Abstract Chronic wounds and fibrotic scarring represent a significant unmet clinical need, affecting millions of patients annually and resulting in impaired tissue function, pain, and reduced quality of life. Current therapeutic approaches lack the spatiotemporal precision needed to modulate the wound microenvironment dynamically, devoid of delivering anti-inflammatory signals early and anti-fibrotic signals later in response to the wound’s own molecular cues. This project proposes the design and experimental validation of a two-stage, NF-κB/STAT3-responsive synthetic gene circuit encoded in a piggyBac transposon vector, engineered for stable integration into dermal fibroblasts. The circuit is designed to first sense early inflammatory NF-κB signaling and secrete IL-10 (an anti-inflammatory cytokine), then switch to a STAT3/NF-κB dual-input logic gate that drives decorin secretion (an anti-fibrotic proteoglycan) as the wound transitions to the proliferative phase. A dual Bxb1 serine integrase and PhiC31 integrase-based irreversible switching mechanism ensures the circuit can irreversibly switch off each phase sequentially, preventing vulnerability to infection or inability to properly heal. mCherry (Stage 1) and EGFP (Stage 2) have been incorporated to enable real-time monitoring of circuit state for testing circuit function and logic. Ideally, this genetic circuit would be transfected in patient-derived fibroblasts, which would be seeded into sutures used at the wound edge.
Subsections of Projects
Individual Final Project: Bioengineered Sutures for Resolving Fibrotic Scarring
Abstract
Chronic wounds and fibrotic scarring represent a significant unmet clinical need, affecting millions of patients annually and resulting in impaired tissue function, pain, and reduced quality of life. Current therapeutic approaches lack the spatiotemporal precision needed to modulate the wound microenvironment dynamically, devoid of delivering anti-inflammatory signals early and anti-fibrotic signals later in response to the wound’s own molecular cues. This project proposes the design and experimental validation of a two-stage, NF-κB/STAT3-responsive synthetic gene circuit encoded in a piggyBac transposon vector, engineered for stable integration into dermal fibroblasts. The circuit is designed to first sense early inflammatory NF-κB signaling and secrete IL-10 (an anti-inflammatory cytokine), then switch to a STAT3/NF-κB dual-input logic gate that drives decorin secretion (an anti-fibrotic proteoglycan) as the wound transitions to the proliferative phase. A dual Bxb1 serine integrase and PhiC31 integrase-based irreversible switching mechanism ensures the circuit can irreversibly switch off each phase sequentially, preventing vulnerability to infection or inability to properly heal. mCherry (Stage 1) and EGFP (Stage 2) have been incorporated to enable real-time monitoring of circuit state for testing circuit function and logic. Ideally, this genetic circuit would be transfected in patient-derived fibroblasts, which would be seeded into sutures used at the wound edge.
Project Aims
Aim 1: Experimental Aim
The first aim of my final project is to design a genetic circuit that will sufficiently sense and respond to fibrotic wound environments by utilizing tools such as Benchling.
Aim 2: Developmental Aim
After designing the appropriate genetic circuit, the next phase of this project would be to design the suture material such that the suture would dissolve over time to prevent excess tension on the wound closure (also contributing to fibrosis) as well as a hydrogel coating that would slowly release the genetically engineered fibroblasts as the layer degrades.
Aim 3: Visionary Aim
Ultimately, I aim to evaluate this suture design through in vitro, in vivo, and eventually clinical models to determine whether the genetic construct could lessen the burden of fibrotic scarring. Fibrotic scarring occurs in 30% of surgical scarring and can affect patient quality of life as well as contribute to further surgical complications in certain applications. The development and success of this suture design could help to improve patient lives as well as relieve the healthcare system from avoidable complications.
