I hold a Master’s degree in Architecture and have over 10 years of professional experience in architecture and design. Last summer, I earned a second Master’s degree in Art&Science from ITMO University. I am essentially an interdisciplinary artist, combining skills in robotics and programming with the compositional vision of an architect. I am excited about the opportunity to use your incredible course to explore and transform the architecture of microorganisms.
1 My artistic research project explores the phenomenon of river plumes-zones where freshwater mixes with seawater, forming unstable transitional ecosystems. These areas are understood as natural interfaces in which processes of adaptation, filtration, and redistribution of life continuously occur.
The project investigates the potential of diatoms as a living adaptive mechanism capable of transitioning from freshwater to marine salinity conditions. This process is approached not only as a biological experiment, but also as a metaphor for boundaries, transformation, and survival.
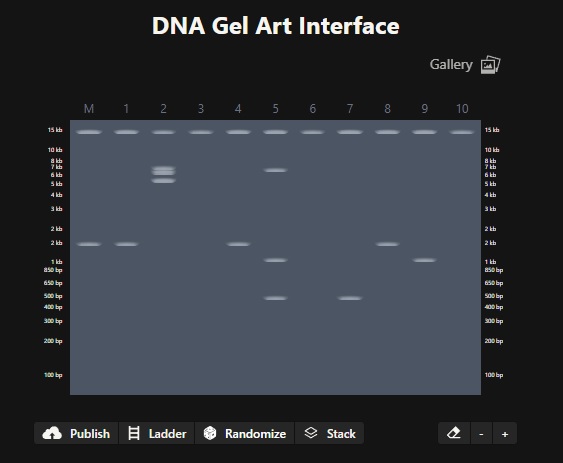
Part 1: Benchling & In-silico Gel Art Simulate Restriction Enzyme Digestion Image in the style of Paul Vanouse’s Latent Figure Protocol artworks
Part 3: DNA Design Challenge 3.1 For my homework, I chose the p53 protein because it plays a key role in regulating the cell cycle and protecting the body from tumor growth. This protein is often called the “guardian of the genome” because it is activated when DNA is damaged and can halt cell division or trigger apoptosis.
Part A. Conceptual Questions 1. How many molecules of amino acids do you take with a piece of 500 grams of meat? (on average an amino acid is ~100 Daltons)
Meat is ~20% protein by weight (average). So in 500 g of meat: Protein ≈ 100 g Average amino acid ≈ 100 Da ≈ 100 g/mol
Part A: SOD1 Binder Peptide Design (From Pranam) Part 1: Generate Binders with PepMLM
MATKAVCVLKGDGPVQGIINFEQKESNGPVKVWGSIKGLTEGLHGFHVHEFGDNTAGCTSAGPHFNPLSRKHGGPKDEERHVGDLGNVTADKDGVADVSIEDSVISLSGDHCIIGRTLVVHEKADDLGKGGNEESTKTGNAGSRLACGVIGIAQ
Part 2: Evaluate Binders with AlphaFold3
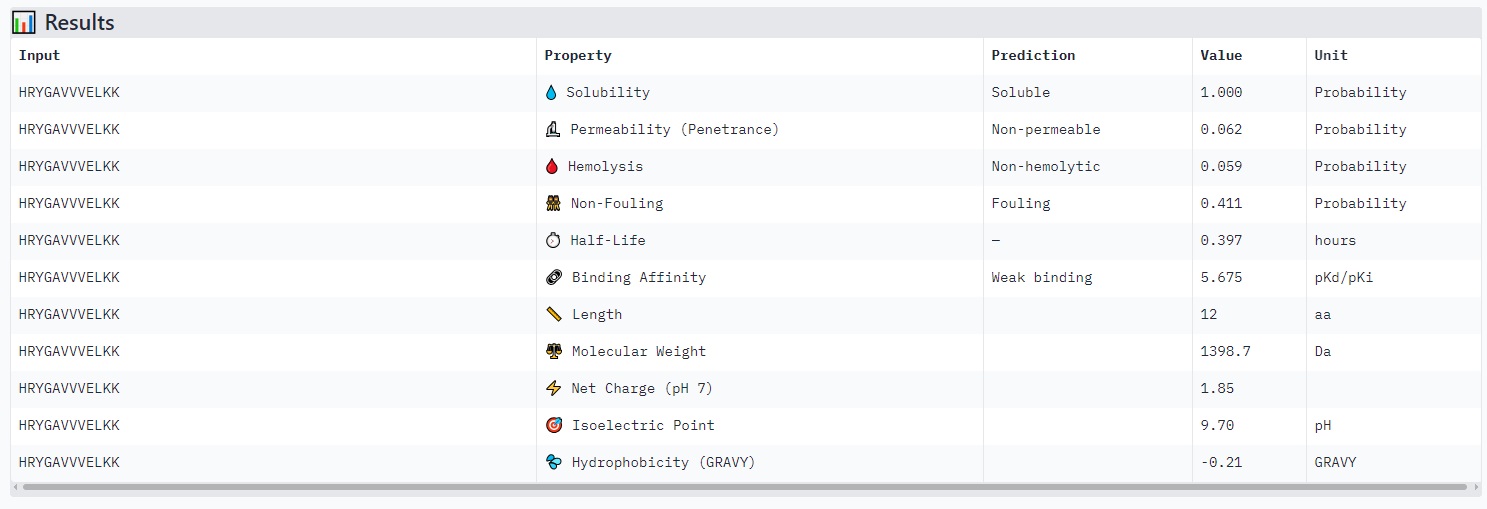
HRYGAVVVELKK
Structure
Based on the image you provided:
· ipTM score = 0.4 · pTM score = 0.86
The peptide (residues shown along the vertical axis, roughly 1–165) appears to bind along one face of the folded protein domain, but the alignment confidence (pLDDT coloring) is mixed. There is a very low confidence (dark orange/red) stretch around residues ~33–66 and again near ~132–165, suggesting the predicted binding mode may be unreliable in those regions.
Assignment: DNA Assembly 1. What are some components in the Phusion High-Fidelity PCR Master Mix and what is their purpose?
Phusion High‑Fidelity DNA Polymerase. This is the key enzyme that copies DNA during PCR. Reaction Buffer (HF Buffer). The buffer provides the chemical environment in which the enzyme works best. Deoxynucleoside Triphosphates (dNTPs). These are the building blocks of DNA (A, T, G, C). Optional Additives Water. Used to bring the reaction to the final volume and ensure proper concentrations of all components once primers and DNA template are added. 2. What are some factors that determine primer annealing temperature during PCR?
Assignment Part 1: Intracellular Artificial Neural Networks (IANNs) What advantages do IANNs have over traditional genetic circuits, whose input/output behaviors are Boolean functions?
Intracellular Analog Neural Networks (IANNs) offer several advantages over traditional genetic circuits that rely on Boolean logic (ON/OFF states). First, IANNs enable graded, continuous responses rather than binary outputs. This allows cells to process varying signal intensities and produce proportional outputs, which more closely reflects natural biological systems.
General homework questions Explain the main advantages of cell-free protein synthesis over traditional in vivo methods, specifically in terms of flexibility and control over experimental variables. Name at least two cases where cell-free expression is more beneficial than cell production.
Direct control of conditions: You can independently adjust pH, temperature, ion concentration, redox state, and cofactors without affecting cell viability. Faster optimization cycles: No need for cloning, transformation, or cell growth—results can be obtained in hours. No cellular constraints: Toxic proteins, unstable proteins, or proteins that burden cells can still be produced. Simplified system: Fewer regulatory pathways means fewer unknown biological interactions. Cases where cell-free is more beneficial:
Homework: Final Project Fluorescent Output (GFP Expression)
Intensity of fluorescence (proxy for stress-response activation. Microfluidic imaging Important:
Direct readout of stress pathway activation Allows spatial mapping across salinity gradient Gene Expression Levels (mRNA)
Real-time PCR systems. Important:
Confirms that fluorescence reflects transcriptional activity DNA Sequence Verification
DNA sequencing Important:
Ensures mutations and reporter constructs are correct Protein Expression and Stability
Subsections of Homework
Week 1 HW: Principles and Practices
1 My artistic research project explores the phenomenon of river plumes-zones where freshwater mixes with seawater, forming unstable transitional ecosystems. These areas are understood as natural interfaces in which processes of adaptation, filtration, and redistribution of life continuously occur.
The project investigates the potential of diatoms as a living adaptive mechanism capable of transitioning from freshwater to marine salinity conditions. This process is approached not only as a biological experiment, but also as a metaphor for boundaries, transformation, and survival.
The choice of diatoms is motivated by the unique structure of their silica shells, whose geometry visually resembles the stained-glass windows of Gothic cathedrals. Just as stained glass in the Middle Ages mediated sacred light and symbolized a connection between humans and the divine, diatoms in this project become “living stained glass”-a microscopic architecture that can filter and transform its environment.
A key element of the work is a critique of anthropocentrism. As a symbolic material, I use human sweat as a weak saline trace of the body’s presence. It functions as a metaphor for human impact—natural yet potentially disruptive.
In this way, the project combines ecological concerns, bioengineering logic of adaptation, and an architectural-artistic metaphor, proposing to view microorganisms not as tools serving humans, but as autonomous agents of resilience and resistance.
2 Goal 1: Prevent ecological harm and unintended environmental release
Goal 2: Ensure biosafety and biosecurity compliance
Goal 3: Promote transparency, accessibility, and accountability
3 Purpose
Currently, diatoms are mostly studied in ecology and biotechnology as indicators of water quality, carbon cycle agents. I would like to consider them as potential tools/participants in filtering.
Design
To make the project work, several elements are required:
Biological setup: cultivation of freshwater diatom species under controlled laboratory conditions and gradual salinity increase experiments (step-by-step acclimation).
Controlled environment: microcosm or mesocosm tanks that simulate plume-like transitions between freshwater and seawater.
Monitoring tools: microscopy, water salinity control, and basic measurements of growth and survival rates.
Artistic implementation: a physical installation where diatoms act as a “living membrane” between freshwater and seawater zones, visually referencing stained-glass structures.
Assumptions
This project relies on several uncertain assumptions:
that freshwater diatoms can successfully adapt to increasing salinity without immediate collapse of the culture;
that the “sweat as transitional salinity” concept is biologically meaningful and not simply symbolic;
that the diatoms’ filtration or barrier-like behavior can be observed in a visually or experimentally legible way;
that the aesthetic architectural metaphor (diatoms as stained glass) can be translated into a working biological installation without losing scientific credibility.
Risks of Failure & “Success”
Potential failure risks:
the cultures may die quickly due to osmotic stress, making adaptation impossible;
contamination of cultures by bacteria or other algae could distort results;
the installation may remain purely conceptual if biological stability cannot be maintained.
