Week 4 HW: Protein Design Part 1
Part A. Conceptual Questions
1. How many molecules of amino acids do you take with a piece of 500 grams of meat? (on average an amino acid is ~100 Daltons)
Assuming 500 g of meat contains about 20–25% protein by weight, that piece has roughly:
- 100–125 g of protein
- 100 daltons = 100 g/mol
- so 100–125 g of amino acid residues = about 1.0–1.25 moles
- 1 mole = 6.022 × 10^23 molecules So the total is about:
- 6.0 × 1023 to 7.5 × 1023 amino acid molecules
2. Why do humans eat beef but do not become a cow, eat fish but do not become fish?
Because human body digests and obsorbs. It breaks food down into raw materials and then rebuilds those materials into human body. After absorption, cells use those pieces according to human DNA, human enzymes, and human metabolism. So even if the amino acids came from cow muscle, human body reassembles them into muscle, enzymes, skin, and so on.
- Proteins are digested into amino acids
- Fats are digested into fatty acids and glycerol
- Carbohydrates are digested into simple sugars
3. Why are there only 20 natural amino acids?
There are 20 canonical amino acids in the standard genetic code because evolution seems to have settled on a set that is chemically versatile enough to build functional proteins while still being simple enough for reliable biosynthesis and translation.
5. Where did amino acids come from before enzymes that make them, and before life started?
The main idea is that early Earth and space already had simple carbon-containing molecules like water, carbon dioxide, methane, ammonia, hydrogen cyanide, and related compounds. With energy sources such as lightning, UV radiation, heat, impacts, and mineral surfaces, those molecules could react abiotically and make amino acids. That is the basic lesson of the classic Miller-type experiments and newer prebiotic chemistry work.
6. If you make an α-helix using D-amino acids, what handedness (right or left) would you expect?
A helix made from D-amino acids would generally be left-handed.
7. Can you discover additional helices in proteins?
Yes, but unlikely.
8. Why are most molecular helices right-handed?
In biology, most proteins are built from L-amino acids. That stereochemistry makes a right-handed α-helix lower in energy than a left-handed one, mainly - -because it gives:
- better backbone bond geometry
- better hydrogen-bond alignment
- fewer steric clashes between backbone and side chains
So for ordinary proteins, right-handed helices are favored because they fit the geometry of L-amino acids better.
9. Why do β-sheets tend to aggregate?
- What is the driving force for β-sheet aggregation?
Beta-sheet aggregation happens because exposed beta-strands can strongly bond to each other through backbone hydrogen bonds, while also hiding hydrophobic parts from water, making the growing clump very stable.
10. Why do many amyloid diseases form β-sheets?
- Can you use amyloid β-sheets as materials?
Amyloid diseases often involve β-sheets because β-sheet-like cross-β fibrils are a generic low-energy aggregation state available to many proteins, not just one special sequence.
Amyloid β-sheets can be used as materials. Researchers use amyloid or amyloid-like fibrils as biomaterials, hydrogels, coatings, scaffolds, filtration media, and nanostructured building blocks because they are stiff, high-aspect-ratio, self-assembling, chemically robust, and sequence-programmable.
Part B: Protein Analysis and Visualization
1. Briefly describe the protein you selected and why you selected it.
- Skeletal aspartic acid-rich protein 1 Acropora millepora (Staghorn coral)
2. Identify the amino acid sequence of your protein.
How long is it? What is the most frequent amino acid? You can use this Colab notebook to count the frequency of amino acids.
- The length of the protein is: 392 aminoacids.
- The most common amino acid is: D, which appears 74 times.
How many protein sequence homologs are there for your protein? Hint: Use Uniprot’s BLAST tool to search for homologs.
- 250
Does your protein belong to any protein family?
- SAARP1 belongs to the coral acid-rich skeletal organic matrix protein group (SAARP/CARP-related SOMPs), rather than to a large universal protein family with a standard conserved domain architecture.
3. Identify the structure page of your protein in RCSB
- AF_AFB3EWY6F1 is the RCSB AlphaFold structure page for skeletal aspartic acid-rich protein 1 from Acropora millepora. This is not an experimentally solved structure but a computed AlphaFold model, so it has no experimental resolution. Its overall confidence is moderate (global pLDDT about 77.99), with a more reliable structured core and several lower-confidence, likely disordered regions. The model contains only a single protein chain (A) and no other molecules. No clear standard structural classification family is listed on the page; the protein is more appropriately described as a coral skeletal acid-rich biomineralization protein.
