Week 5 HW: Protein Design Part II
Part A: SOD1 Binder Peptide Design (From Pranam)
Part 1: Generate Binders with PepMLM
MATKAVCVLKGDGPVQGIINFEQKESNGPVKVWGSIKGLTEGLHGFHVHEFGDNTAGCTSAGPHFNPLSRKHGGPKDEERHVGDLGNVTADKDGVADVSIEDSVISLSGDHCIIGRTLVVHEKADDLGKGGNEESTKTGNAGSRLACGVIGIAQ
Part 2: Evaluate Binders with AlphaFold3
HRYGAVVVELKK
Structure
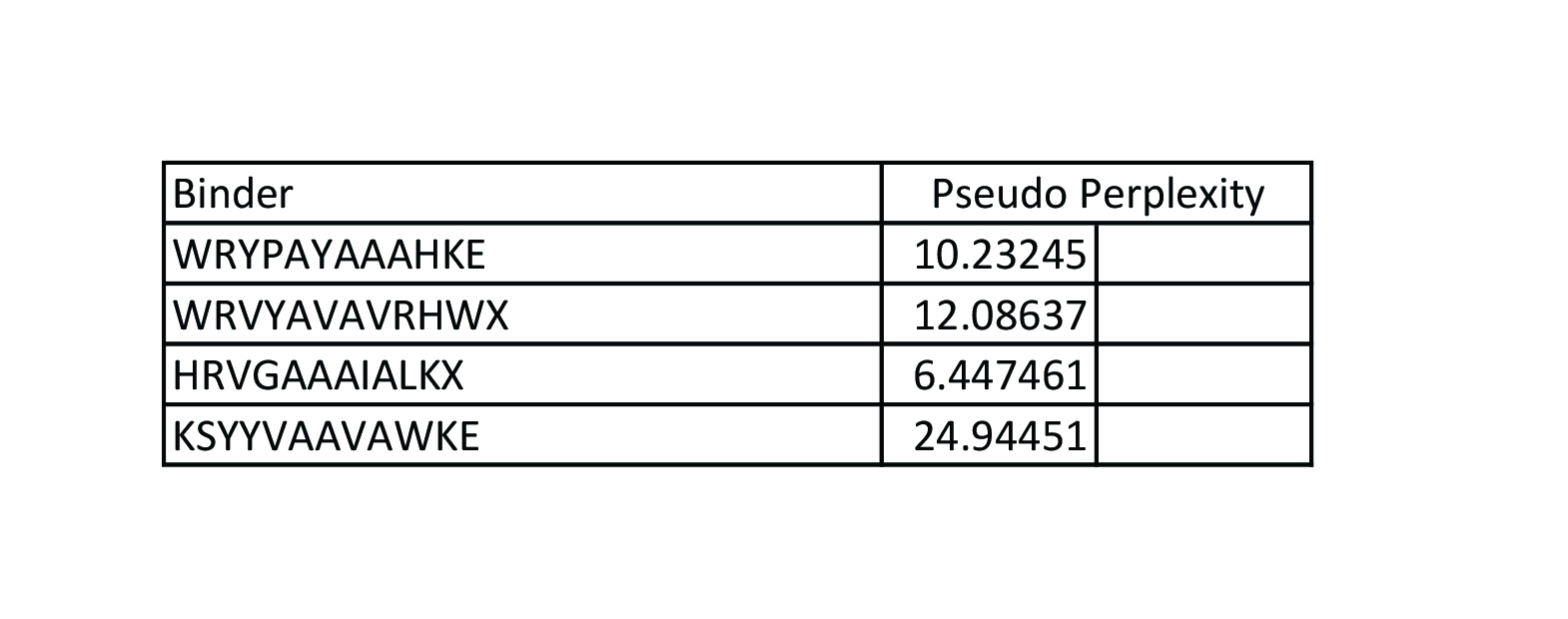
Based on the image you provided:
· ipTM score = 0.4 · pTM score = 0.86
The peptide (residues shown along the vertical axis, roughly 1–165) appears to bind along one face of the folded protein domain, but the alignment confidence (pLDDT coloring) is mixed. There is a very low confidence (dark orange/red) stretch around residues ~33–66 and again near ~132–165, suggesting the predicted binding mode may be unreliable in those regions.