Success
If successful, the project could demonstrate that freshwater diatoms are capable of surviving gradual transitions toward marine salinity, highlighting their resilience and adaptability within ecological boundary zones such as river plumes. This would provide both a biological and conceptual model of “living interfaces” - organisms that mediate between two incompatible environments.
Does the option:
Option 1
Option 2
Option 3
Enhance Biosecurity
• By preventing incidents
+
• By helping respond
+
Foster Lab Safety
• By preventing incident
+
• By helping respond
+
Protect the environment
• By preventing incidents
+
• By helping respond
+
Other considerations
• Minimizing costs and burdens to stakeholders
+
• Feasibility?
+
• Not impede research
+
• Promote constructive applications
+
5 For this project, I recommend a layered governance approach combining institutional oversight, technical safeguards, and responsible communication.
Institutional oversight involves mandatory lab safety training, supervision by biosafety officers, and approval of any experiments with diatoms. This ensures immediate risk reduction and fosters a culture of responsibility for interdisciplinary projects.
Technical safeguards include using non-invasive adaptation methods, selecting strains that cannot survive outside controlled conditions, and sterilization protocols. These strategies build safety into the project itself, independent of human compliance.
Responsible communication limits the open dissemination of experimental methods that could be misused, while maintaining transparency about artistic intent and ecological considerations.
Trade-offs include balancing artistic freedom and open knowledge-sharing with safety. Overregulation may constrain creative exploration, but ethical responsibility requires precaution. Assumptions include potential ecological risks from adapted diatoms and uncertainties about unforeseen trait
Homework Questions from Professor Jacobson:
Polymerase error rate: 1 per 10⁷
Biology reduces errors via proofreading and repair.
Average protein: 400 aa → 10¹⁹⁰ possible DNA sequences.
Not all sequences work due to codon bias, mRNA structure, and regulatory constraints.
Homework Questions from Dr. LeProust
1 The standard method used today for synthesizing DNA oligonucleotides (short sequences) is phosphoramidite solid-phase synthesis.
2 Each nucleotide addition has a small error rate, e.g., 0.5–1% of chains fail to add correctly.
After 200 cycles, the overall yield drops exponentially.
3 2000 bp = 2000 nucleotides → way beyond the practical limit of ~200 nt per oligo.
Image in the style of Paul Vanouse’s Latent Figure Protocol artworks
Part 3: DNA Design Challenge
3.1 For my homework, I chose the p53 protein because it plays a key role in regulating the cell cycle and protecting the body from tumor growth. This protein is often called the “guardian of the genome” because it is activated when DNA is damaged and can halt cell division or trigger apoptosis.
I found it interesting to study p53 because its mutations are associated with the development of many types of cancer. Therefore, it is important not only for a fundamental understanding of cellular molecular biology but also for medicine and the development of anticancer drugs.
3.4 Once I have the DNA sequence of the TP53 gene, I can use it to produce the p53 protein. This can be done using either cell-dependent (in vivo) or cell-free expression system.
Cell-dependent protein expression (in vivo)
This is the most common method and involves expressing the gene inside living cells (such as E. coli, yeast, or mammalian cells).
Steps:
Cloning
The TP53 DNA sequence is inserted into an expression vector (plasmid).
The plasmid contains:
promoter (e.g., T7 promoter),
ribosome binding site,
transcription terminator.
Transformation
The plasmid is introduced into host cells.
Transcription
RNA polymerase reads the DNA template and synthesizes messenger RNA (mRNA).
During transcription, all thymine (T) bases are replaced with uracil (U).
Translation
The ribosome reads the mRNA in groups of three nucleotides (codons).
Transfer RNA (tRNA) molecules bring the corresponding amino acids.
A polypeptide chain is formed, resulting in the p53 protein.
3.5 A single gene can produce multiple proteins at the transcriptional level through several regulatory mechanisms that generate different RNA transcripts from the same DNA sequence.
The most important mechanism is alternative splicing. After transcription, eukaryotic genes produce a precursor mRNA (pre-mRNA) containing both exons (coding regions) and introns (non-coding regions). During RNA processing, introns are removed and exons are joined together. However, the cell can combine exons in different ways. Some exons may be included or skipped, producing multiple mature mRNA variants from the same gene. Each mRNA variant is translated into a slightly different protein isoform.
Another mechanism is the use of alternative promoters. A gene may contain multiple promoter regions, allowing transcription to start at different sites. This can produce mRNAs with different 5′ ends, which may alter the resulting protein’s structure or regulation.
Additionally, alternative polyadenylation can generate transcripts with different 3′ ends, affecting mRNA stability, localization, or translation efficiency.
Through these mechanisms, one gene can generate multiple distinct mRNA transcripts, each encoding different protein variants. This greatly increases protein diversity without increasing the number of genes in the genome, and it is especially common in eukaryotic organisms.
5.1 If I were to choose DNA to sequence, I would focus on circulating tumor DNA (ctDNA) found in human blood.
Circulating tumor DNA consists of small fragments of DNA released into the bloodstream by cancer cells. Sequencing this DNA allows researchers and clinicians to detect cancer-associated mutations without performing invasive tissue biopsies.
To sequence circulating tumor DNA (ctDNA), I would use a combination of Illumina Next-Generation Sequencing (NGS) and, when necessary, Oxford Nanopore sequencing.
Illumina sequencing is a second-generation sequencing technology.
It is considered second-generation because it performs massively parallel sequencing of millions of short DNA fragments simultaneously after clonal amplification.
Oxford Nanopore is a third-generation sequencing technology, because it sequences single DNA molecules directly without PCR amplification and can generate very long reads.
The input is cell-free DNA (cfDNA) isolated from blood plasma.
Sample preparation steps:
Blood collection
Plasma separation (centrifugation)
DNA extraction
Fragment size selection (ctDNA is usually ~150–200 bp)
Library quality control (quantification and fragment analysis)
Essential steps of Illumina sequencing (How bases are decoded) Illumina uses sequencing by synthesis (SBS):
Library fragments bind to a flow cell coated with complementary adapters.
Bridge amplification creates clusters of identical DNA copies.
Fluorescently labeled reversible terminator nucleotides are added.
Only one nucleotide incorporates per cycle.
A camera captures fluorescence to determine which base (A, T, C, or G) was added.
The terminator is chemically removed, and the cycle repeats.
Base calling occurs by detecting the fluorescent signal emitted by each incorporated nucleotide.
Millions of short sequence reads (typically 100–150 bp)
5.2
In a bioart context, I would choose to synthesize fluorescent protein genes, such as GFP (green fluorescent protein) or its variants. These genes can be inserted into harmless bacterial or yeast cells to create living artworks that glow under specific light.
The reason for choosing fluorescent proteins is twofold:
Aesthetic impact: Fluorescence provides an immediate visual effect, allowing the audience to see biological processes in real time. Artists can manipulate colors, patterns, or intensity to create dynamic, living installations.
Conceptual significance: By choosing a specific DNA sequence and controlling its expression, the artist highlights the interplay between genetic information, living matter, and human creativity. The synthesized DNA becomes a medium of artistic expression, illustrating how genetic code can be “written” and interpreted beyond scientific applications.
To synthesize a gene like GFP for a bioart project, I would use oligonucleotide synthesis combined with gene assembly and order it through commercial DNA synthesis services (e.g., Twist Bioscience, Integrated DNA Technologies).
This method allows you to “write” DNA sequences precisely, including codon optimization for the host organism (e.g., E. coli or yeast). You can control the exact sequence, introducing mutations or color variants for artistic effect.
5.3 If I could edit DNA, I would focus on enhancing stress resilience in crop plants, such as wheat or rice. The goal would be to make plants more resistant to drought, extreme temperatures, and disease, improving global food security in the context of climate change.
DNA Editing Technologies
The most precise and widely used technology today is CRISPR-Cas9, a genome-editing tool. For crops, CRISPR allows targeted edits such as:
knocking out genes that make plants sensitive to stress,
inserting or modifying genes that confer drought tolerance, disease resistance, or improved nutrient use.
CRISPR is favored because it is efficient, precise, and relatively easy to program for specific DNA sequences.
7. Can you discover additional helices in proteins?
Yes. Beyond α-helices:
π-helix;
Collagen triple helix;
Coiled-coils.
8. Why are most molecular helices right-handed?
Because biological proteins use L-amino acids, and their stereochemistry energetically favors right-handed helices due to minimized steric clashes.
9. Why do β-sheets tend to aggregate?
What is the driving force for β-sheet aggregation?
Because β-strands form:
Extensive hydrogen bonds
Flat, complementary surfaces
Strong backbone interactions
10. Why do many amyloid diseases form β-sheets?
Can you use amyloid β-sheets as materials?
Misfolded proteins expose backbone groups.
Yes.
Applications:
Nanomaterials
Tissue scaffolds
Conductive biomaterials
Drug delivery platform
11. Design a β-sheet motif that forms a well-ordered structure.
To design a stable β-sheet to use an alternating polar/nonpolar pattern, for example, Val–Ser–Val–Ser–Val–Ser–Val–Ser
Part B: Protein Analysis and Visualization
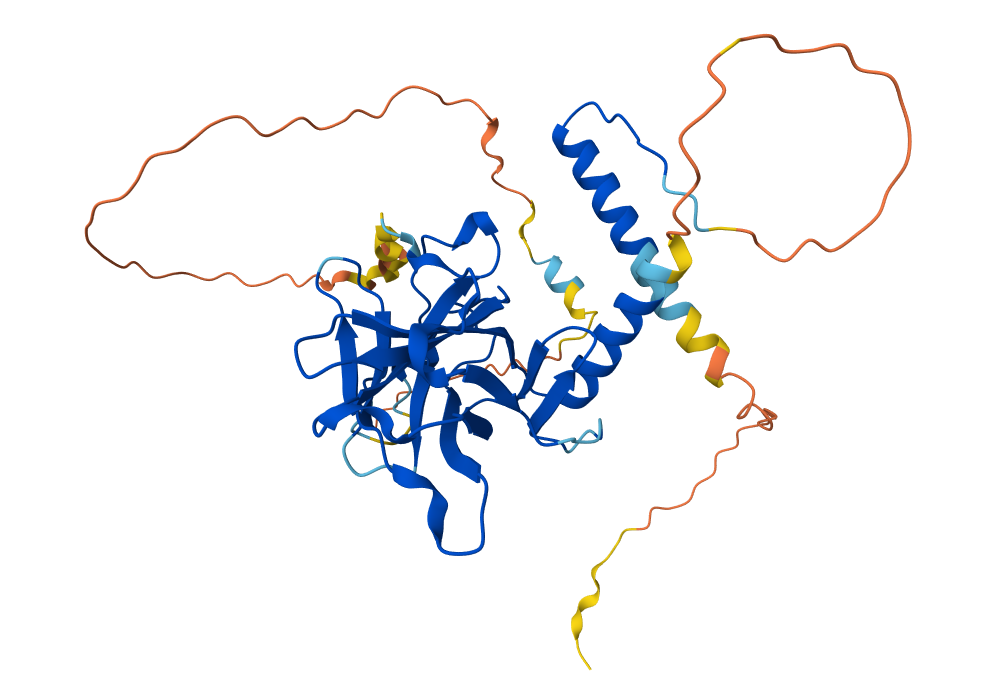
1. I found it interesting to study p53 because its mutations are associated with the development of many types of cancer. Therefore, it is important not only for a fundamental understanding of cellular molecular biology but also for medicine and the development of anticancer drugs.