4. Open the structure of your protein in any 3D molecule visualization software:
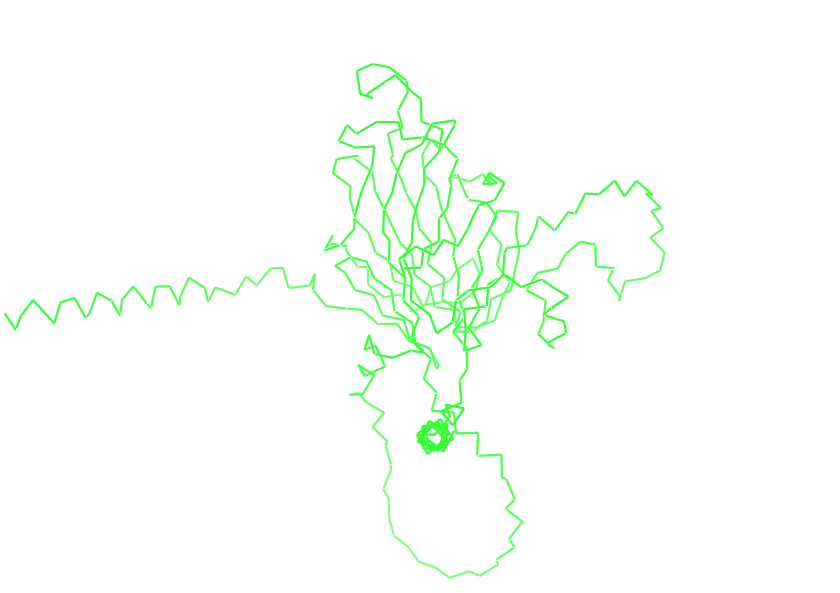
- Visualize the protein as “cartoon”, “ribbon” and “ball and stick”.
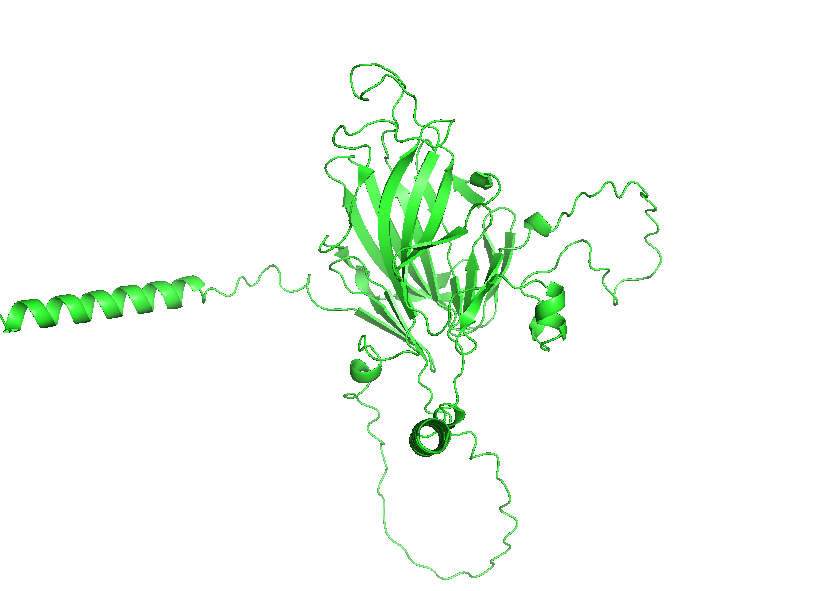

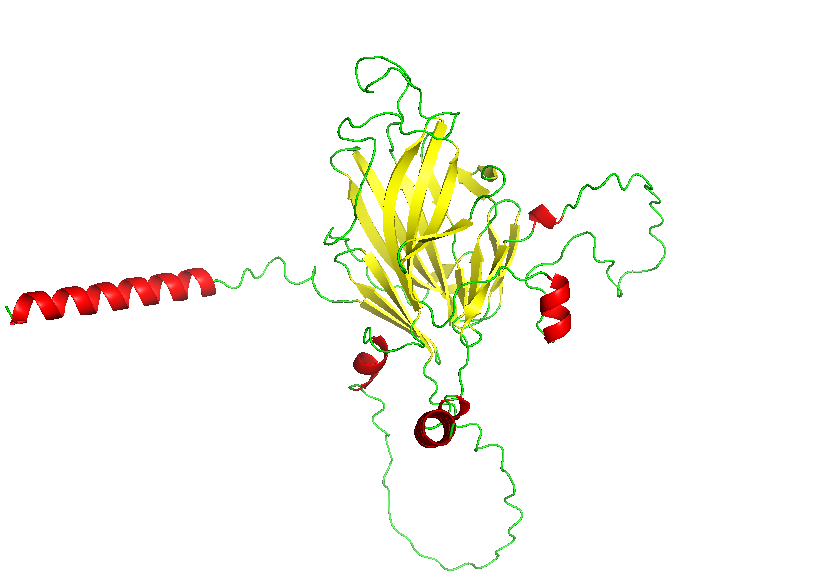
- Color the protein by secondary structure. Does it have more helices or sheets?