Background
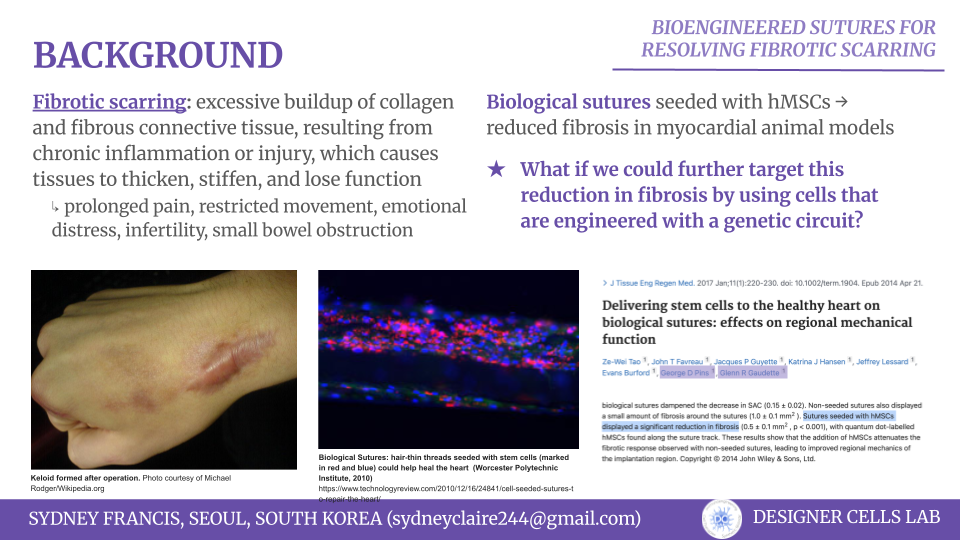
Normal wound healing proceeds through four phases–hemostasis, inflammation, proliferation, and remodeling–with orchestrated cues from a variety of cell types. However, this concerted effort can become disorganized, resulting in impaired healing. One example of this is the formation of hypertrophic and keloid scarring, which is caused by overinflammation of the wound microenvironment leading to the overproduction of fibrotic factors [1][2]. Some of the potential contributors of these conditions are IL-6, tension present at the wound site [3], and overproduction of fibroblast proteins [4]. In a study by Tao et al., it was demonstrated that biological sutures seeded with hMSCs could significantly reduce fibrosis at the wound side in myocardial animal models. While other therapies had failed to deliver and retain the stem cells to the proper sites, it was also documented that the hMSCs were found consistenly along the suture track, allowing for more spatial control of the cells [5].
This project aims to adapt the biological sutures designed by Tao et al. in order to further reduce fibrosis by replacing the hMSCs with genetically engineered fibroblasts. The introduction of a genetic circuit into patient-derived fibroblasts will allow the specific targeting and counteraction of the factors contributing to the inflammation and pro-fibrotic environment and will provide spatiotemporal control of such expression, reducing the chances of off-target effects and complimenting the complex orchestration of the wound healing process. This application challenges the existing standard of wound treatment by autonomously responding the the environment itself.
Each year, over 100 million patients develop a dermal scar as a result of a surgical procedure and fibrotic scarring is estimated to result in 30% of surgical scarring cases [6]. The treatment of these scares is approximated to be around $4 billion per year in the United States healthcare system, yet despite this budget, the condition remains underresearched and the industry still lacks a definitive treatment that effectively resolves fibrotic scarring [2]. While fibrotic scarring can cause discomfort and mental distress, it also can reduce mobility, cause pain, and lead to further surgical complications [2][6]. This project presents a unique opportunity to utilize synthetic biology and genetic engineering to address fibrotic scarring at the suture level, decreasing the risk kof further complications. A targeted, effective intervention, such as bioengineered sutures represents the chance to improve patient outcomes, reduce systemic healthcare costs, and address a gap that has persisted largely unchanged despite decades of surgical advancement.