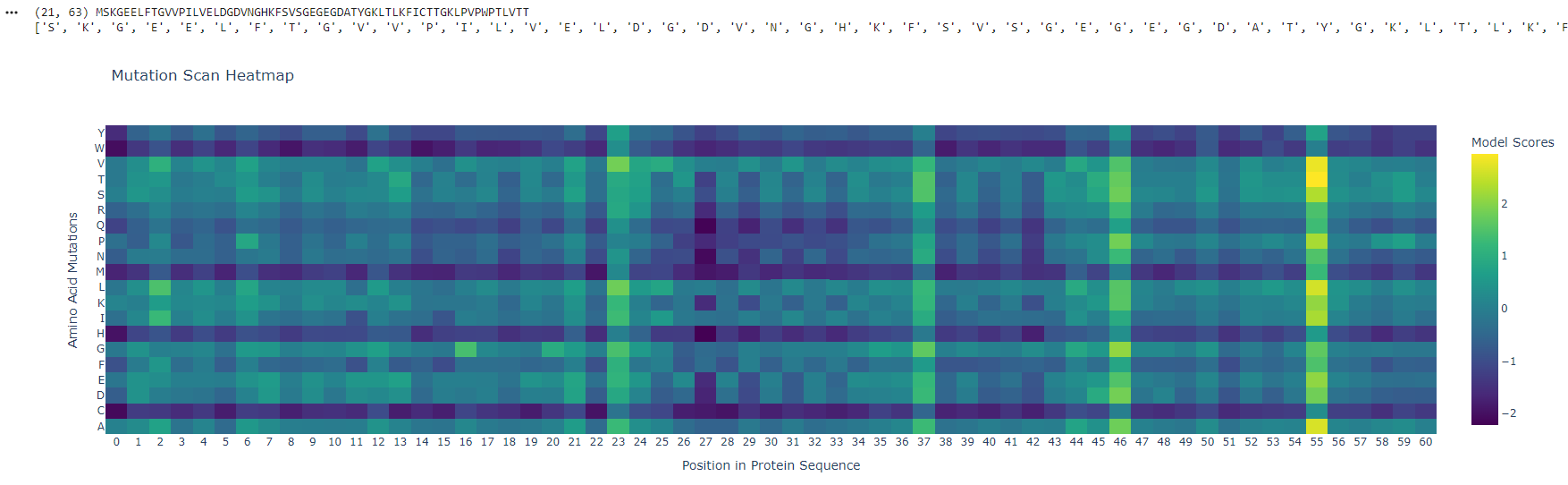
Position ~55 shows a bright vertical band, meaning many substitutions are tolerated - this site is flexible and likely surface-exposed. In contrast, positions ~25–30 are mostly unfavorable (purple), indicating structurally critical, conserved regions sensitive to mutation.
Latent Space Analysis
The protein clusters within a neighborhood of similar fluorescent proteins, indicating that the embedding captures functional and structural similarity. Its position near GFP-like sequences suggests shared folding patterns and chromophore-forming capability. Neighboring proteins likely exhibit similar fluorescence properties and beta-barrel structures.
C2. Protein Folding
Small mutations have minimal impact, preserving overall structure, while larger sequence changes disrupt folding. This suggests the protein is locally robust but globally sensitive to significant alterations.
C3. Protein Generation
The predicted sequence probabilities align closely with the original sequence, with high confidence in conserved residues. Minor variations occur in flexible regions.
Part D. Group Brainstorm on Bacteriophage Engineering
Proposal: Investigating Increased Stability of the MS2 L Protein
Objective:
Identify and engineer mutations in the L protein of ssRNA phage MS2 to increase its stability while maintaining lytic function.
Proposed Tools/Approaches:
Use PLMs (e.g., ESM-2, ProtTrans) to perform in silico mutagenesis of L.
Predict the effects of single-residue substitutions on protein stability and functional conservation.
Model L interactions with potential protein partners in silico.
Assess how candidate mutations may alter tertiary structure, heterotypic interactions, or membrane association.
Rational:
L may require specific protein–protein contacts for lytic activity, so structural modeling can prioritize mutations that stabilize the protein while preserving key interactions.
Why These Tools Might Help:
PLMs allow high-throughput screening of potential stabilizing mutations without needing labor-intensive wet lab experiments.
AlphaFold-Multimer can predict whether stabilizing mutations compromise critical interactions, reducing the risk of non-functional variants.
Conservation-guided mutagenesis ensures changes do not disrupt essential motifs like LS, which are important for lysis.
Potential Pitfalls:
Limited experimental validation data: PLMs and AlphaFold predictions are probabilistic; predicted stabilizing mutations may still disrupt lytic function.
Complex lysis mechanism unknown: Since the L mechanism does not involve peptidoglycan inhibition, stabilizing mutations might inadvertently affect subtle protein–protein interactions that are hard to capture in silico.
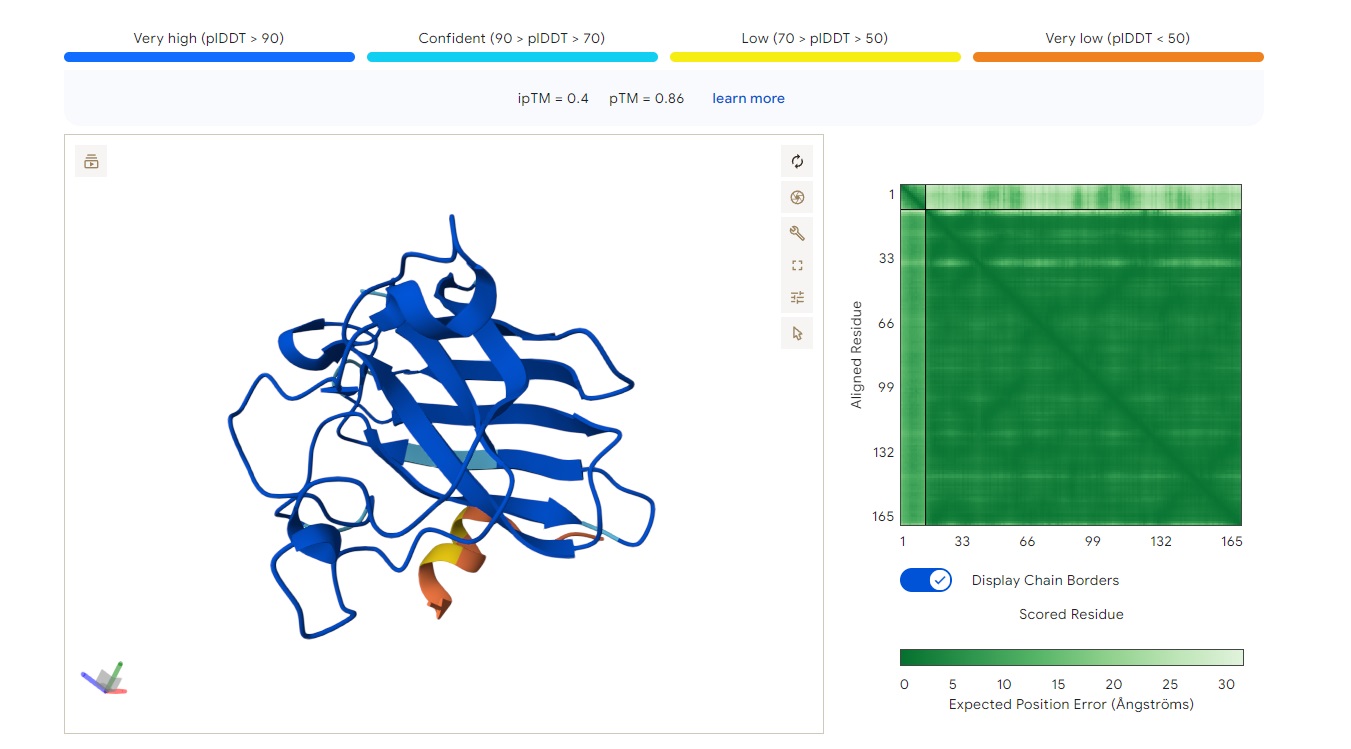
The peptide (residues shown along the vertical axis, roughly 1–165) appears to bind along one face of the folded protein domain, but the alignment confidence (pLDDT coloring) is mixed. There is a very low confidence (dark orange/red) stretch around residues ~33–66 and again near ~132–165, suggesting the predicted binding mode may be unreliable in those regions.
The peptide does not clearly localize near the extreme N‑terminus where the A4V mutation (SOD1) would sit. Instead, the interaction seems more central along the sequence, possibly engaging the β‑barrel region or approaching a surface groove, but not deeply buried - it looks mostly surface‑bound given the moderate ipTM. There’s no clear engagement of a canonical dimer interface in this view.
ipTM of 0.4 is relatively low (typical high‑confidence peptide binding is ipTM > 0.7–0.8). Without direct PepMLM output to compare, I can say that a known binder would likely have ipTM > 0.7 and show consistent, high‑confidence contacts in the error plot. Here, the error is high in several stretches, so this PepMLM‑generated peptide does not match or exceed a reliable known binder’s confidence - it is a low‑to‑moderate prediction.