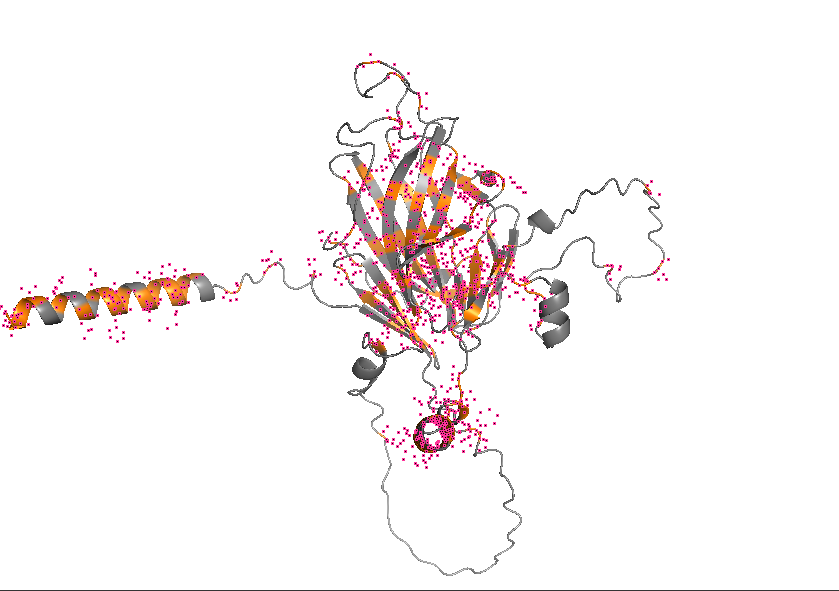
- Color the protein by residue type. What can you tell about the distribution of hydrophobic vs hydrophilic residues?
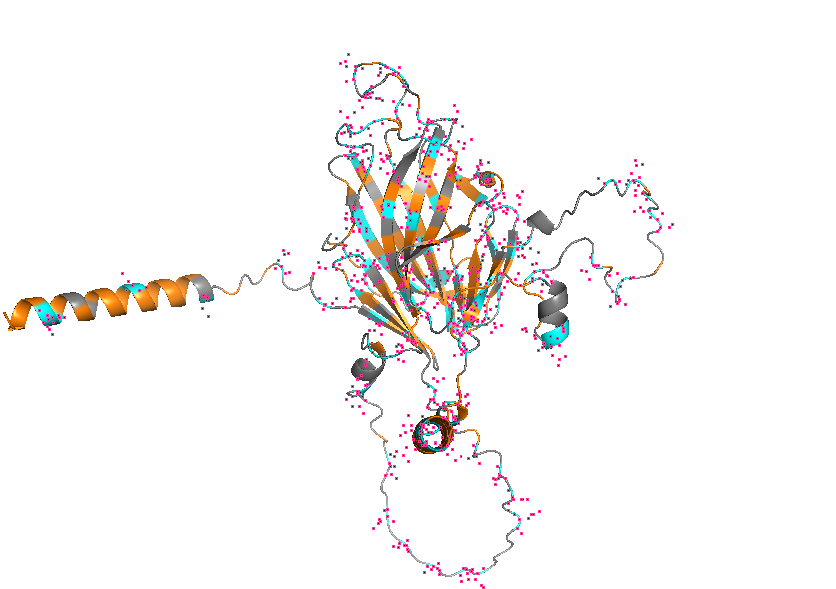
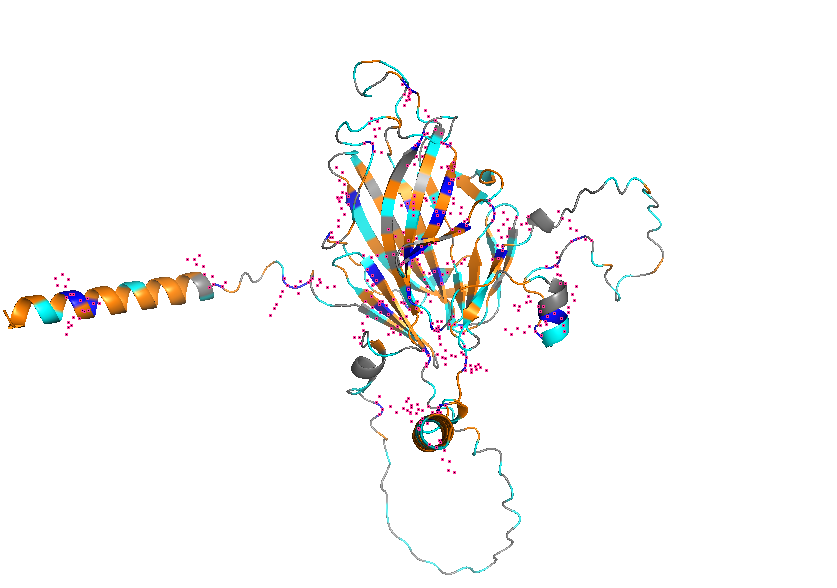
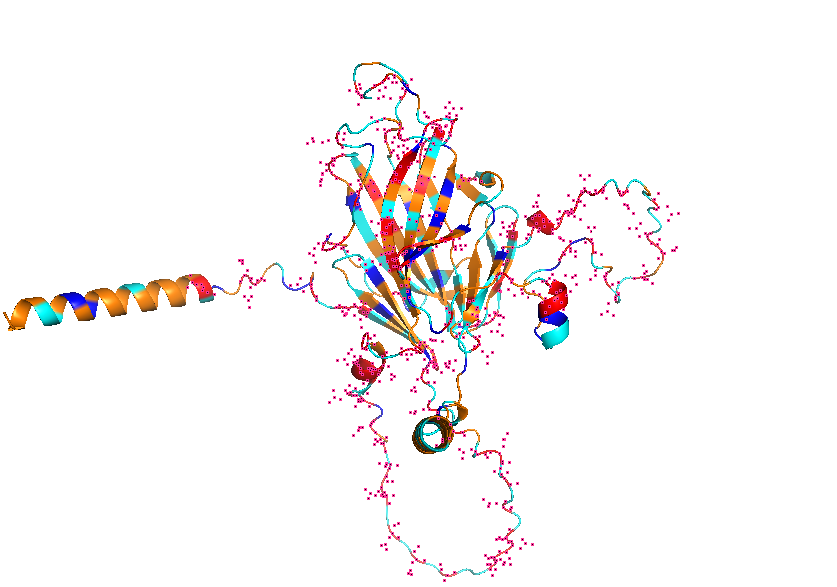
- Visualize the surface of the protein. Does it have any “holes” (aka binding pockets)?