Since this project is a medical application, it is imperative that it is non-maleficent. beneficient, and is acted upon responsibly. First and foremost, this project aims to introduce genetically mofied cells into a patient’s body, which inherently risks introducing off-target gene editing effects, uncontrolled cell proliferation, immune rejection, and potential mutagenesis. However, this tool is being developed as a response to genuine and well-documented clinical need to reduce suffering, disability, and economic burden to surgical patients. Autonomy and informed consent will play a critical role, since patients should be transparently informed of the experimental nature of any genetically engineered intervention and their right to decline the treatment as such.
In order to ensure that this project is conducted as ethically and responsibly as possible, there are various measures that can be adopted at both the research and societal levels. Firstly, all experimental protocols involving genetically engineered cells should first be rigorously validated in in vitro and animal models before any human application. As the application moves towards human testing and participation, informed consent should be thouroughly ensured and accessible with the acknowledgement that unintended consequences are possible even with precaution. With the production of new data, safety should be re-evaluated in a timely manner.
Experimental Design, Techniques, Tools, and Technology
Step 1: define final construct architecture and annotate in Benchling
Purpose: finalize all genetic elements, their order, and regulatory logic before synthesis
Method: annotate the fulle piggyBac construct in Benchling, confirming ITR orientation, promoter-CDS-terminator order for all three phases, attB/attP site pleacement for the two integrases, reporter fusion desing (IL-10-P2A-mCherry; dedcorin-P2A-EGFP)
Expected result: fully annotated GenBank file ready for Twist submission
Timeline: Day 1-2
Step 2: order whole-plasmid synthesis from Twist Bioscience
Purpose: obtain sequence-verified, ready-to-transfect plasmid DNA without requiring in-house assembly
Method: submit annotated GenBank file (~8kb) to Twist Bioscience as a whole-plasmid synthesis order with kanamycin resistance, pUC ori
Expected result: sequence-verified plasmid delivered within 7-10 business days
If unable to order through Twist, can alternatively assemble the plasmid using a round of Golden Gate assembly (SapI) and Gibson assembly (to insert the final PhiC31 integrase)
Timeline: Day 2-12
Step 3: resuspend and QC Twist plasmid by Nanodrop and gel
Purpose: confirm plasmid integrity and concentration before transfection
Method: resuspend plasmid in TE buffer to 100ng/uL, measuring the A260/A280 on Nanodrop; run 1% agarose gel to condirm supercoiled band at expected size
Expected result: A260/A280 should be more than or equal to 1.8 with a single band at ~8kb
Timeline: Day 12-13
Step 4: culture HEK293T cells
Purpose: prepare healthy, proliferating human cells to test and optimize the logic of the designed circuit
Timeline: Day 1-14 (alongside Twist order)
Step 5: transfect piggyBac construct into HEK293T cells
Purpose: deliver the circuit construct and piggyBac transposase into HEK293T cells for stable integration
Method: co-transfect HEK293T cells with the Twist plasmid (circuit) + a piggyBac transposase expression plasmid (ratio 4:1); 48 hours post-transfection, begin selection
Expected result: stable integrants within 7-10 days of selection
Timeline: Day 13-23
Step 6: confirm stable integration by genomic PCR
Purpose: verify that the piggyBac construct has integrated into the HEK293T genome
Method: extract genomic DNA from selected cells; design primers spanning the piggyBac ITR-genome junction; run PCR on a thermocycler and resolve on 2% agarose gel
Expected result: junction band at expected size (~500bp); absent in untransfected controls
Timeline: Day 23-25
Step 7: seed engineered HEK293T cells into 384-well plate for assay
Purpose: prepare high-throughput assay-ready plates with uniform cell density
Method: dilute to 2,000 cells/well; incubate overnight at 37 C
Expected result: uniform cell monolayer across all wells
Purpose: establish baseline mCherry and EGFP levels before any stimulation to confirm reporter silence at rest
Method: transfer plates to plate reader; read mCherry (Ex 587/Em 610) and EGFP (Ex 488/Em 507) across all wells
Expected result: low baseline fluorescece in both channels; confirms the circuit is off at rest
Timeline: Day 26
Step 9: prepare stimulation reagent plates
Purpose: precisely dispense nanoliter volume of TNF-a (stage 1 trigger) and STAT3 agonist + NF-kB inhibitor (stage 2 trigger) into assay plates
Timeline: Day 26
Step 10: assay plate layout
Purpose: define experimental conditions, controls, and replicates across the 384-well plate
384-Well Plate Layout (Example)
Columns 1–4: Unstimulated control (media only) — n=16 wells
Columns 5–8: TNF-α only (Stage 1 trigger) — n=16 wells
Columns 9–12: IL-6 + Bay 11-7082 (Stage 2 trigger) — n=16 wells
Columns 13–16: TNF-α → Stage 2 switch (sequential stimulation) — n=16 wells
Columns 17–20: Dose-response TNF-α (0.1, 1, 10, 100 ng/mL) — n=4 per dose
Columns 21–24: Positive control (constitutive CMV-mCherry + CMV-EGFP) — n=16 wells
Rows A–P used for all conditions above.
