Week 4 HW: Protein Design Part I
Part A. Conceptual Questions
1. How many molecules of amino acids do you take with a piece of 500 grams of meat? (on average an amino acid is ~100 Daltons)
Meat is ~20% protein by weight (average). So in 500 g of meat: Protein ≈ 100 g Average amino acid ≈ 100 Da ≈ 100 g/mol
Thus: 100 g protein÷100 g/mol=1 mol amino acids
1 mole = 6.022 × 10²³ molecules that’s why 6 × 10²³ amino acid molecules
3. Why are there only 20 natural amino acids?
Evolution selected a chemically diverse but efficient set. They provide sufficient variation in:
- charge;
- polarity;
- size;
- hydrophobicity
5. Where did amino acids come from before enzymes that make them, and before life started?
- Prebiotic chemistry (Miller–Urey type reactions)
- Hydrothermal vents
- Interstellar chemistry (found in meteorites like Murchison)
- Abiotic synthesis from simple gases (CO₂, NH₃, CH₄)
6. If you make an α-helix using D-amino acids, what handedness (right or left) would you expect?
Natural L-amino acids → right-handed α-helix D-amino acids → left-handed α-helix
7. Can you discover additional helices in proteins?
Yes. Beyond α-helices:
- π-helix;
- Collagen triple helix;
- Coiled-coils.
8. Why are most molecular helices right-handed?
Because biological proteins use L-amino acids, and their stereochemistry energetically favors right-handed helices due to minimized steric clashes.