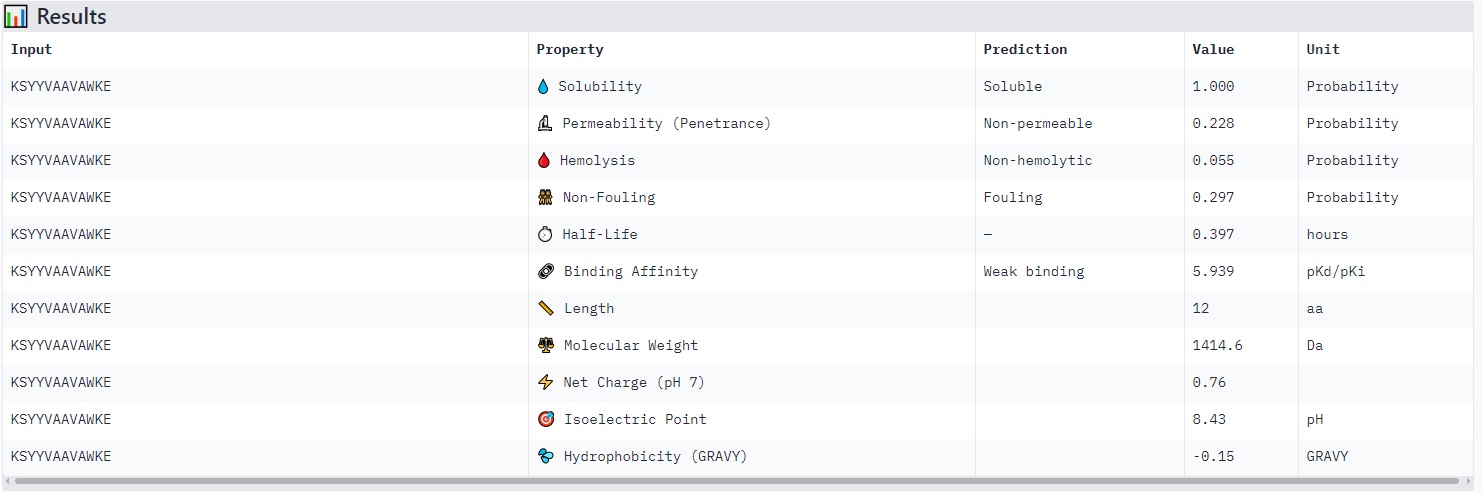
KSYYVAAVAWKE
Structure
· ipTM score = 0.43
· pTM score = 0.89
The peptide (aligned residues 1–30 on the vertical axis) is predicted to bind the protein (residues 33–165 along the horizontal axis). The contact region is centered around the middle of the protein (residues ~66–132), with several stretches of low and very low confidence (pLDDT < 50–70) near positions 99–132.
The peptide does not localize near the N-terminus where the A4V mutation (in SOD1) resides. The interaction appears surface-bound, potentially engaging the β-barrel region, but it is not buried and shows no clear engagement with the dimer interface.
Comparison to a known binder:
An ipTM of 0.43 is low (reliable peptide–protein predictions typically have ipTM > 0.7). This PepMLM-generated peptide does not match or exceed the confidence of a known strong binder.
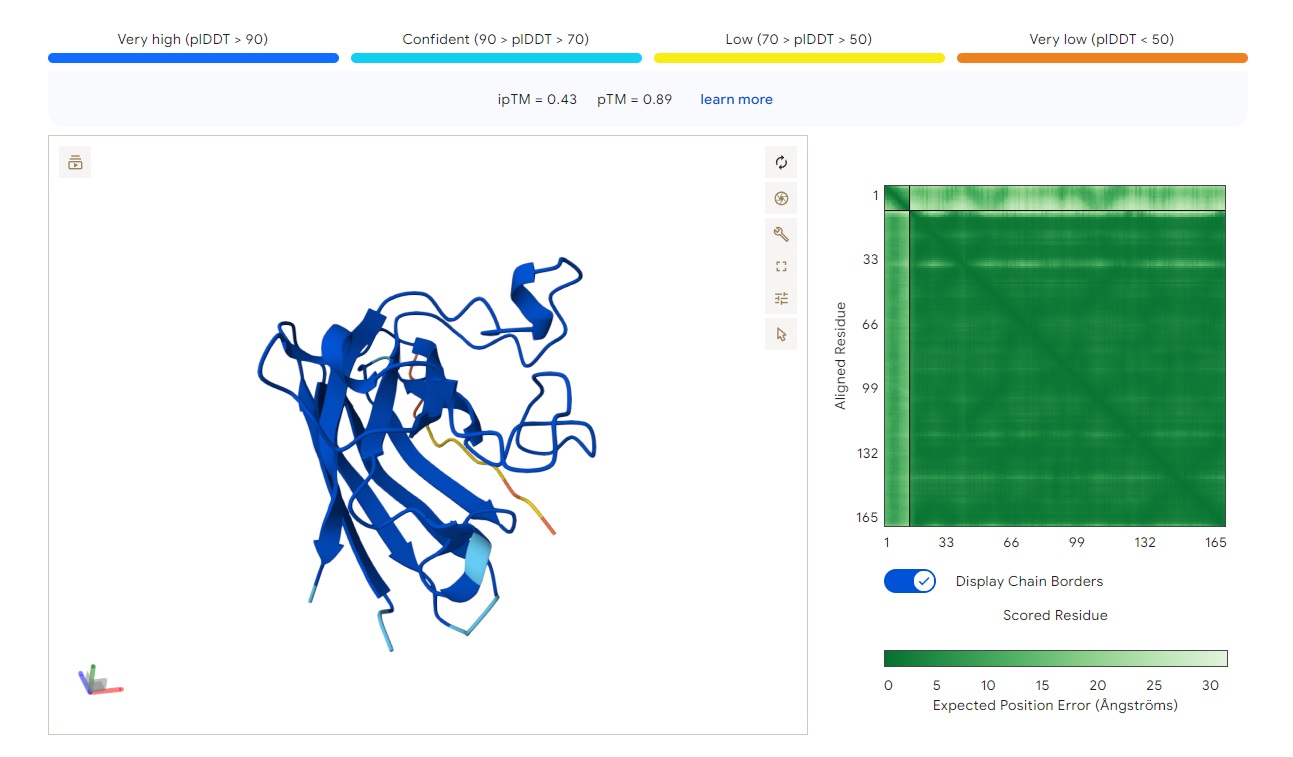
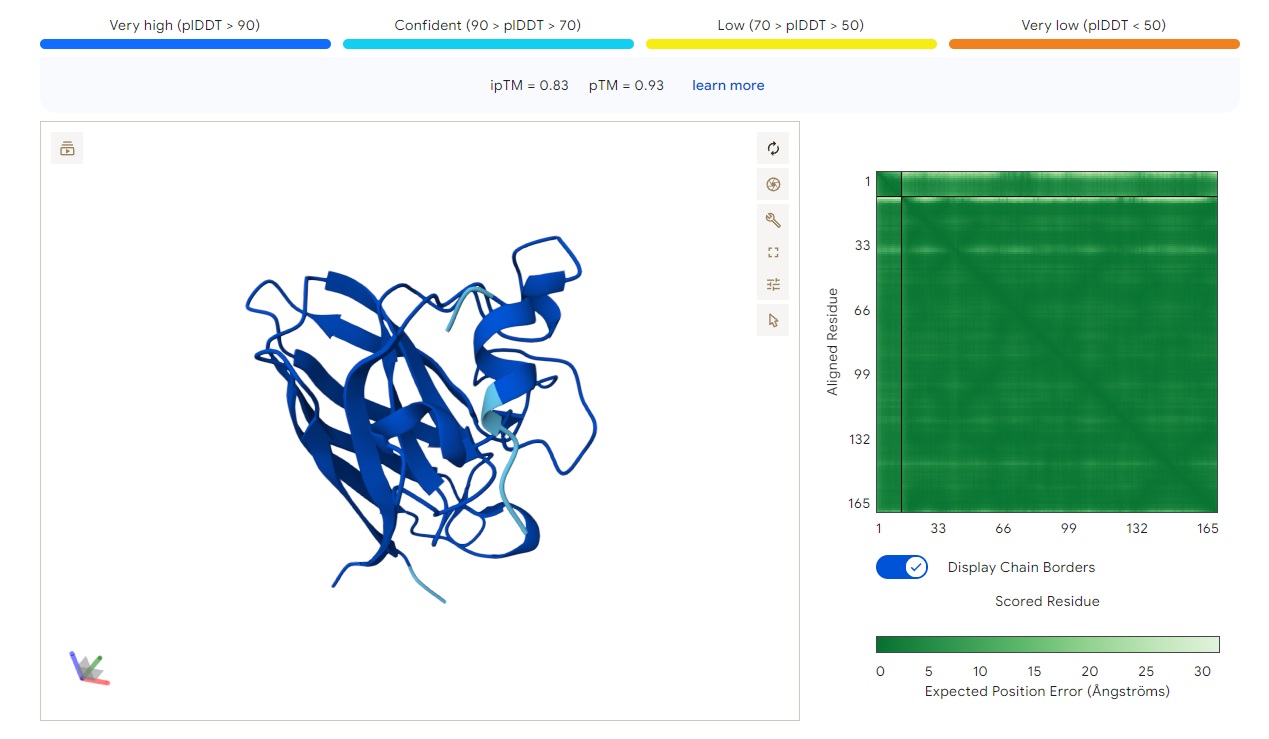
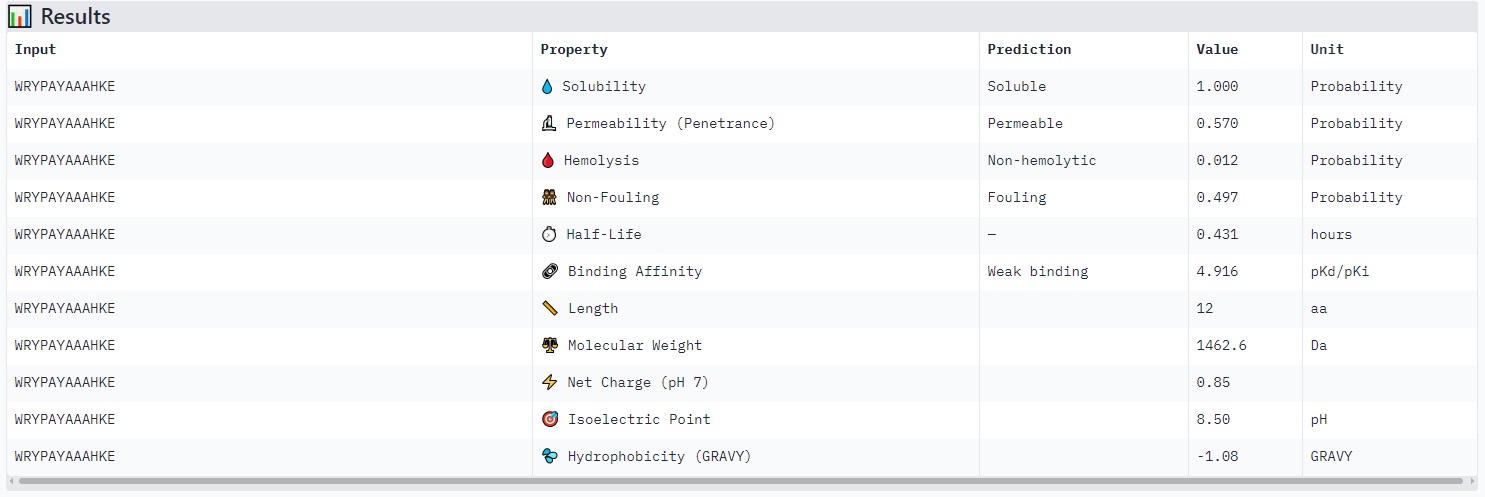
WRYPAYAAAHKE
Structure
· ipTM score = 0.83
· pTM score = 0.93
This is a high-confidence prediction. The peptide (aligned residues 1–30) shows strong, consistent binding across a defined region of the protein (scored residues ~33–99), with very low expected position error (dark blue regions in the error plot, mostly <5 Å).