The peptide does not clearly localize near the extreme N‑terminus where the A4V mutation (SOD1) would sit. Instead, the interaction seems more central along the sequence, possibly engaging the β‑barrel region or approaching a surface groove, but not deeply buried - it looks mostly surface‑bound given the moderate ipTM. There’s no clear engagement of a canonical dimer interface in this view.
ipTM of 0.4 is relatively low (typical high‑confidence peptide binding is ipTM > 0.7–0.8). Without direct PepMLM output to compare, I can say that a known binder would likely have ipTM > 0.7 and show consistent, high‑confidence contacts in the error plot. Here, the error is high in several stretches, so this PepMLM‑generated peptide does not match or exceed a reliable known binder’s confidence - it is a low‑to‑moderate prediction.
KSYYVAAVAWKE
Structure
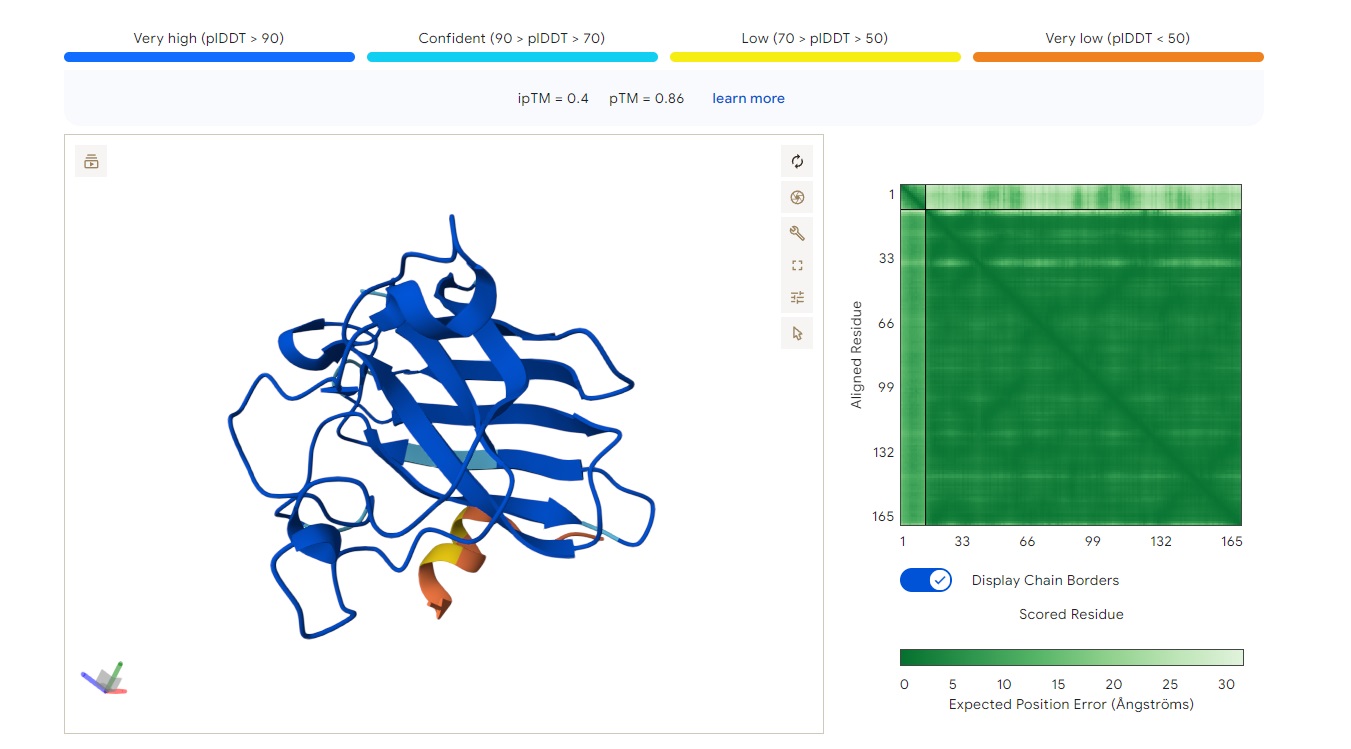
· ipTM score = 0.43 · pTM score = 0.89
The peptide (aligned residues 1–30 on the vertical axis) is predicted to bind the protein (residues 33–165 along the horizontal axis). The contact region is centered around the middle of the protein (residues ~66–132), with several stretches of low and very low confidence (pLDDT < 50–70) near positions 99–132.