9. Why do β-sheets tend to aggregate? What is the driving force for β-sheet aggregation?
Because β-strands form:
- Extensive hydrogen bonds
- Flat, complementary surfaces
- Strong backbone interactions
10. Why do many amyloid diseases form β-sheets? Can you use amyloid β-sheets as materials?
Misfolded proteins expose backbone groups.
Yes. Applications:
- Nanomaterials
- Tissue scaffolds
- Conductive biomaterials
- Drug delivery platform
11. Design a β-sheet motif that forms a well-ordered structure.
To design a stable β-sheet to use an alternating polar/nonpolar pattern, for example, Val–Ser–Val–Ser–Val–Ser–Val–Ser
Part B: Protein Analysis and Visualization
1. I found it interesting to study p53 because its mutations are associated with the development of many types of cancer. Therefore, it is important not only for a fundamental understanding of cellular molecular biology but also for medicine and the development of anticancer drugs.
2.
- Protein length: 121 amino acids
- Most frequent: L (16 times)
- E-value 5e-75
3.
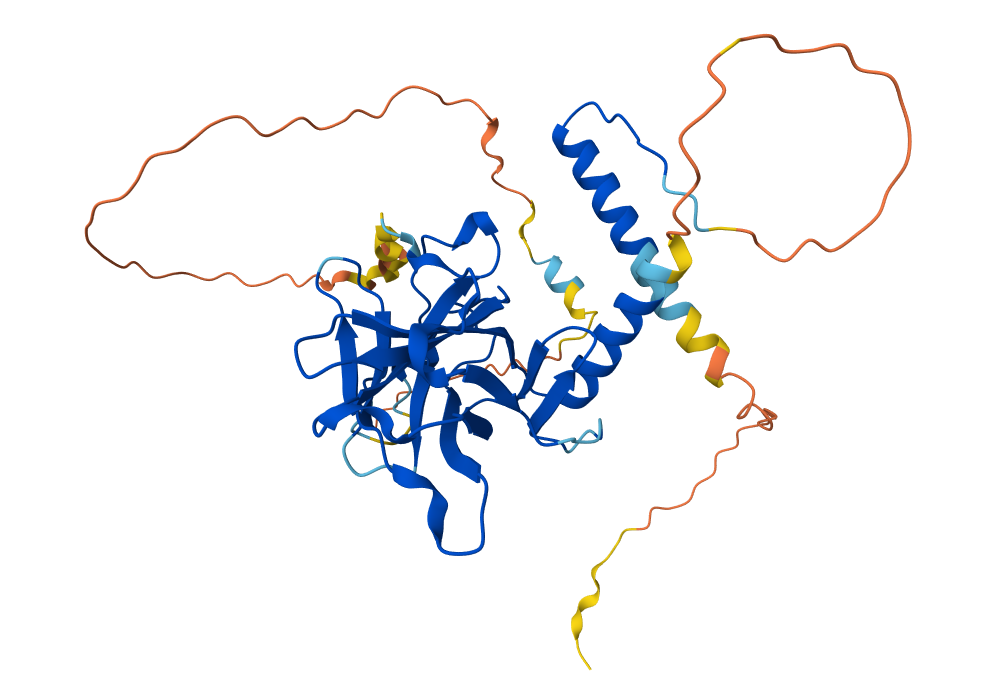
- Released in AlphaFold DB: 2021-07-01
- Not found
- p53 tetramerization domain, p53 DNA-binding domain-like, transactivation domain (TAD)
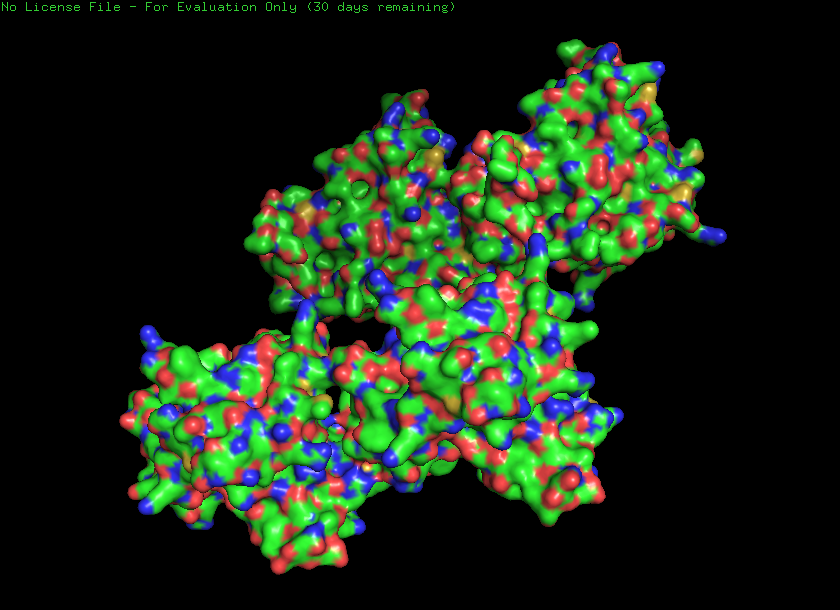
4.
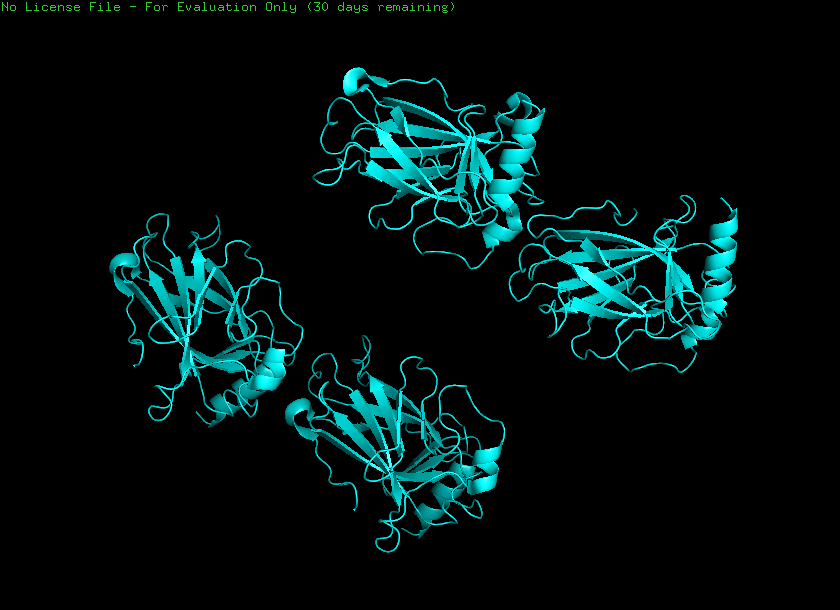
Ribbon

Cartoon
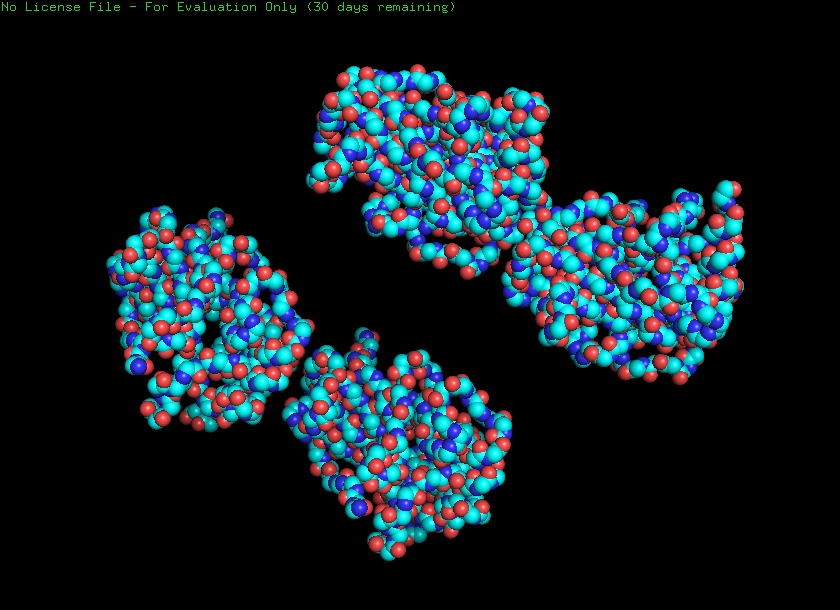
Ball and stick
Helices and sheets
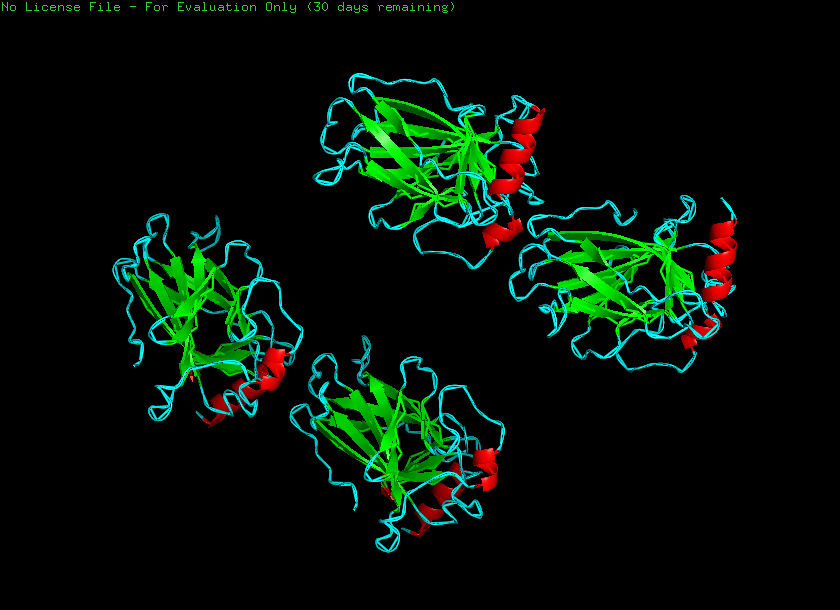
Helices - 1222 atoms Sheets - 3769 atoms