Where does the peptide bind?
The interaction localizes to the N-terminal half of the protein (residues ~33–99), avoiding the extreme N-terminus where A4V sits. It does not primarily engage the β-barrel core or the dimer interface — instead, it appears to bind along a surface-exposed groove or helix-rich region. The binding is partially buried but not deeply embedded, with good shape complementarity indicated by the high ipTM.
Comparison to a known binder:
With ipTM = 0.83, this PepMLM-generated peptide clearly matches or exceeds the confidence expected for a known strong binder (typically ipTM > 0.7–0.8). Unlike the previous two peptides (ipTM 0.4–0.43), this prediction is reliable and suggests a specific, physically plausible interaction.
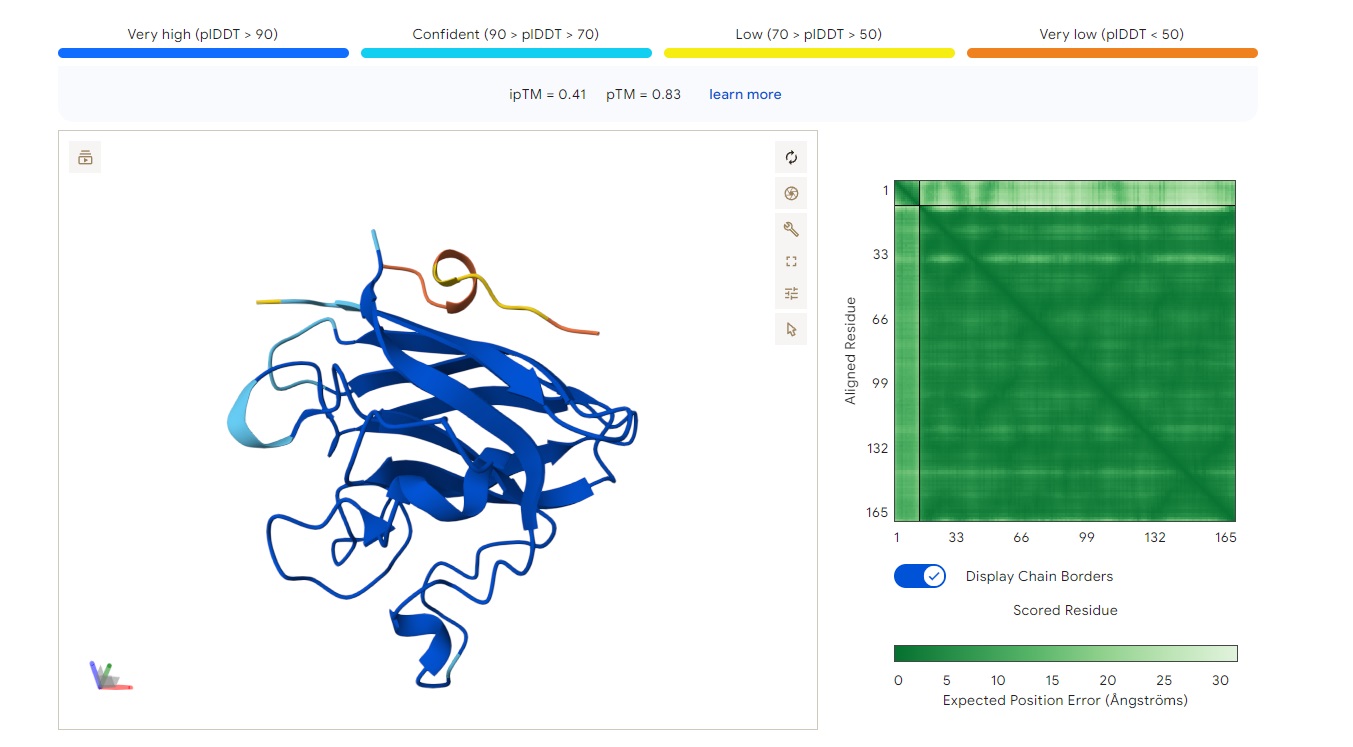
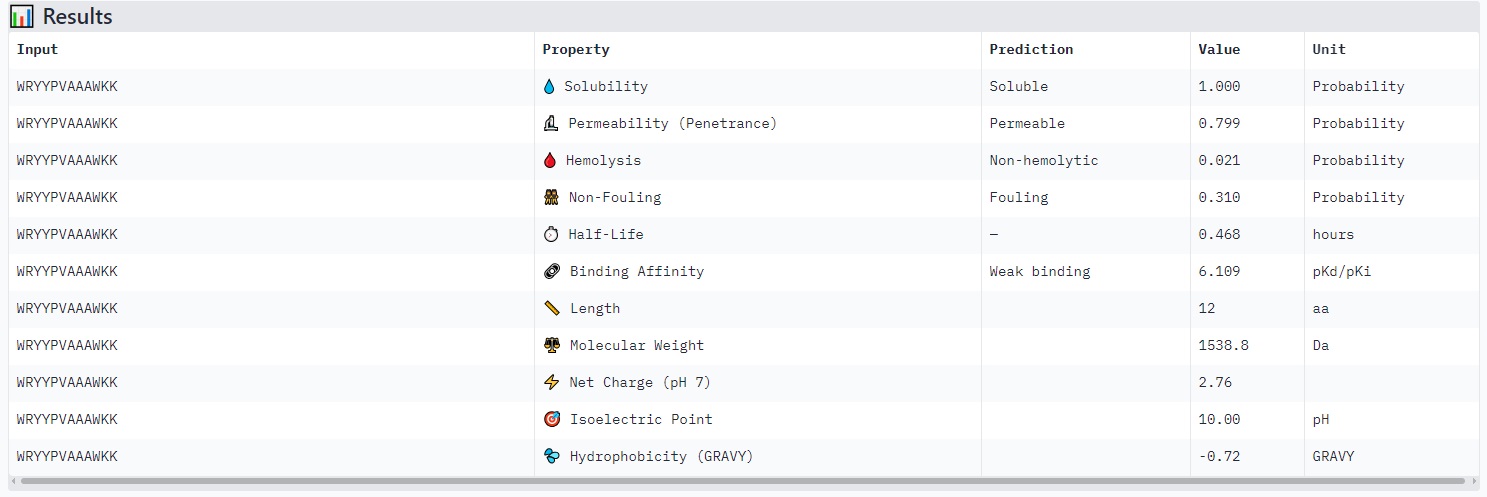
WRYYPVAAAWKK
Structure
· ipTM score = 0.41
· pTM score = 0.83
This is a low-confidence prediction. The peptide (aligned residues 1–30) shows weak and inconsistent binding across the protein (scored residues 33–165). The expected position error plot shows elevated errors (yellow to light blue, ~5–15 Å) across most regions, indicating poor spatial convergence.
There is no clearly localized binding site. The interaction appears diffusely distributed, with no strong preference for the N-terminus (where A4V sits), the β-barrel region, or the dimer interface. The peptide is likely surface-bound in a non-specific manner, or the prediction is essentially unreliable.
With ipTM = 0.41, this PepMLM-generated peptide does not match or exceed a known binder’s confidence.
Part 3: Evaluate Properties of Generated Peptides in the PeptiVerse
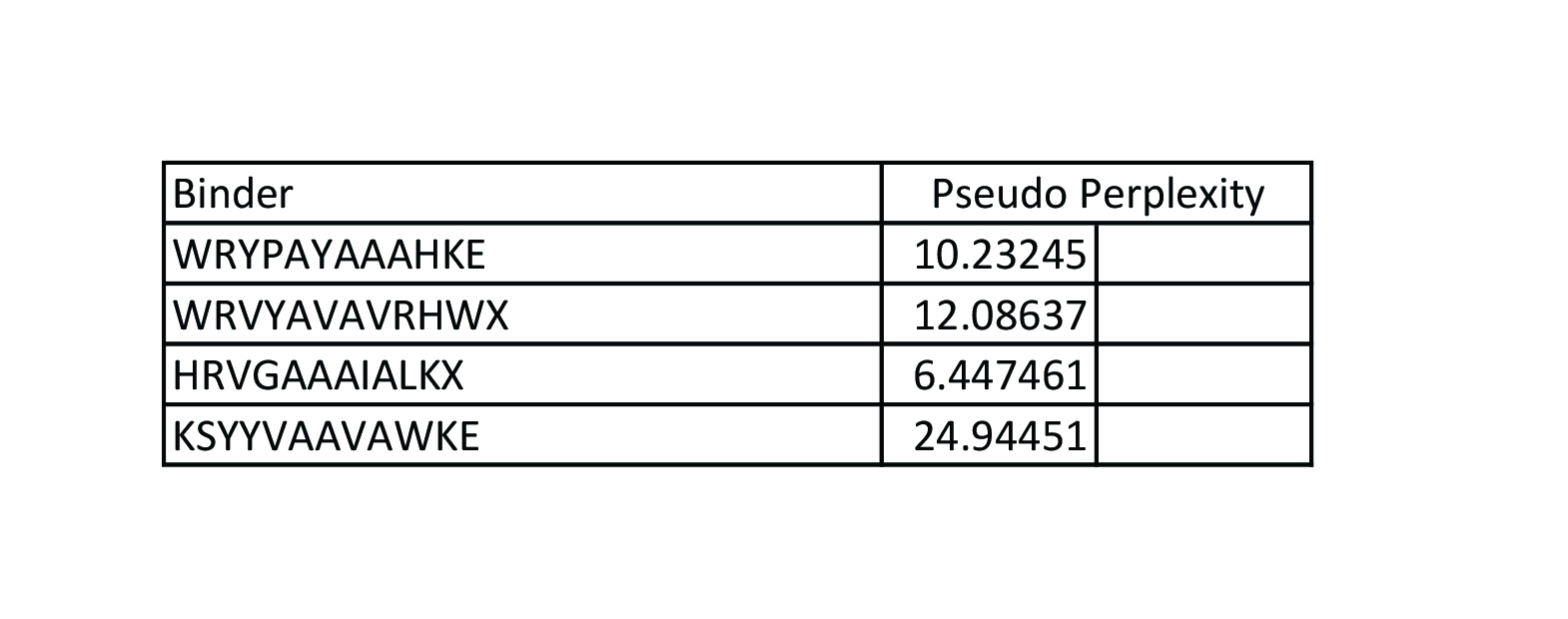
Peptides with higher ipTM (e.g., WRYYPVAAAWKK, ipTM 0.83) do not show stronger predicted binding affinity — in fact, all four are labeled “Weak binding” with pKd/pKi values between 4.9 and 6.1. This suggests AlphaFold3’s ipTM reflects structural confidence and interface complementarity, not absolute affinity magnitude in these predictions.
