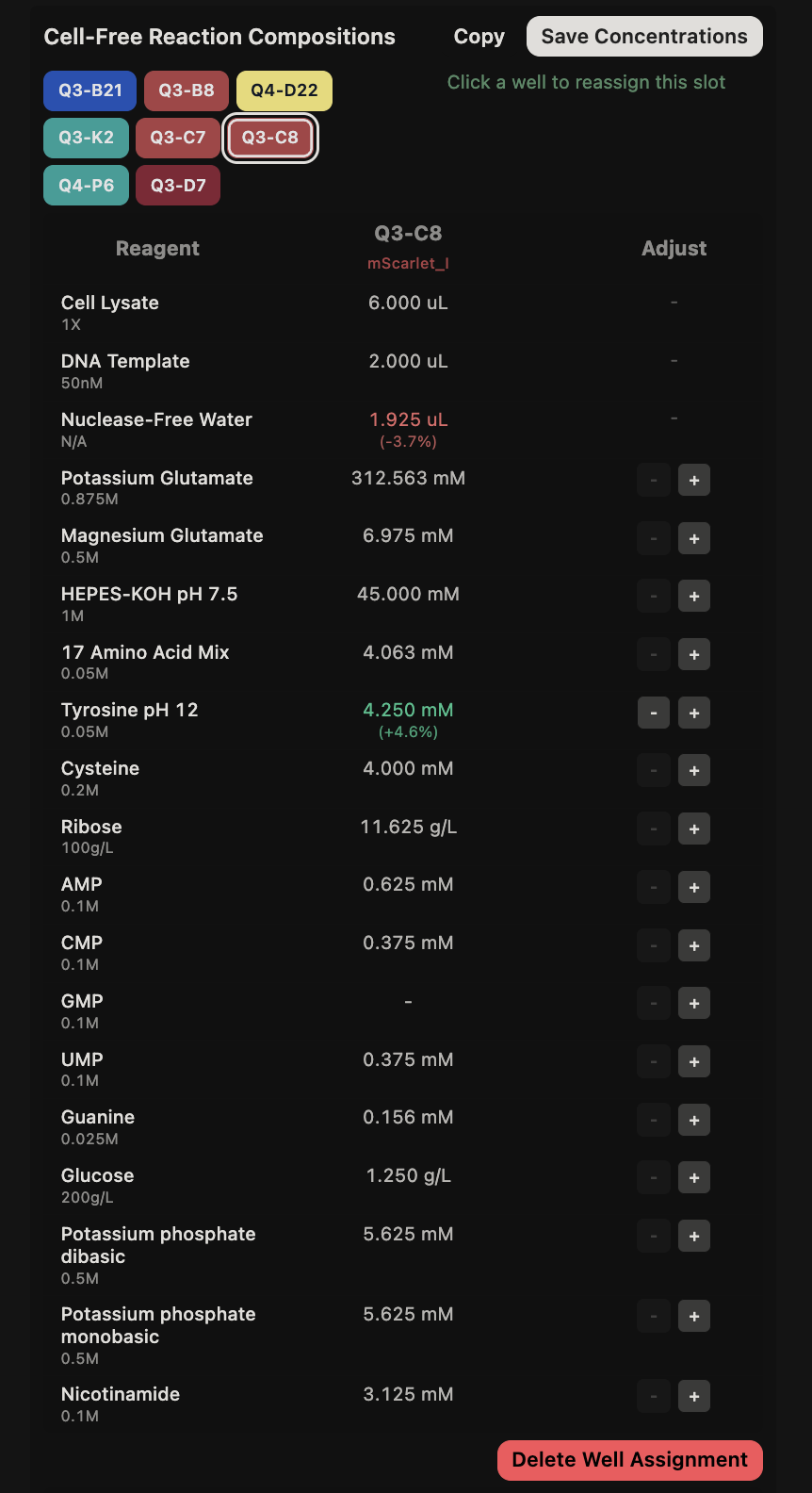
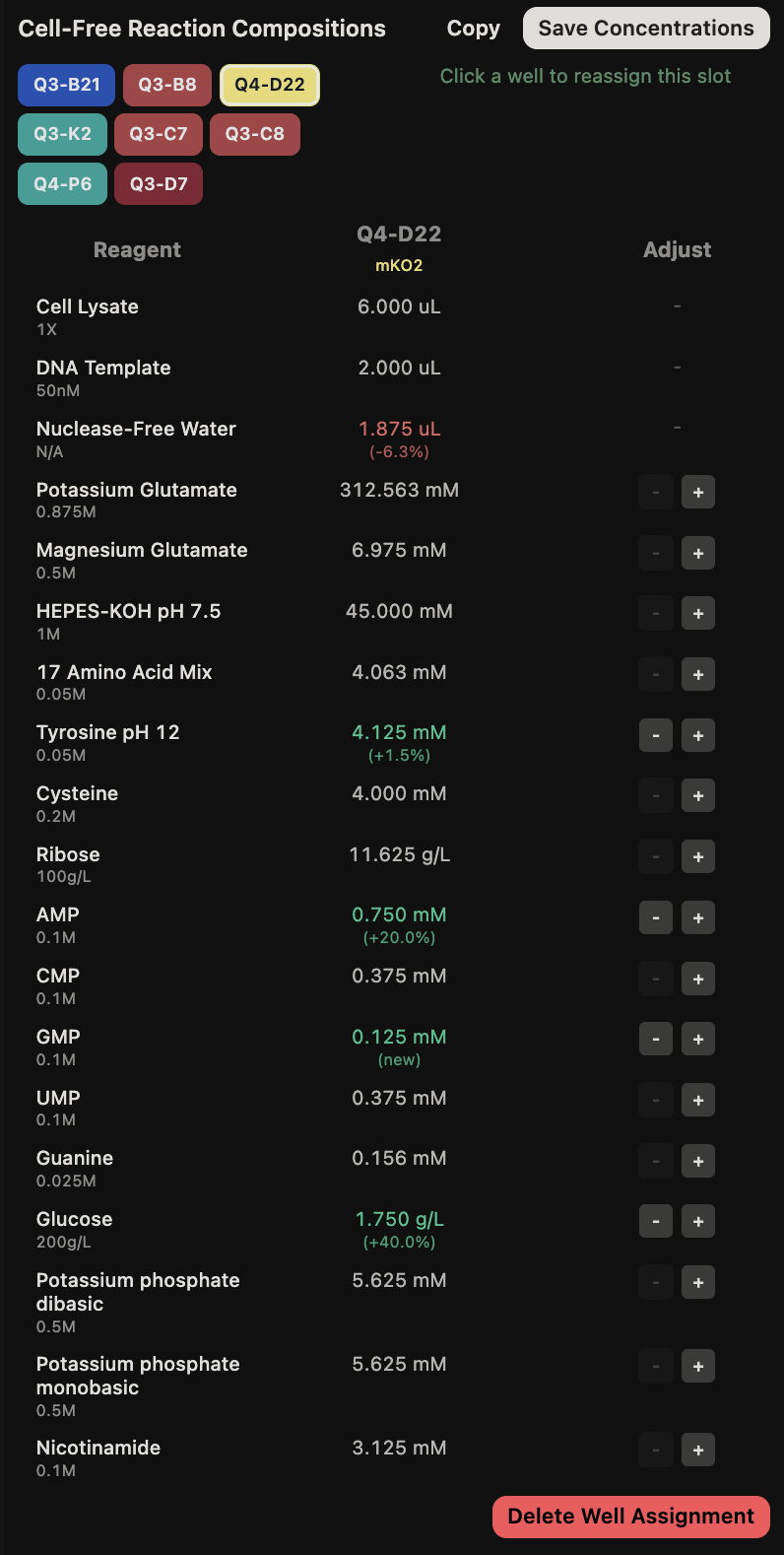
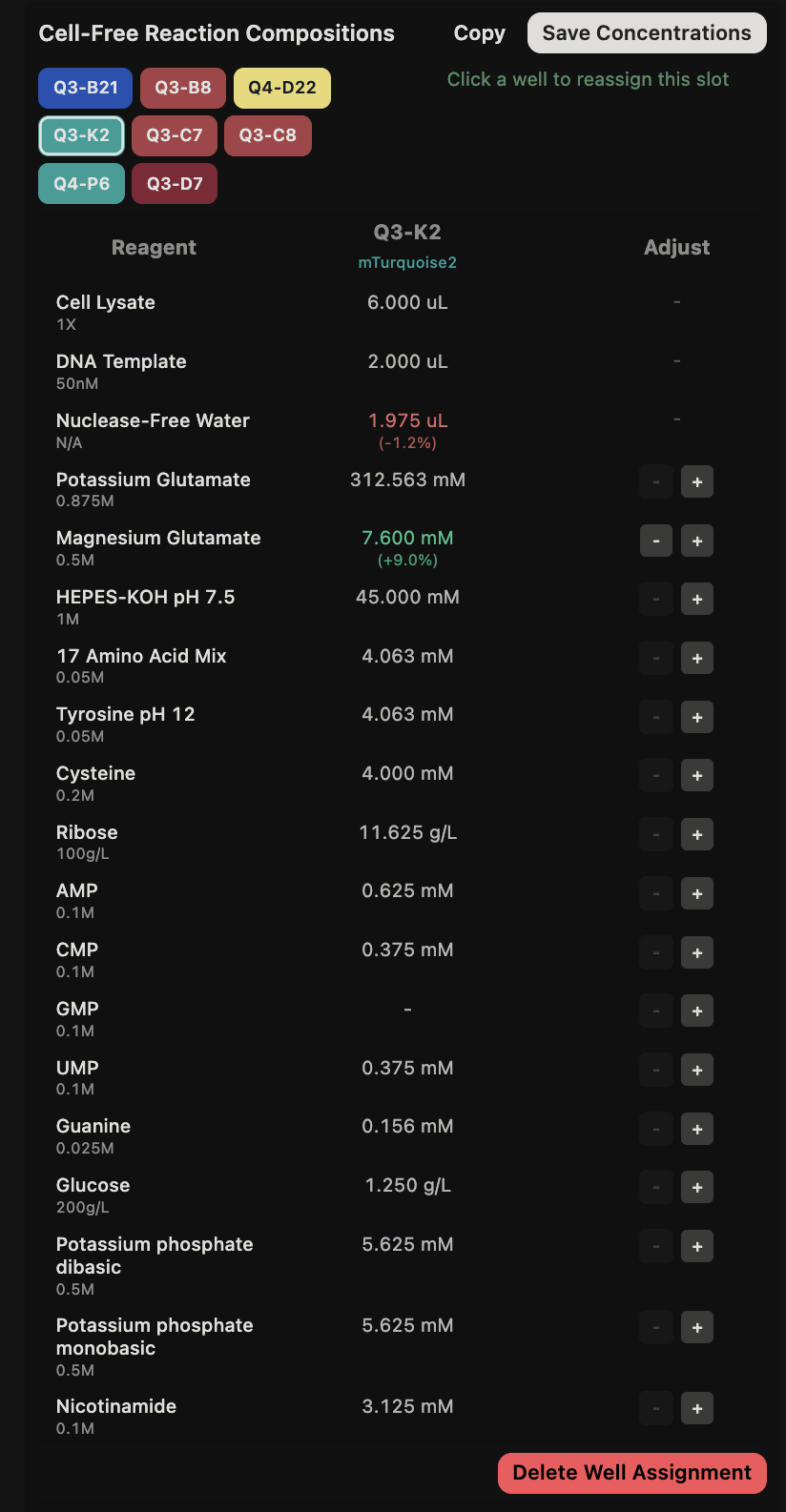
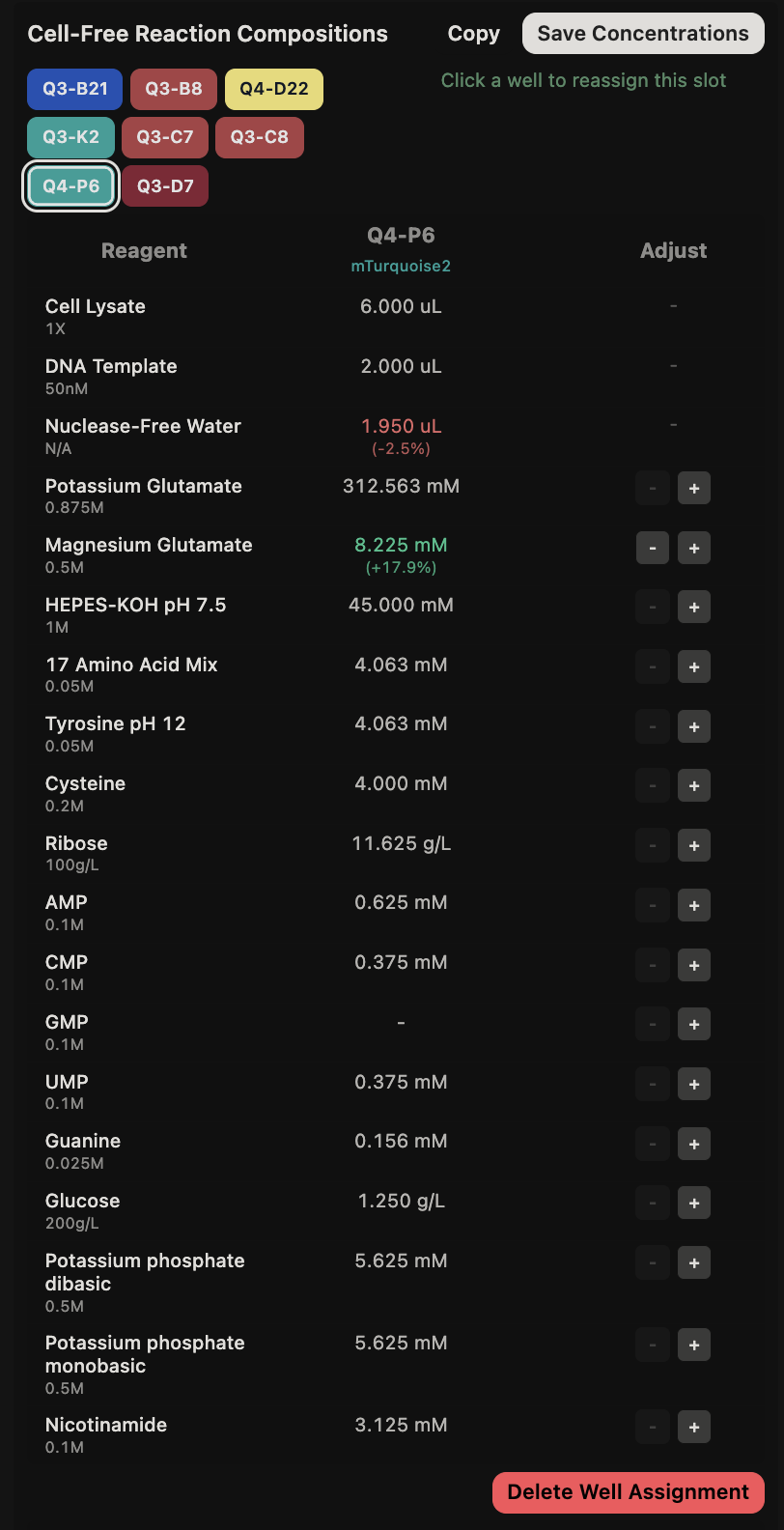
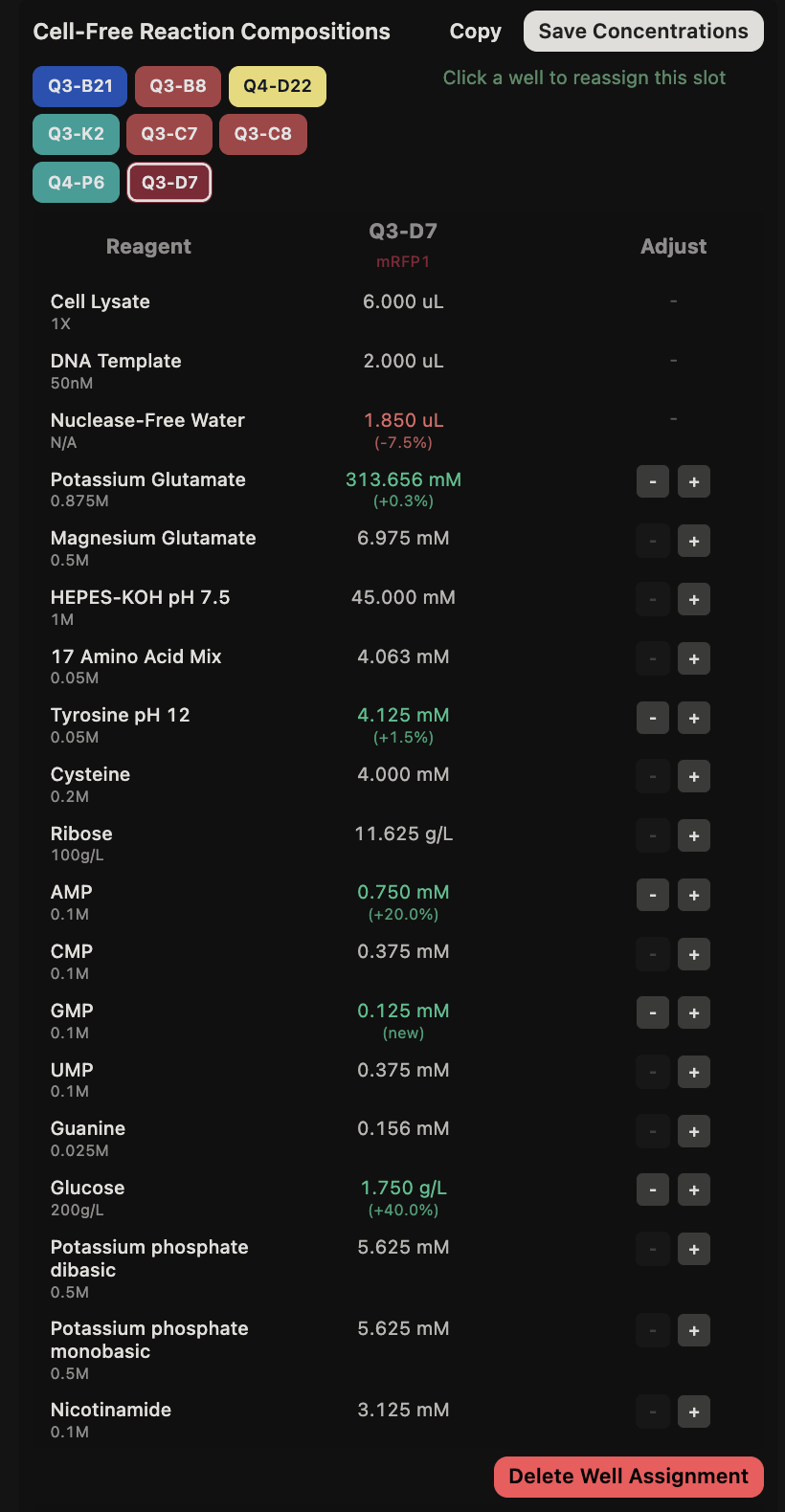
General questions (Question 1) Since the cell-free protein synthesis system eliminates the cell membrane, this means that the environment that the reaction is performed in is less limited by what can enter or exit the cell as it alters the dependence of the reaction on other cellular constraints. For example, the energy source and the chaperone/cofactor concentrations can be altered independently of the cell’s own needs. This poses a particularly interesting environment for cases such as the incorporation of non-standard amino acids, in which cells may not contain the machinery necessary to incorporate but contain machinery that would resist the incorporation of such amino acids. Another intriguing application would be the prototyping of vaccine antigen production. Due to the speed that cell-free systems can perform at, the system would be able to produce a functional antigen from a gene sequence much quicker without the need to engineer a stable cell line to express the desired antigen.