Hydrophobic - 5319 atoms Hydrophilic - 7166 atoms
Holes
Part C. Using ML-Based Protein Design Tools
C1. Protein Language Modeling
Deep Mutational Scans
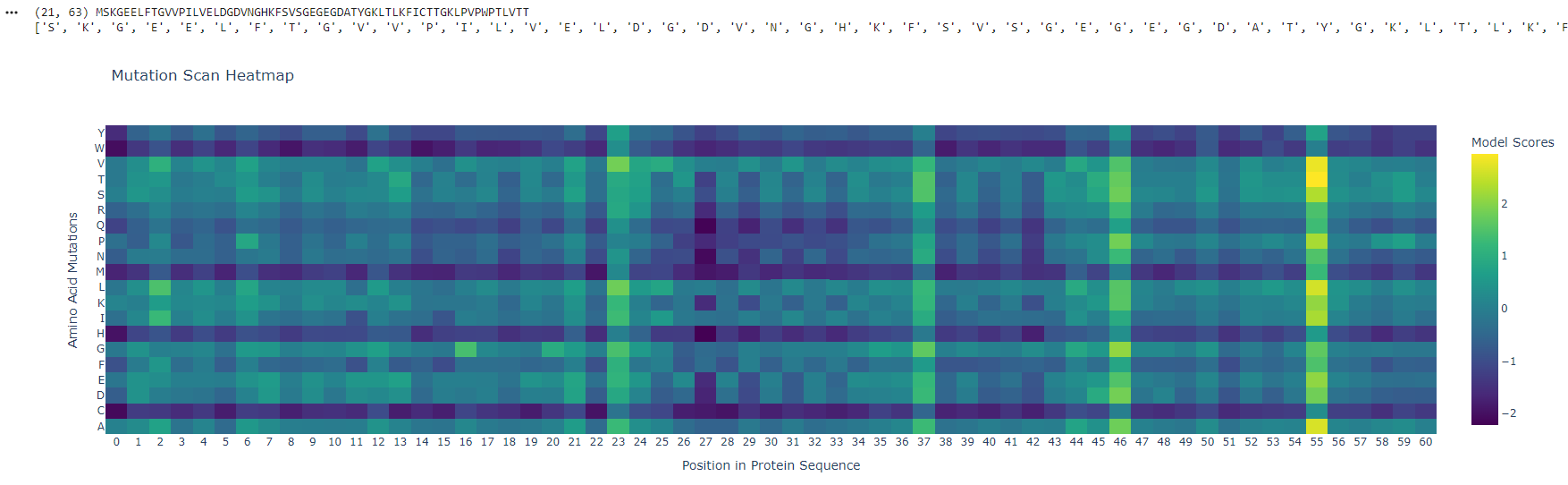
Position ~55 shows a bright vertical band, meaning many substitutions are tolerated - this site is flexible and likely surface-exposed. In contrast, positions ~25–30 are mostly unfavorable (purple), indicating structurally critical, conserved regions sensitive to mutation.
Latent Space Analysis
The protein clusters within a neighborhood of similar fluorescent proteins, indicating that the embedding captures functional and structural similarity. Its position near GFP-like sequences suggests shared folding patterns and chromophore-forming capability. Neighboring proteins likely exhibit similar fluorescence properties and beta-barrel structures.
C2. Protein Folding
Small mutations have minimal impact, preserving overall structure, while larger sequence changes disrupt folding. This suggests the protein is locally robust but globally sensitive to significant alterations.
C3. Protein Generation
The predicted sequence probabilities align closely with the original sequence, with high confidence in conserved residues. Minor variations occur in flexible regions.
Part D. Group Brainstorm on Bacteriophage Engineering
Proposal: Investigating Increased Stability of the MS2 L Protein
Objective: Identify and engineer mutations in the L protein of ssRNA phage MS2 to increase its stability while maintaining lytic function.
Proposed Tools/Approaches:
- Use PLMs (e.g., ESM-2, ProtTrans) to perform in silico mutagenesis of L.
- Predict the effects of single-residue substitutions on protein stability and functional conservation.
- Model L interactions with potential protein partners in silico.
- Assess how candidate mutations may alter tertiary structure, heterotypic interactions, or membrane association.
Rational: L may require specific protein–protein contacts for lytic activity, so structural modeling can prioritize mutations that stabilize the protein while preserving key interactions.
Why These Tools Might Help:
PLMs allow high-throughput screening of potential stabilizing mutations without needing labor-intensive wet lab experiments. AlphaFold-Multimer can predict whether stabilizing mutations compromise critical interactions, reducing the risk of non-functional variants. Conservation-guided mutagenesis ensures changes do not disrupt essential motifs like LS, which are important for lysis.
Potential Pitfalls:
Limited experimental validation data: PLMs and AlphaFold predictions are probabilistic; predicted stabilizing mutations may still disrupt lytic function. Complex lysis mechanism unknown: Since the L mechanism does not involve peptidoglycan inhibition, stabilizing mutations might inadvertently affect subtle protein–protein interactions that are hard to capture in silico.