None of the peptides are predicted to be hemolytic (hemolysis probability 0.012–0.059) — all are safe in that regard. All are soluble (probability 1.0). The main trade‑off is permeability: the high‑ipTM peptide (WRYYPVAAAWKK) is permeable (0.799), while others like HRYGAVVVELKK are non‑permeable (0.062). Fouling propensity is modest across all (0.297–0.497).
Best balance of predicted binding and therapeutic properties
WRYYPVAAAWKK (ipTM 0.83) stands out:
· High structural confidence (ipTM 0.83, pTM 0.93)
· Soluble, non‑hemolytic, permeable
· Weak binding (pKd ~6.1), but the structural model suggests a specific, burying interaction — affinity might be improvable while keeping properties
Part 4: Generate Optimized Peptides with moPPIt
We applied moPPIt (Multi-Objective Guided Discrete Flow Matching, MOG-DFM) to shift from purely probabilistic sampling toward a more controlled, motif-driven strategy for peptide design. In contrast to PepMLM, which relies on conditioning over the entire target sequence, moPPIt enables precise specification of target residues on SOD1 while concurrently optimizing multiple therapeutic criteria.
Part C: Final Project: L-Protein Mutants
Mutation Selection Strategy for MS2 L-Protein
I first cross-checked candidate mutations against the experimental L-Protein mutant dataset to evaluate whether LLR scores correlate with lysis phenotypes. Similar to previous observations, overlap between computationally favorable mutations and experimentally validated lytic variants was limited, suggesting that LLR scores alone are insufficient to predict lysis function. This is likely due to additional constraints such as membrane insertion, oligomerization, and host-factor (e.g., DnaJ) interactions.
Thus, LLR scores were used primarily as a hypothesis-generation tool, complemented with structural and biochemical reasoning.
Mutation Design Constraints
2 mutations in the soluble region (positions 1–40)
3 mutations in the transmembrane (TM) region (positions 41–75)
Rationale:
Conservative polar substitution (Gln → Asn) that may subtly alter hydrogen-bonding networks in the soluble domain without destabilizing the fold. Could modulate interaction with DnaJ.
Rationale:
Charge reversal (negative → positive) may significantly alter electrostatic surface properties and disrupt chaperone recognition or binding interfaces.
Rationale:
C-terminal TM region mutation increasing hydrophobicity, potentially stabilizing oligomerization and pore formation.
Summary
This alternative set of mutations was selected based on:
positive LLR scores
maintaining structural integrity (conservative substitutions where needed)
improving membrane compatibility in TM regions
altering charge/polarity in the soluble domain to modulate DnaJ interaction
Week 6 HW: Genetic Circuits Part I: Assembly Technologies
Assignment: DNA Assembly
1. What are some components in the Phusion High-Fidelity PCR Master Mix and what is their purpose?
Phusion High‑Fidelity DNA Polymerase. This is the key enzyme that copies DNA during PCR.
Reaction Buffer (HF Buffer). The buffer provides the chemical environment in which the enzyme works best.
Deoxynucleoside Triphosphates (dNTPs). These are the building blocks of DNA (A, T, G, C).
Optional Additives
Water. Used to bring the reaction to the final volume and ensure proper concentrations of all components once primers and DNA template are added.
2. What are some factors that determine primer annealing temperature during PCR?
The primer annealing temperature (Ta) in PCR is critical because it determines how specifically your primers bind to the target DNA. If the temperature is too low, primers may bind nonspecifically; if too high, they may not bind efficiently.
Primer sequence composition
Primer melting temperature
Salt concentration in the reaction
Primer-target complementarity
Presence of additives
PCR polymerase used
Some polymerases are more tolerant to mismatches or high GC content; high-fidelity polymerases like Phusion often work better with slightly higher annealing temperatures.
3. What are some components in the Phusion High-Fidelity PCR Master Mix and what is their purpose?
Protocol differences
Aspect
PCR
Restriction Enzyme Digest
Input material
Template DNA containing the target sequence
DNA containing one or more recognition sites for the enzyme
Single incubation at optimal temperature for enzyme (usually 37°C)
Time
Typically 1–3 hours depending on fragment length and cycles
Usually 30 min–2 hours
When one method is preferable
Use PCR
to need highly specific fragments from a complex genome
to want to amplify from very little starting material
to want to add custom sequences, like restriction sites, tags, or mutations
Use Restriction Enzyme Digest
to have large amounts of DNA and want fragments exactly defined by natural restriction sites
to want to cut plasmids or genomic DNA into predictable pieces for cloning, mapping, or analysis
to prefer a simpler, single-temperature protocol without thermal cycling
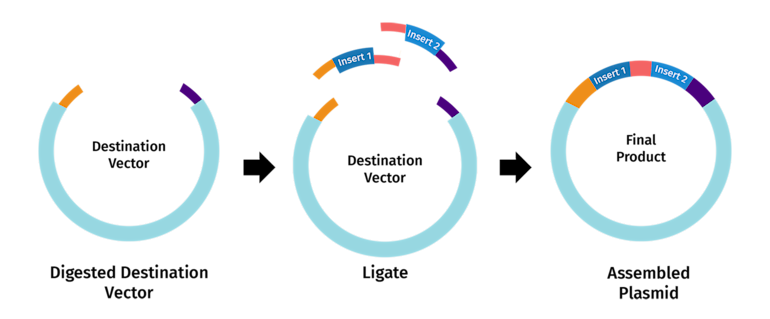
4. How can you ensure that the DNA sequences that you have digested and PCR-ed will be appropriate for Gibson cloning?
Understand the requirements of Gibson Assembly
Gibson Assembly joins two or more DNA fragments in a single isothermal reaction using:
Exonuclease – chews back the 5′ ends to create single-stranded 3′ overhangs. DNA polymerase – fills in the gaps after fragments anneal. DNA ligase – seals the nicks to make a continuous DNA molecule.
Designing PCR products for Gibson cloning
When using PCR to generate fragments:
Forward primer for fragment A should include a 5′ extension that is complementary to the start of fragment B.
Reverse primer for fragment B should include a 5′ extension complementary to the end of fragment A.
These extensions become the overlapping regions for Gibson Assembly.
5. How does the plasmid DNA enter the E. coli cells during transformation?
Plasmid DNA enters E. coli during transformation because the cells are made temporarily permeable (competent).
In chemical transformation, DNA binds to the membrane and slips in during heat shock.
In electroporation, DNA is driven through pores formed by a brief electric pulse.
Once inside, plasmid DNA replicates and can express its genes, including selectable markers.
6. Describe another assembly method in detail
Golden Gate Assembly is a modular DNA assembly method that allows multiple DNA fragments to be joined in a defined order in a single reaction. It uses type IIS restriction enzymes, which cut outside of their recognition sequences, creating custom overhangs. Each fragment is designed with specific overhang sequences that match the adjacent fragment, so they ligate in the correct order. The reaction is performed simultaneously with a DNA ligase, so the fragments are cut and joined in cycles, and the recognition sites are eliminated in the final product, leaving seamless DNA. This method is very efficient for constructing multi-gene pathways or synthetic constructs because it allows precise assembly without leaving extra sequences. Golden Gate is often faster and more flexible than traditional cloning, especially when assembling more than 3–4 fragments. By careful design of overhangs, you can assemble fragments in a predefined order with high fidelity.
Assignment: Asimov Kernel
I don’t have access to the Asimov Kernel Software yet
Week 7 HW: Genetic Circuits Part II: Neuromorphic Circuits
Assignment Part 1: Intracellular Artificial Neural Networks (IANNs)
What advantages do IANNs have over traditional genetic circuits, whose input/output behaviors are Boolean functions?
Intracellular Analog Neural Networks (IANNs) offer several advantages over traditional genetic circuits that rely on Boolean logic (ON/OFF states). First, IANNs enable graded, continuous responses rather than binary outputs. This allows cells to process varying signal intensities and produce proportional outputs, which more closely reflects natural biological systems.
Describe a useful application for an IANN; include a detailed description of input/output behavior, as well as any limitations an IANN might face to achieve your goal.