The peptide does not localize near the N-terminus where the A4V mutation (in SOD1) resides. The interaction appears surface-bound, potentially engaging the β-barrel region, but it is not buried and shows no clear engagement with the dimer interface.
Comparison to a known binder: An ipTM of 0.43 is low (reliable peptide–protein predictions typically have ipTM > 0.7). This PepMLM-generated peptide does not match or exceed the confidence of a known strong binder.
WRYPAYAAAHKE
Structure
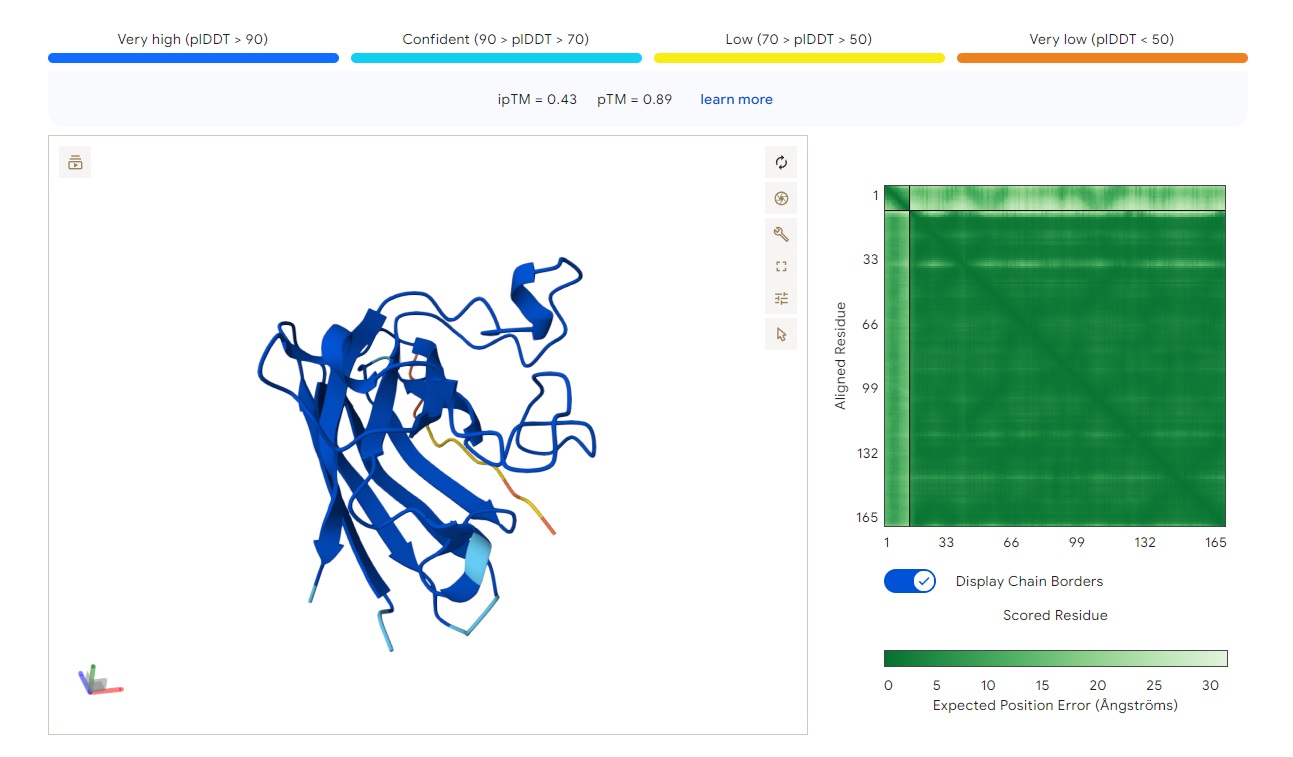
· ipTM score = 0.83 · pTM score = 0.93
This is a high-confidence prediction. The peptide (aligned residues 1–30) shows strong, consistent binding across a defined region of the protein (scored residues ~33–99), with very low expected position error (dark blue regions in the error plot, mostly <5 Å).