Week 10: Advanced Imaging and Measurement Technology
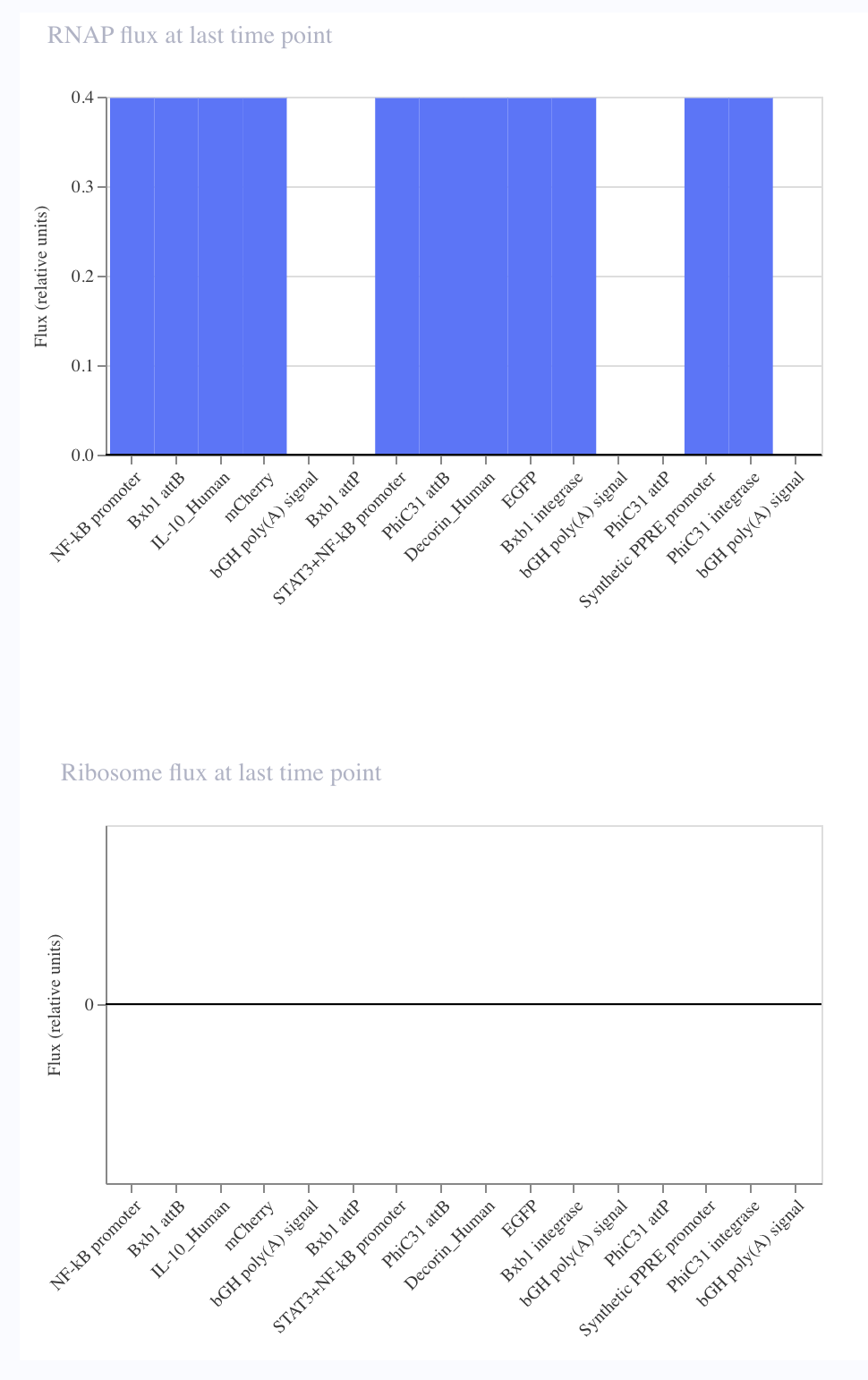
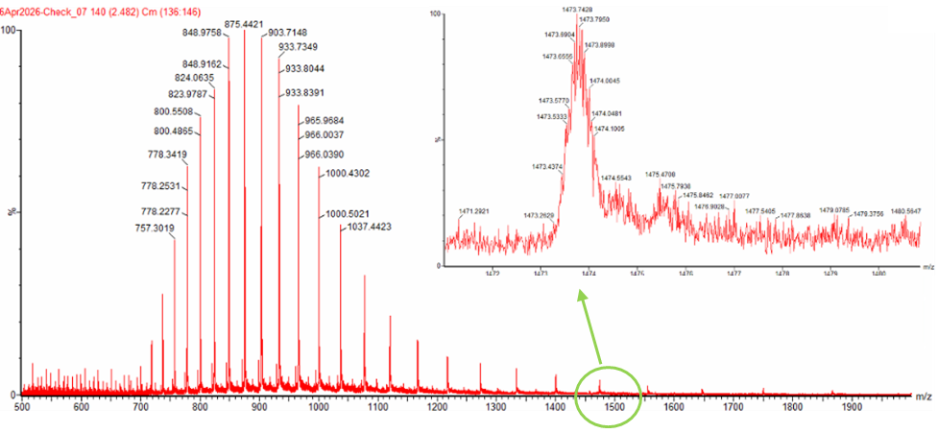
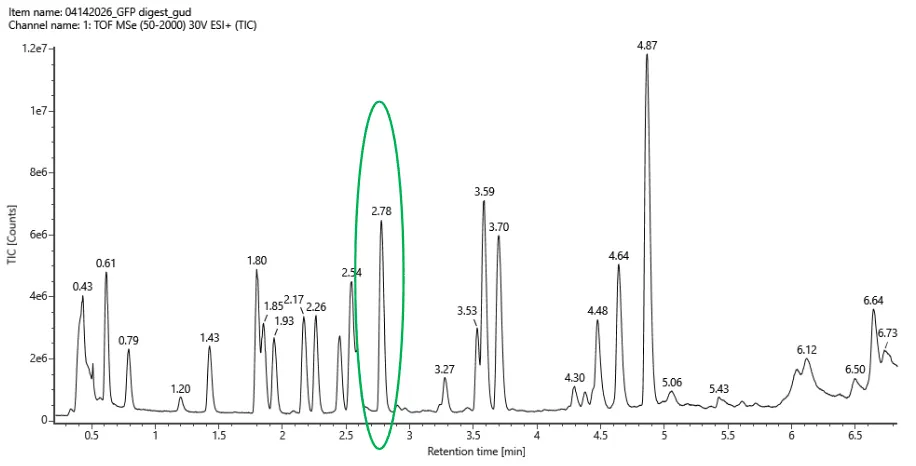
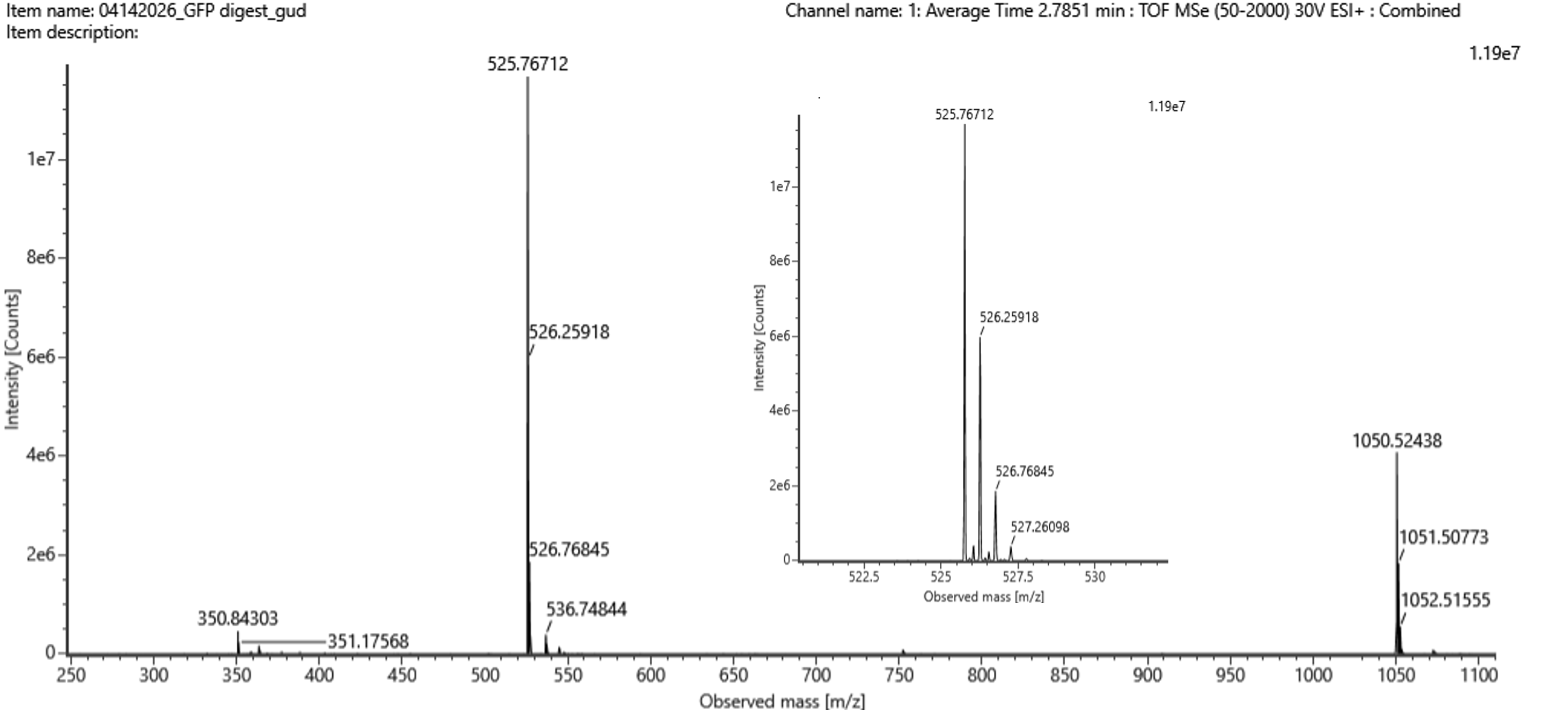
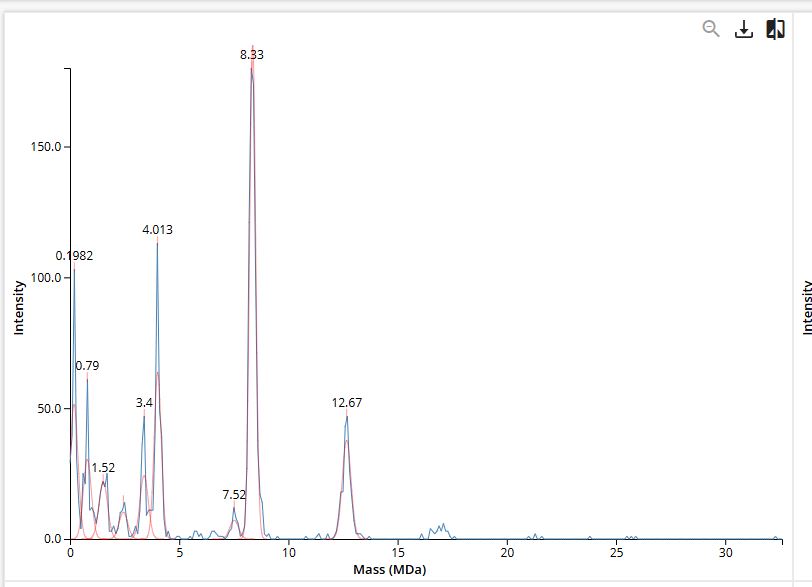
Final Project For my final project, I will need to sequence the genetic circuit that I ultimately construct as well as the concentration of the IL-10, Decorin, and Bxb1 and PhiC31 integrase that is produced by the circuit. In order to sequence the genertic circuit, the most common method would be to use Sanger Sequencing, which utilizes electrophoresis after the synthesis in order to properly sort and sequence the circuit based on lengths and the base that terminated sequencing In order to measure the concentrations of the IL-10, Decorin, and Bxb1 and PhiC31 integrase produced by my genetic circuit, I can use Mass Spectroscopy. After harvesting the expression cells at the appropriate time points, I will use the spike-in standards strategy and then calculate the ratio of my endogenous peptide signal to the heavy standard signal, calculating the concentration based on the moles of the protein measured divided by the volume of my original sample. Waters Pt. 1: Molecular Weight For the following calculations, I will be using the provided eGFP sequence
Week 11: Bioproduction and Cloud Labs
The 1,536 Pixel Artwork Canvas I ended up contributing 6 pixels of various colors to the canvas, which were mostly made on the border, but didn’t end up in the final artwork. I really enjoyed that this assignment was a play on other iterations of the collaborative pixel artwork challenges across various platforms, and felt like a fun way to be able to interact with the entire HTGAA community. I think that a lower cooldown time was needed (and I heard that it was implemented towards the end), as I would often click onto another tab while waiting (and then would get distracted…). Overall though, it was fun to see what came out of the community and what ended up on the final canvas.