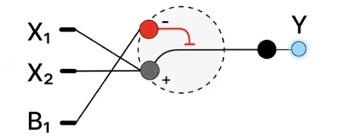
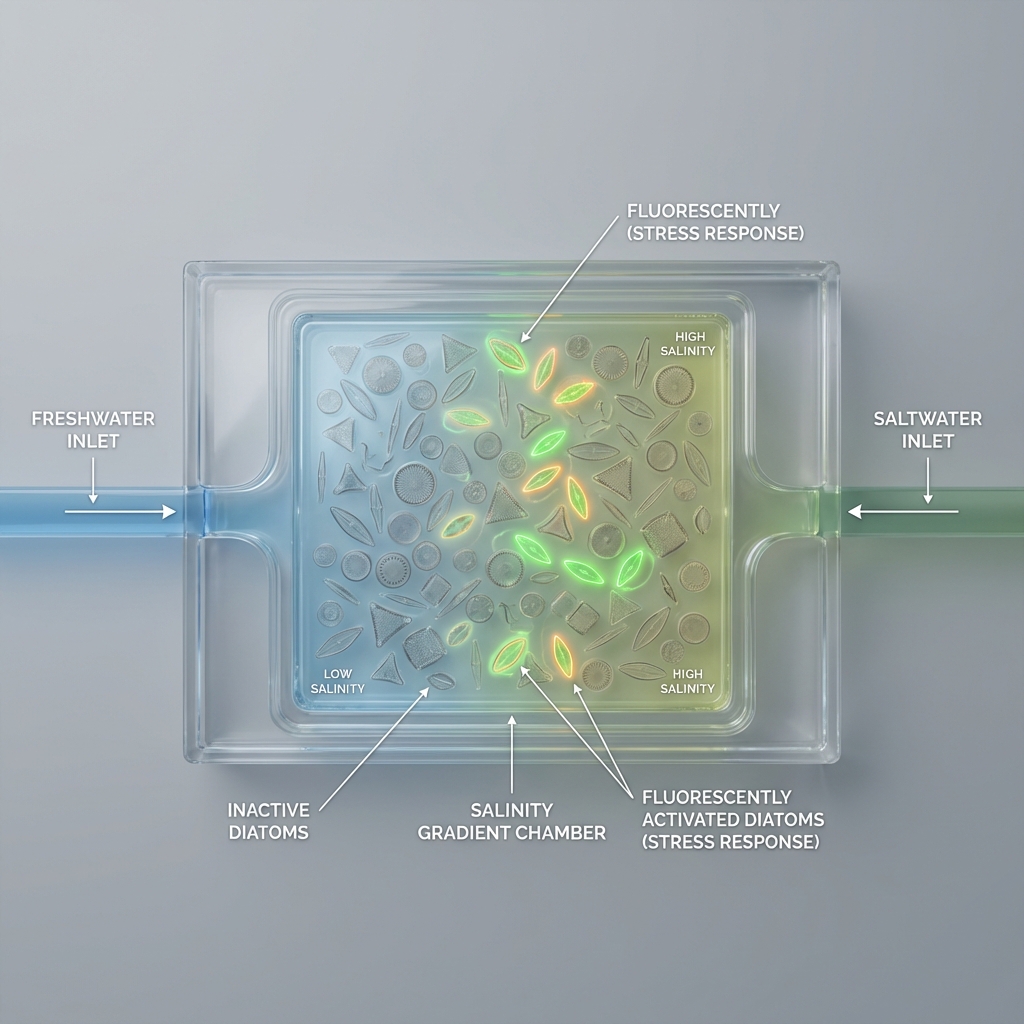
Application Example: Salinity Stress Sensing in Diatoms
A useful application of an IANN is in designing a salinity-responsive system in diatoms that produces a graded fluorescent output (e.g., GFP) based on environmental stress levels.
Input/Output Behavior:
X₁ (inhibitory input): expression of Csy4 endoribonuclease
X₂ (activating input): expression of a fluorescent reporter gene (e.g., GFP mRNA) Csy4 cleaves or destabilizes the mRNA of the reporter
Output (Y): fluorescence intensity
Low X₁ (low inhibition) + high X₂ → strong fluorescence
High X₁ → reduced fluorescence (due to mRNA cleavage)
Intermediate levels → graded fluorescence output
Advantages:
Enables real-time sensing of environmental stress gradients
Produces quantitative readouts instead of simple ON/OFF signals
Can be integrated into microfluidic systems for visualization
Limitations:
Biological noise and variability
Gene expression is inherently stochastic
Precise control of promoter strength and degradation rates is complex
Crosstalk and unintended interactions
Assignment Part 2: Fungal Materials
What are some examples of existing fungal materials and what are they used for? What are their advantages and disadvantages over traditional counterparts?
Mycelium composites (e.g., Ecovative). Grown from agricultural waste into rigid boards or foam-like fills. Used for packaging, insulation, acoustic panels, and furniture. Advantages: biodegradable, low energy to produce, fire-resistant. Disadvantages: lower strength than plastics/wood, moisture sensitivity, batch variability.
Mycelium leather. Sheet-grown or processed mycelium. Used for bags, shoes, watch straps. Advantages: faster to grow than animal leather, tunable texture, no heavy metal tanning. Disadvantages: less durable than synthetic leathers, requires finishing layers.
Fungal foams. Flexible, sponge-like mycelium. Used for shoe midsoles, cushioning, wound dressings. Advantages: breathable, hypoallergenic, compostable. Disadvantages: lower rebound resilience than petroleum foams.
What might you want to genetically engineer fungi to do and why?
Produce novel enzymes (e.g., cellulases, lignin peroxidases) for bioremediation (breaking down plastics, oil, dyes) or biofuel production from crop waste.
Make fungal materials stronger or waterproof – Expressing hydrophobins or crosslinking enzymes to replace petrochemical coatings.
Self-coloring mycelium – Engineer pigment biosynthesis so materials grow with color, eliminating dyeing steps.
Living sensors – Fungi that change color or emit light when exposed to toxins or pathogens.
What are the advantages of doing synthetic biology in fungi as opposed to bacteria?
Protein secretion – Filamentous fungi are industrial champions of secretion (grams per liter), while bacteria often form inclusion bodies.
Larger DNA capacity – Fungi tolerate larger genetic payloads and multi-gene pathways (e.g., for secondary metabolites).
Safety – Non-pathogenic lab strains (e.g., Aspergillus nidulans, Saccharomyces cerevisiae) have GRAS status and are easier to contain than many bacteria.
Week 9 HW: Cell-Free Systems
General homework questions
Explain the main advantages of cell-free protein synthesis over traditional in vivo methods, specifically in terms of flexibility and control over experimental variables. Name at least two cases where cell-free expression is more beneficial than cell production.
Direct control of conditions: You can independently adjust pH, temperature, ion concentration, redox state, and cofactors without affecting cell viability.
Faster optimization cycles: No need for cloning, transformation, or cell growth—results can be obtained in hours.
No cellular constraints: Toxic proteins, unstable proteins, or proteins that burden cells can still be produced.
Simplified system: Fewer regulatory pathways means fewer unknown biological interactions.
Cases where cell-free is more beneficial:
Expression of toxic proteins
Rapid protein prototyping
Production of proteins requiring non-standard conditions or non-natural amino acids.
Describe the main components of a cell-free expression system and explain the role of each component.
Main components of a cell-free expression system and their roles:
Cell-free protein synthesis template DNA or mRNA: Encodes the protein of interest.
Energy system: Supplies ATP/GTP for transcription and translation.
Amino acids: Building blocks for protein synthesis.
Salts and ions (Mg²⁺, K⁺): Stabilize ribosomes and enzyme activity.
Buffer system: Maintains optimal pH.
Why is energy provision regeneration critical in cell-free systems? Describe a method you could use to ensure continuous ATP supply in your cell-free experiment.
Protein synthesis is extremely energy-intensive: ATP and GTP are required for transcription, translation, tRNA charging, and elongation steps. Without regeneration, the system quickly stalls.
In Cell-free protein synthesis systems, ATP depletion is one of the main limiting factors.
Compare prokaryotic versus eukaryotic cell-free expression systems. Choose a protein to produce in each system and explain why.
Prokaryotic system
Fast, cheap, high yield
Best for simple proteins without complex modifications
Example protein: Green fluorescent protein (GFP)
Reason: folds easily, no glycosylation required, expresses efficiently
Eukaryotic system
Supports post-translational modifications
Better for complex human proteins
How would you design a cell-free experiment to optimize the expression of a membrane protein? Discuss the challenges and how you would address them in your setup.
Designing a cell-free system for membrane protein expression:
Include detergents or nanodiscs/liposomes to mimic membranes
Use oxidizing conditions if disulfide bonds are needed
Add molecular chaperones to assist folding
Optimize magnesium and potassium concentrations
Key challenges:
Aggregation of hydrophobic regions
Misfolding due to lack of membrane insertion
Low solubility
Homework question from Kate Adamala
Function: Biosensing of glucose with fluorescent output
Synthetic cell detects glucose in the environment and produces a fluorescent signal (eGFP).
Input: glucose
Output: fluorescence (GFP signal)
Partially, yes. A cell-free transcription/translation (Tx/Tl) system can produce GFP in response to glucose in solution. However, without encapsulation, the system lacks compartmentalization, stability, and control over the local environment.
Yes, this function could be implemented in a genetically engineered bacterium such as Escherichia coli.
The system should produce a fluorescence signal proportional to glucose concentration, with:
low background signal
high sensitivity
fast response
Membrane
The synthetic cell will be a liposome composed of:
phosphatidylcholine (POPC)
cholesterol (for stability)
Tx/Tl system - cell-free extract
Genetic circuit - glucose-responsive promoter, reporter gene: eGFP and egulatory system: glucose-sensitive transcription factor
A bacterial system (E. coli) is sufficient because:
no complex post-translational modifications are required
efficient and widely used
List of components
Lipids:
POPC
cholesterol
Genes:
eGFP (reporter)
glf (glucose transporter)
How to measure function:
fluorescence spectroscopy
plate reader measurements
fluorescence microscopy
Homework question from Peter Nguyen
Integrate freeze-dried Cell-free protein synthesis systems into building materials to create pollution-responsive facades that actively detect and degrade urban air contaminants.
The proposed system embeds freeze-dried cell-free expression components directly into construction materials such as concrete panels or surface coatings. These systems contain DNA templates encoding enzymes capable of degrading airborne pollutants (e.g., NOx or volatile organic compounds).
Societal challenge. Urban air pollution remains a major global challenge, particularly in densely populated cities where traditional mitigation strategies are limited. Current architectural materials are largely passive and do not contribute to environmental remediation.
How do you envision addressing the limitation of cell-free reactions (e.g., activation with water, stability, one-time use)?
Activation (water dependence).
The system is designed to be moisture-activated, using naturally occurring environmental water sources such as rain or humidity, eliminating the need for external energy input.
Homework question from Ally Huang
Вackground
Long-duration space missions expose astronauts to microgravity and increased radiation, which can alter microbial behavior and increase the risk of pathogenicity and antibiotic resistance. Monitoring microbial gene expression in real time is critical for crew health and spacecraft safety. Traditional cell-based assays are difficult to maintain in space due to resource limitations. Freeze-dried Cell-free protein synthesis systems, such as BioBits®, offer a stable, low-resource alternative for detecting specific genetic signatures. This project proposes a portable, rapid-response system to monitor microbial stress responses during spaceflight.
Molecular Target
Stress-response and virulence-associated genes from spacecraft-associated bacteria such as Escherichia coli.
Relevance of Target
Stress-response genes such as recA and rpoS are upregulated under DNA damage and environmental stress, conditions common in space. Monitoring their expression provides insight into how microbes adapt to microgravity and radiation. Increased expression of virulence-related genes (e.g., toxA) may indicate elevated pathogenic potential, posing risks to astronaut health. By targeting these genes, the system can function as an early-warning biosensor, enabling detection of harmful microbial shifts before they lead to infection or system contamination.
Hypothesis
Microgravity and space-related stressors induce measurable increases in microbial stress-response gene expression, which can be detected using a freeze-dried cell-free system. Specifically, BioBits® reactions programmed with DNA templates responsive to target gene sequences will produce a fluorescent signal when these genes are present or amplified.
The goal is to develop a rapid, portable biosensing platform that integrates DNA amplification with cell-free protein expression and fluorescence detection. This system would enable astronauts to monitor microbial adaptation in real time without the need for complex laboratory infrastructure.