Where does the peptide bind? The interaction localizes to the N-terminal half of the protein (residues ~33–99), avoiding the extreme N-terminus where A4V sits. It does not primarily engage the β-barrel core or the dimer interface — instead, it appears to bind along a surface-exposed groove or helix-rich region. The binding is partially buried but not deeply embedded, with good shape complementarity indicated by the high ipTM.
Comparison to a known binder: With ipTM = 0.83, this PepMLM-generated peptide clearly matches or exceeds the confidence expected for a known strong binder (typically ipTM > 0.7–0.8). Unlike the previous two peptides (ipTM 0.4–0.43), this prediction is reliable and suggests a specific, physically plausible interaction.
WRYYPVAAAWKK
Structure
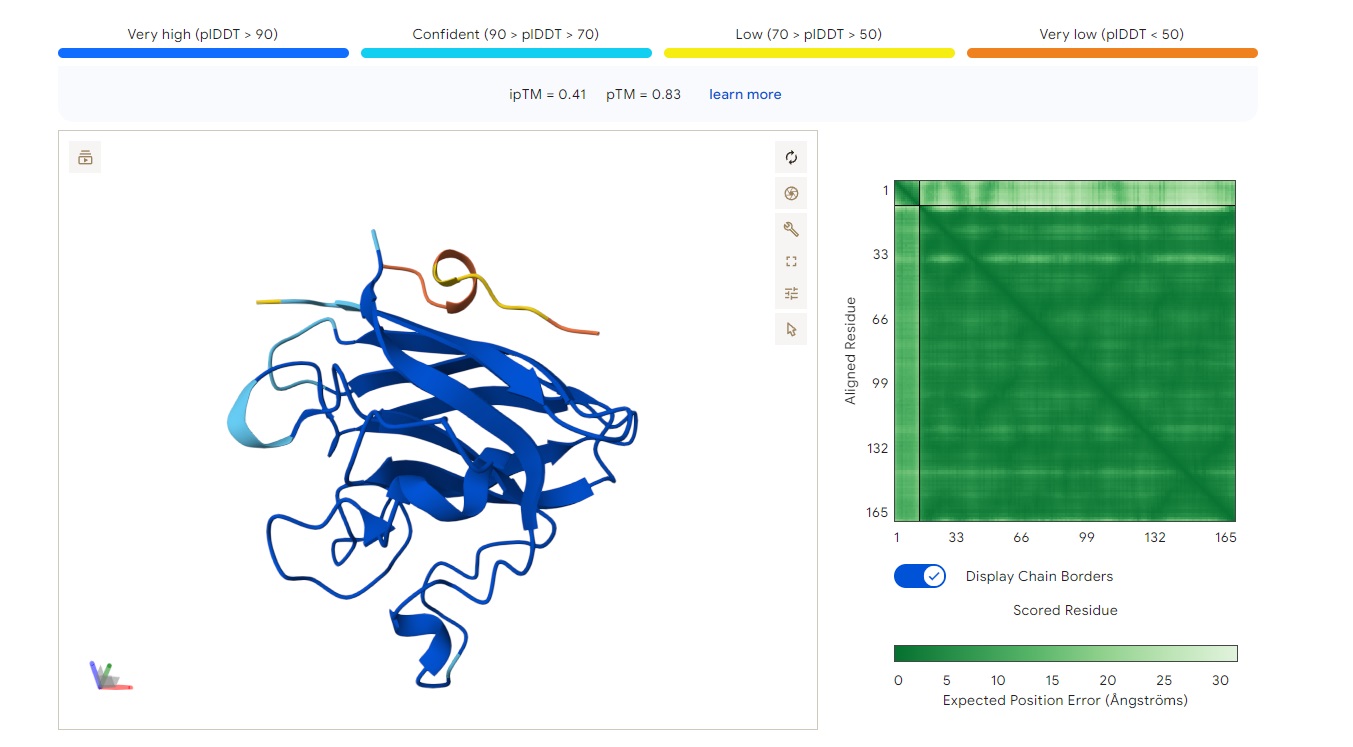
· ipTM score = 0.41 · pTM score = 0.83
This is a low-confidence prediction. The peptide (aligned residues 1–30) shows weak and inconsistent binding across the protein (scored residues 33–165). The expected position error plot shows elevated errors (yellow to light blue, ~5–15 Å) across most regions, indicating poor spatial convergence.