Edge wells (Row A, Row P, Col 1, Col 24) reserved as blank/media controls.
Timeline: Day 26
Step 11: incubate stimulated plates
Purpose: allow sufficient time for NF-kB and STAT3 signaling to activate transcription and produce detectable fluorescent protein
Method: incubate for 24 hours (stage 1 read) and 48 hours (stage 2 read)
Expected result: mCherry signal rises in TNF-a wells by 24 hours; EGFP signal rises in stage 2 wells by 48 hours
Timeline: Day 26-28
Step 12: read post-stimulation fluorescence
Purpose: quantify mCherry and EGFP signal changes after stimulation
Method: read mCherry and EGFP on plate reader at specified time points
Timeline: Day 27-28
Step 13: Normalize data and calculate fold-change
Purpose: account for well-to-well variability in cell seeding density
Method: normalize fluorescence values to Hoechst nuclear stain signal; calculate fold-change relative to unstimulated controls
Expected result: statistically significant (p<0.05) fold-changes in both mCherry and EGFP channels under correct stimulation conditions
Timeline: Day 28-29
Step 14: Confirm Bxb1 and PhiC31 recombination by PCR
Purpose: verify that the integrase-mediated switch has occurred at the DNA level in cells
Method: extract genomic DNA from stimulated cells; design primers flanking the Bxb1 attB/attP and PhiC31 attB/attP recombination site; run PCR on thermocycler; expect size shift from pre- to post-recombination
Exxpected result: amplicon size shift consistent with attL/attR product formation that is absent in unstimulated cells
Step 15: qPCR quantification of IL-10 and decorin transcript levels
Purpose: confirm that the fluorescent reporter signal correlates with therapeutic gene transcription
Method: extract RNA from stage 1 and stage 2 cells; synthesize cDNA; run qPCR with probes for IL-10, decorin, mCherry, and EGFP
Expected result: IL-10 and mCherry transcripts elevated in stage 1 cells; decorin and EGFP transcripts elevated in stage 2 cells; all transcripts absent in post-stimulated cells
Timeline: Day 30-32
After all of these steps have been completed, corrected and adjusted construct can be transfected into human dermal fibroblasts
Relevant techniques
Pipetting
Lab Safety
Bioethical Considerations
DNA Sequencing
DNA editing
DNA construct design
(Possibly) Restriction Enzyme digestion
Gel electrophoresis
DNA purification from gel
Databases
Designing Twist order
Use of Benchling
Plasmid Preparation
Primer design or selection
PCR reactions
(Possibly) Gibson Assembly
(Possibly) Golden Gate Assembly
The primary method of synthesizing my construct will be using Benchling to first design the construct computationally, and then ordering the construct using Twist Biosciences. However, if the construct is unable to be synthesized, then I will need to alternatively use a combination of Golden Gate assembly and Gibson assembly. I would first use Golden Gate (SapI) to assemble the majority of my circuit. Then, due to internal restriction enzyme sites, I would use Gibson Assembly to insert the PhiC31 integrase piece.