Experimental Plan
Bacterial samples will be processed to extract DNA. Target genes will be amplified using miniPCR®. Amplified DNA will be added to BioBits cell-free reactions containing reporter constructs that produce fluorescence upon detection. Fluorescence output will be measured using the P51 Molecular Fluorescence Viewer.
Controls include:
negative control (no DNA);
positive control (known target DNA)
Data will consist of fluorescence intensity over time, indicating gene presence and relative activity.
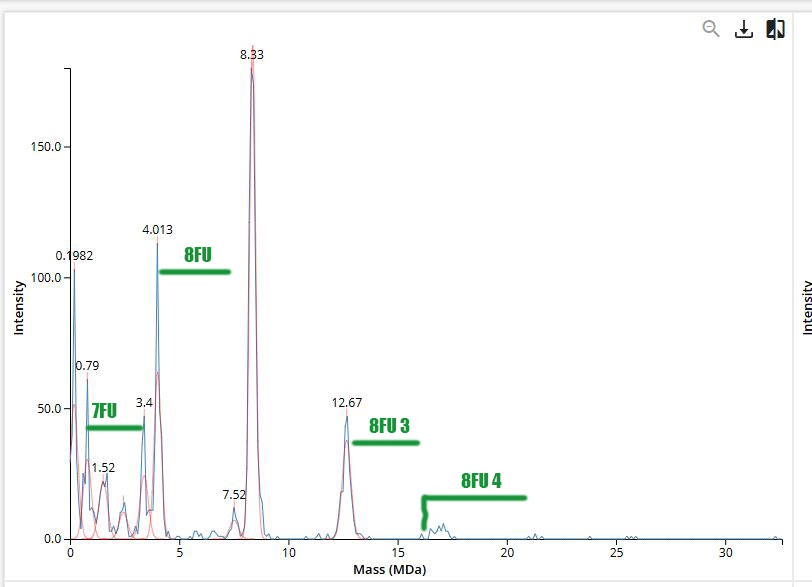
No. To determine the state of charge (z) in LC-MS, several peaks are needed, such as (z), (z+1), (z+2)
Homework: Waters Part II — Secondary/Tertiary structure
1.
In its native state, a protein like eGFP is properly folded into a compact 3D structure stabilized by noncovalent interactions (hydrogen bonds, hydrophobic interactions, ionic interactions). This folded conformation is what gives the protein its biological function.
In the denatured state, the protein is unfolded. These stabilizing interactions are disrupted (e.g., by organic solvent, acid, heat), causing the structure to lose its compact shape and become extended and flexible. Importantly, the primary structure (amino acid sequence) remains intact—only the higher-order structure is lost.
Proteins are typically analyzed using electrospray ionization (ESI), which produces multiply charged ions.
2.
The peak at ~2800 m/z has a +10 charge state, determined from the ~0.1 m/z spacing between isotopic peaks (Δm/z = 1/z)
Homework: Waters Part III — Peptide Mapping - primary structure
1.
peptide sequence
HNIEDGSVQLADHYQQNTPI GDGPVLLPDNHYLSTQSALS K
DHMVLLEFVTAAGITLGMDE LYK
GEELFTGVVPILVELDGDVN GHK
LPVPWPTLVTTLTYGVQCFS R
LEYNYNSHNVYIMADK
FSVSGEGEGDATYGK
SAMPEGYVQER
LEHHHHHH
FEGDTLVNR
EDGNILGHK
QHDFFK
YPDHMK
FICTTGK
DDGNYK
TIFFK
DPNEK
GIDFK
VNFK
IELK
K = 15
R = 3
2.
19 peptides
3.
22 chromatographic peaks
4.
No. There are 3 more peaks in the chromatogram.
5.
The isotope spacing is= 1/z; the peaks are spaced ~0.5 m/z apart (525.76712, 526.25918), therefore z = +2.
M=(m/z×z)−z×1.007
M=(525.767×2)−(2×1.007)
M≈1051.534−2.014=1049.52 Da
[M+H]⁺= = M+1.0073 = 1,049.51964 + 1.0073 = 1,050.52694 Da
ABSTRACT The project explores how diatoms adapt to rapidly changing salinity in transitional environments such as river plumes, where freshwater mixes with seawater. These zones are increasingly important under climate change, as shifting salinity patterns affect microbial survival and ecosystem stability. Understanding how diatoms respond at molecular and structural levels is critical, as they play a key role in global oxygen production and biogeochemical cycles.
Proposal: Increasing Stability of the MS2 L Protein Objective
The goal of this project is to identify and engineer mutations in the L protein of the ssRNA phage MS2 bacteriophage that increase its structural stability while preserving its lytic function in Escherichia coli. Improving protein stability may enhance its functional lifetime and overall efficiency during infection.
Subsections of Projects
Individual Final Project
ABSTRACT
The project explores how diatoms adapt to rapidly changing salinity in transitional environments such as river plumes, where freshwater mixes with seawater. These zones are increasingly important under climate change, as shifting salinity patterns affect microbial survival and ecosystem stability. Understanding how diatoms respond at molecular and structural levels is critical, as they play a key role in global oxygen production and biogeochemical cycles.
The overall objective is to model and visualize adaptive mechanisms in diatoms under osmotic stress. The project hypothesizes that changes in ion transport and stress-response pathways can be linked to observable shifts in cell behavior and morphology, including frustule structure.
To address this, the project will
design a controlled salinity gradient using a microfluidic chip;
simulate adaptive mutations in key proteins using computational tools;
visualize stress-response activation using a fluorescent reporter system such as GFP
The expected outcome is an integrated scientific and visual framework that connects molecular adaptation with environmental transitions, offering both biological insight and a platform for interdisciplinary exploration.
Aim 1: Experimental Aim
The first aim of my final project is to model and visualize stress-response activation in diatoms under controlled salinity gradients by utilizing a microfluidic chip system, fluorescent reporter constructs (e.g., GFP linked to stress-response genes), and computational protein design tools such as ESM-based models and AlphaFold for simulating adaptive mutations in ion transport and stress-related proteins.
Aim 2: Development Aim
The second aim is to extend this system by experimentally validating computationally predicted adaptive mutations, integrating optimized genetic constructs into diatom cells, and improving the microfluidic platform to allow long-term observation of adaptive dynamics and potential evolutionary changes across multiple generations.
Aim 3: Visionary Aim
The third aim is to translate laboratory-based insights into real-world environmental applications by developing strategies to enhance the salinity tolerance of diatoms in transitional ecosystems such as river plumes and estuaries. In the long term, this could contribute to maintaining ecosystem balance, supporting primary productivity, and strengthening the role of diatoms in global carbon and oxygen cycles.
BACKGROUND
Diatoms are key primary producers in aquatic ecosystems and contribute significantly to global carbon fixation and oxygen production. In transitional environments such as river plumes and estuaries, diatoms are exposed to rapid and often extreme fluctuations in salinity, requiring efficient adaptive responses. Previous studies have shown that osmotic stress in microalgae activates ion transport systems and stress-response pathways that regulate intracellular ion balance and protect cellular structures.[1] For example, research on salinity stress in diatoms demonstrates that ion transporters and compatible solute pathways play a central role in maintaining cellular homeostasis under changing environmental conditions.
Additionally, the change in the morphology of diatom frustules during the transition between freshwater and marine conditions is analyzed, as well as its dependence on genetic and environmental factors that determine adaptive mechanisms.[2]
[1] - Diatom community response to inland water salinization: a review, C. Stenger-Kovács, V. B. Béres, K. Buczkó, K. Tapolczai, J. Padisák, G. B. Selmeczy & E. Lengyel/Published: 26 April 2023, pages 4627–4663, (2023)
[2] - Kamakura, S. et al. Morphological plasticity in response to salinity change in the euryhaline diatom Pleurosira laevis (Bacillariophyta). J. Phycol.58, 631–642 (2022)
Group Final Project
Proposal: Increasing Stability of the MS2 L Protein
Objective
The goal of this project is to identify and engineer mutations in the L protein of the ssRNA phage MS2 bacteriophage that increase its structural stability while preserving its lytic function in Escherichia coli. Improving protein stability may enhance its functional lifetime and overall efficiency during infection.
Proposed Tools and Approaches
To systematically explore the mutational landscape of the L protein, we propose a computational pipeline combining sequence-based and structure-based methods.
First, we will use protein language models (PLMs), including ESM-2, to perform in silico mutagenesis. The model enable high-throughput evaluation of single-residue substitutions and provide likelihood-based scores (e.g., LLR) that reflect the compatibility of mutations with evolutionary constraints. This allows us to identify mutations that are potentially stabilizing while maintaining overall sequence plausibility.
Next, we will employ AlphaFold-Multimer to predict the structural consequences of selected mutations. This step will be used to assess whether candidate variants preserve proper folding, tertiary structure, and potential interaction interfaces. In particular, we will examine how mutations affect protein conformation, heterotypic interactions, and membrane association.
Rationale
The L protein likely relies on specific protein–protein interactions and membrane-associated behavior to mediate lysis. Therefore, increasing stability alone is insufficient; mutations must also preserve these functional interactions. By integrating PLM-based predictions with structural modeling, we aim to prioritize mutations that improve stability without disrupting key biological functions.
Selected Mutations and Rationale
Five candidate mutations were selected based on positive LLR scores and additional structural considerations:
Q13N (soluble region): A conservative polar substitution (Gln → Asn) that may subtly modify hydrogen-bonding networks without destabilizing the fold. This mutation could influence interactions with host factors such as DnaJ.
E24K (soluble region): A charge-reversal mutation (negative → positive) that significantly alters surface electrostatics, potentially affecting chaperone binding or interaction interfaces.
I48L (TM region): A highly conservative hydrophobic substitution expected to stabilize helix packing within the membrane while preserving structural integrity.
S51A (TM region): Removal of a polar residue within the transmembrane domain, likely improving membrane compatibility and reducing unfavorable interactions.
Q68L (TM region): Increased hydrophobicity in the C-terminal TM region, potentially enhancing oligomerization and pore-forming capability.
Membrane association analysis
Since L is likely membrane-associated:
predict transmembrane or hydrophobic regions;
avoid destabilizing mutations in these domains
Expected Outcome
Identification of candidate stabilizing mutations
A ranked list of variants for future experimental validation
Improved understanding of L protein structure–function relationship
Potential Pitfalls
Despite the strengths of this approach, several limitations remain. First, both PLMs and structure prediction tools are inherently probabilistic, and predicted stabilizing mutations may still disrupt lytic function. Second, the mechanism of L-mediated lysis is not fully understood and does not follow classical pathways such as peptidoglycan inhibition. As a result, subtle but critical interactions may not be captured by current computational models.