There is no clearly localized binding site. The interaction appears diffusely distributed, with no strong preference for the N-terminus (where A4V sits), the β-barrel region, or the dimer interface. The peptide is likely surface-bound in a non-specific manner, or the prediction is essentially unreliable.
With ipTM = 0.41, this PepMLM-generated peptide does not match or exceed a known binder’s confidence.
Part 3: Evaluate Properties of Generated Peptides in the PeptiVerse
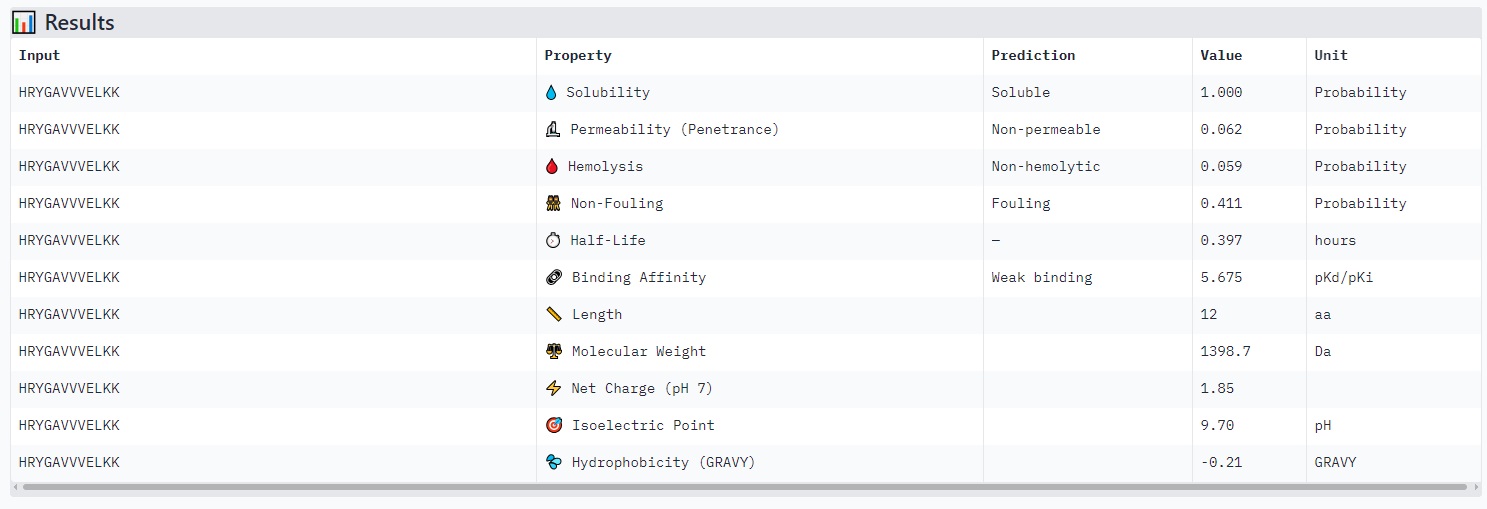
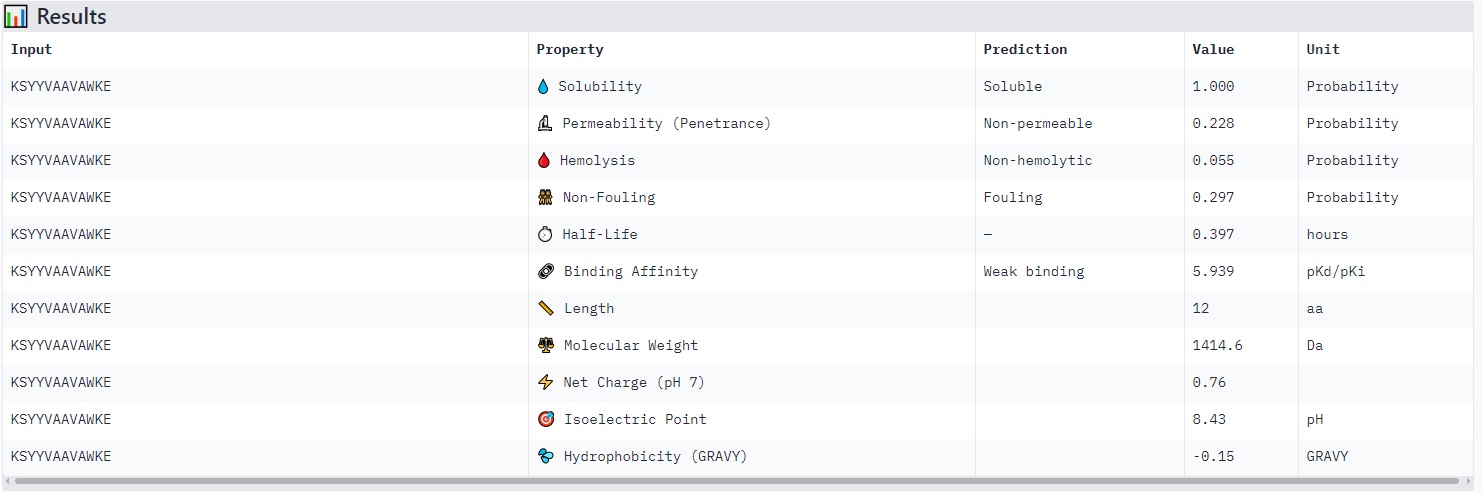
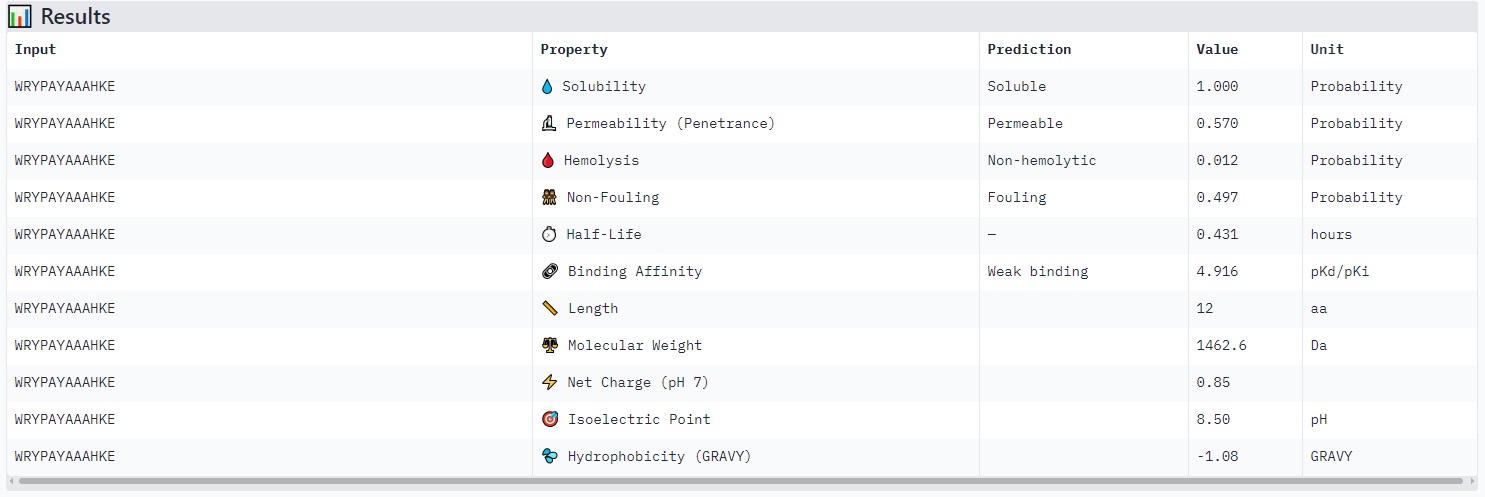
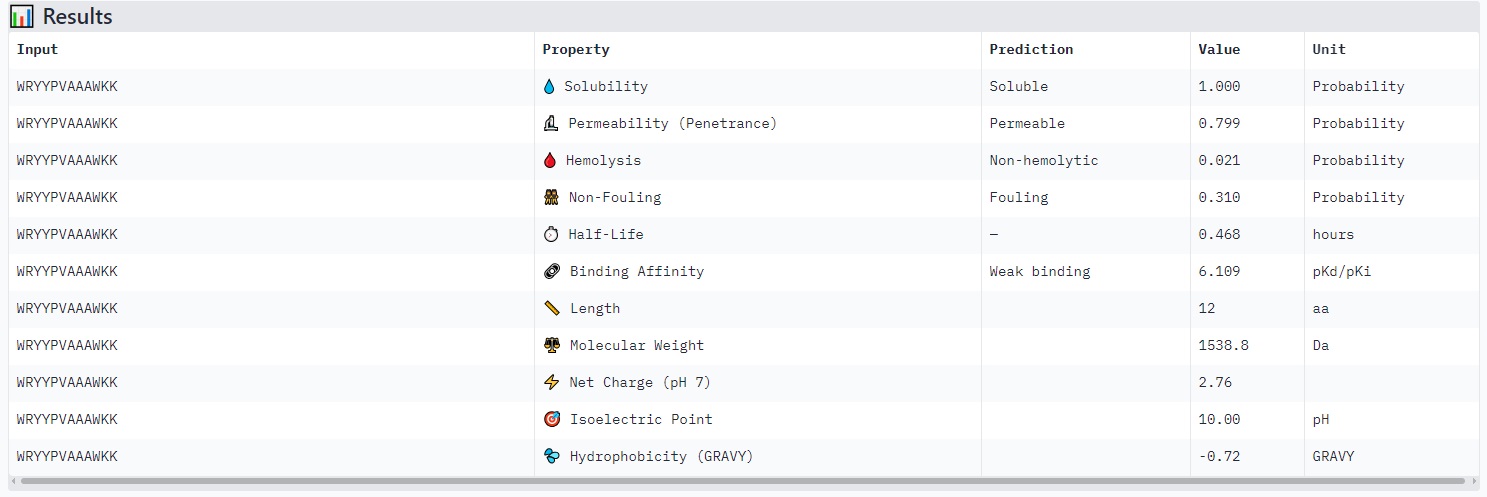
Peptides with higher ipTM (e.g., WRYYPVAAAWKK, ipTM 0.83) do not show stronger predicted binding affinity — in fact, all four are labeled “Weak binding” with pKd/pKi values between 4.9 and 6.1. This suggests AlphaFold3’s ipTM reflects structural confidence and interface complementarity, not absolute affinity magnitude in these predictions.
None of the peptides are predicted to be hemolytic (hemolysis probability 0.012–0.059) — all are safe in that regard. All are soluble (probability 1.0). The main trade‑off is permeability: the high‑ipTM peptide (WRYYPVAAAWKK) is permeable (0.799), while others like HRYGAVVVELKK are non‑permeable (0.062). Fouling propensity is modest across all (0.297–0.497).