Results and Quantitative Expectations
For this project, I was able to complete the computational design of the genetic circuit by utilizing Benchling [7]. This genetic circuit was designed by targeting the main contributors of fibrotic scarring, which are over-inflammation (targeted using IL-10) and the pro-fibrotic factors (targeted using decorin)
When designing this circuit, I had a few main goals in mind.
In wound healing, inflammation comes before the remodeling phase, so this should be the sequential order of the gene sets
The two gene sets should be irreversibly inactivated at appropriate times in order to prevent the wound becoming susceptible to infection and non-healing
The genetic circuit should autonomously sense and trigger the expression of the correct gene sets using endogenous cues relevant to the wound microenvironment
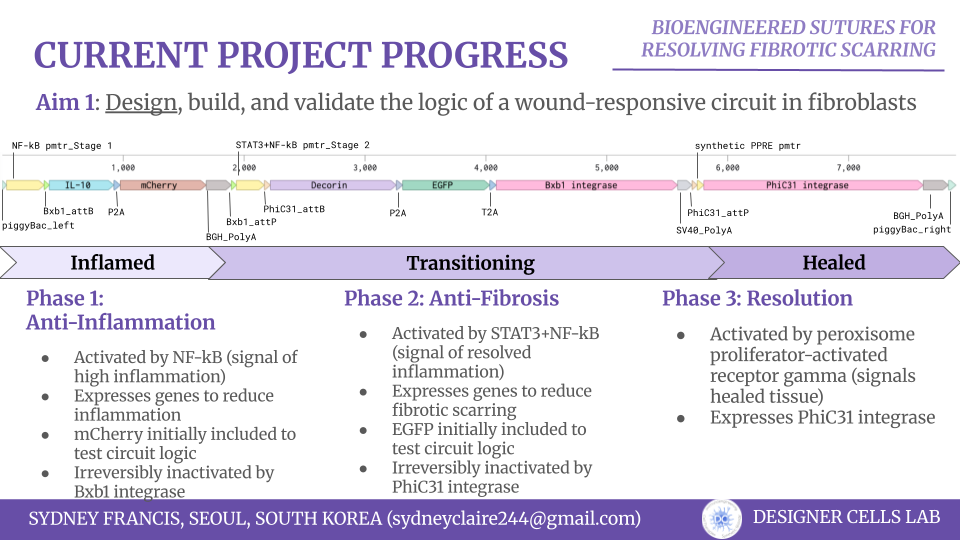
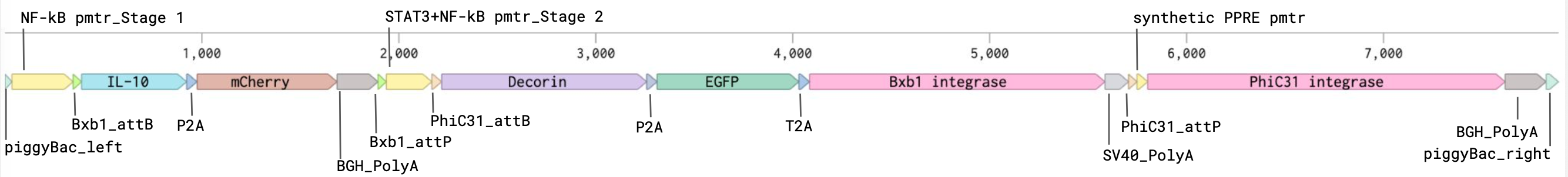
Keeping this in mind, I began to assemble the circuit by splitting the genetic circuit into three phases.
The first phase (NF-kB promoter–Bxb1 attB–IL-10–P2A–mCherry–BGH PolyA–Bxb1 attP) is aimed at sensing and responding to the inflammation. High levels of NF-kB is associated with inflammatory wound microenvironments, which would trigger an anti-inflammatory gene set, which I chose as IL-10. I also included the mCherry fluorescent reporter as a visual validation for early iterations of the circuit. The IL-10 and mCherry is surrounded by Bxb1 integrase sites, which would allow the inflammatory gene set to be irreversibly inactivated by Bxb1 integrase, which is encoded in the second phase of the circuit.