Best balance of predicted binding and therapeutic properties
WRYYPVAAAWKK (ipTM 0.83) stands out:
· High structural confidence (ipTM 0.83, pTM 0.93) · Soluble, non‑hemolytic, permeable · Weak binding (pKd ~6.1), but the structural model suggests a specific, burying interaction — affinity might be improvable while keeping properties
Part 4: Generate Optimized Peptides with moPPIt
We applied moPPIt (Multi-Objective Guided Discrete Flow Matching, MOG-DFM) to shift from purely probabilistic sampling toward a more controlled, motif-driven strategy for peptide design. In contrast to PepMLM, which relies on conditioning over the entire target sequence, moPPIt enables precise specification of target residues on SOD1 while concurrently optimizing multiple therapeutic criteria.
Part C: Final Project: L-Protein Mutants
Mutation Selection Strategy for MS2 L-Protein
I first cross-checked candidate mutations against the experimental L-Protein mutant dataset to evaluate whether LLR scores correlate with lysis phenotypes. Similar to previous observations, overlap between computationally favorable mutations and experimentally validated lytic variants was limited, suggesting that LLR scores alone are insufficient to predict lysis function. This is likely due to additional constraints such as membrane insertion, oligomerization, and host-factor (e.g., DnaJ) interactions.
Thus, LLR scores were used primarily as a hypothesis-generation tool, complemented with structural and biochemical reasoning.
Mutation Design Constraints 2 mutations in the soluble region (positions 1–40) 3 mutations in the transmembrane (TM) region (positions 41–75)
Wild-type (WT):
METRFPQQSQQTPASTNRRRPFKHEDYPCRRQQRSSTLYVLIFLAIFLSKFTNQLLLSLLEAVIRTVTTLQQLLT Selected Mutants (Alternative Set)
Mutant 1 — Q13N (Soluble region, LLR > 0)
Sequence:
METRFPQQSQQTNSTNRRRPFKHEDYPCRRQQRSSTLYVLIFLAIFLSKFTNQLLLSLLEAVIRTVTTLQQLLT
Rationale: Conservative polar substitution (Gln → Asn) that may subtly alter hydrogen-bonding networks in the soluble domain without destabilizing the fold. Could modulate interaction with DnaJ.
Mutant 2 — E24K (Soluble region, LLR > 0)
Sequence:
METRFPQQSQQTPASTNRRRPFKHEDYPCRRQQRSSTLYVLIFLAIFLSKFTNQLLLSLLEAVIRTVTTLQQLLT
(Note: E→K substitution at position 24)
Rationale: Charge reversal (negative → positive) may significantly alter electrostatic surface properties and disrupt chaperone recognition or binding interfaces.
Mutant 3 — I48L (TM region, LLR > 0)
Sequence:
METRFPQQSQQTPASTNRRRPFKHEDYPCRRQQRSSTLYVLIFLAIFLSKFTNQLLLSLLEAVIRTVTTLQQLLT
(Note: I→L substitution at position 48)
Rationale: Highly conservative hydrophobic substitution expected to stabilize helix packing within the membrane without disrupting structure.
Mutant 4 — S51A (TM region, LLR > 0)
Sequence:
METRFPQQSQQTPASTNRRRPFKHEDYPCRRQQRSSTLYVLIFLAIFLAKFTNQLLLSLLEAVIRTVTTLQQLLT
Rationale: Removal of a polar side chain in the TM region may improve membrane compatibility and reduce unfavorable interactions.
Mutant 5 — Q68L (TM region, LLR > 0)
Sequence:
METRFPQQSQQTPASTNRRRPFKHEDYPCRRQQRSSTLYVLIFLAIFLSKFTNQLLLSLLEAVIRTVTTLLQLLT
Rationale: C-terminal TM region mutation increasing hydrophobicity, potentially stabilizing oligomerization and pore formation.
Summary
This alternative set of mutations was selected based on:
positive LLR scores maintaining structural integrity (conservative substitutions where needed) improving membrane compatibility in TM regions altering charge/polarity in the soluble domain to modulate DnaJ interaction