The second phase (STAT3+NF-kB promoter–PhiC31 attB–Decorin–P2A–EGFP–T2a–Bxb1 integrase–SV40 PolyA–PhiC31 attP) is aimed at sensing and responding to the lowered inflammation triggered from phase one and prevents the overexpression of pro-fibrotic factors. STAT3 and low levels of NF-kB is a biomarker of lowered wound inflammation, so I chose to use an AND promoter of these two genes to trigger the expression of decorin, an anti-fibrotic gene. I also included the EGFP fluorescent reporter as another visual validation signal. The Bxb1 integrase is also included in this section of the genetic circuit. The decorin, EGFP, and Bxb1 integrase are surrounded by PhiC31 integrase sites, which allow the anti-fibrotic gene set to be irreversibly inactivated by PhiC31 integrase, which is encoded in the third phase of the circuit.
The third phase (Synthetic PPRE promoter–PhiC31 integrase–BGH PolyA) is aimed solely at inactivated the second phase when the wound microenvironment signals it is healed. The Synthetic PPRE (peroxisome proliferator-response element) promoter senses peroxisome proliferator-activated receptor gamma, a biomarker of healed tissue. This triggers the expression of PhiC31 integrase, which subsequently inactivates the second phase.
In order to achieve this design, I had to consider a few techniques to computationally assemble the circuit as well as logically figure out how to synthesize it. I used Benchling to design the DNA construct and label the appropriate pieces. Ideally, I would use Twist Biosciences to synthesize the construct, however, due to it’s large size (~8kb), this may not be possible. As such, I tried to simulate how the circuit would be assembled using the various assembly techniques we used, ultimately landing on a combination of Golden Gate Assembly and Gibson Assembly.
By simulating the Golden Gate assembly, I was able to find that SapI could assemble the majority of my circuit, with the exception of the PhiC31 integrase, which included internal restriction enzyme sites. I would start by first assembling my entire circuit using Golden Gate and then clone in the PhiC31 integrase using Gibson assembly, thus constructing my entire construct.
One of the potential pitfalls that I anticipate with this final project is the correct balancing of the promoters and the gene sets in order to truly elucidate therapeutic benefits in the wound environment. While this can only be realized through in vitro and in vivo models, these tests can be used to observe if the circuit is performing the logic correctly, if the promoters have the correct sensitivity, or if the genes are beneficial to the purposes that I would like them to achieve. For example, binding sites of the promoters’ targets can be added/removed as needed in order to adjust the activation of the subsequential gene sets. The modularity of the circuit allows each of these to be modified appropriately without undoing the overall logic of the circuit.
Additional Information
Sources
[1] Limandjaja, Grace C., et al. “Hypertrophic scars and keloids: Overview of the evidence and practical guide for differentiating between these abnormal scars.” Experimental Dermatology, vol. 30, no. 1, 6 July 2020, pp. 146–161, https://doi.org/10.1111/exd.14121.
[4] Gauglitz, Gerd G., et al. “Hypertrophic scarring and keloids: Pathomechanisms and current and emerging treatment strategies.” Molecular Medicine, vol. 17, no. 1–2, 5 Oct. 2010, pp. 113–125, https://doi.org/10.2119/molmed.2009.00153.
[5] Tao, Ze-Wei, et al. “Delivering stem cells to the healthy heart on biological sutures: Effects on regional mechanical function.” Journal of Tissue Engineering and Regenerative Medicine, vol. 11, no. 1, 21 Apr. 2014, pp. 220–230, https://doi.org/10.1002/term.1904.
[6] Mokos, Zrinka Bukvić, et al. “Current therapeutic approach to hypertrophic scars.” Frontiers in Medicine, vol. 4, 20 June 2017, https://doi.org/10.3389/fmed.2017.00083.