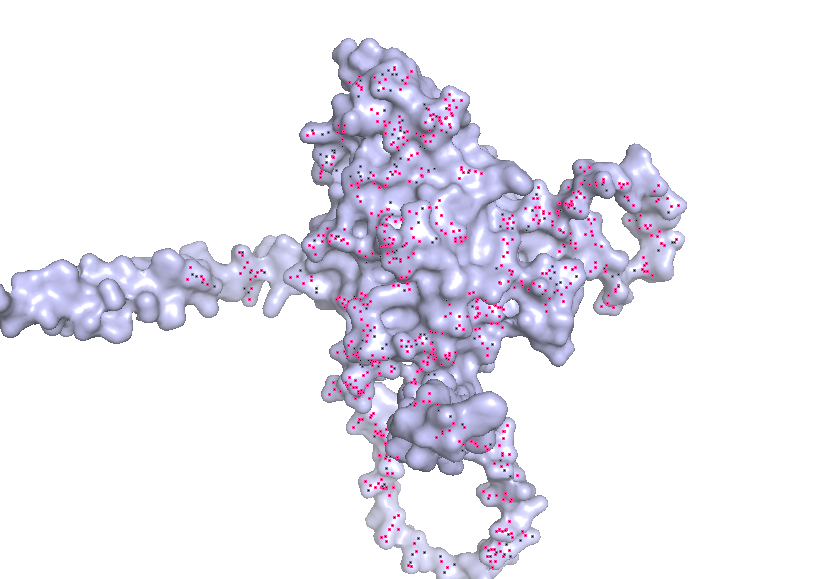
Part C. Using ML-Based Protein Design Tools
1BG2 | pdb_00001bg2, HUMAN UBIQUITOUS KINESIN MOTOR DOMAIN
C1. Protein Language Modeling
1. Deep Mutational Scans
a. Use ESM2 to generate an unsupervised deep mutational scan of your protein based on language model likelihoods.
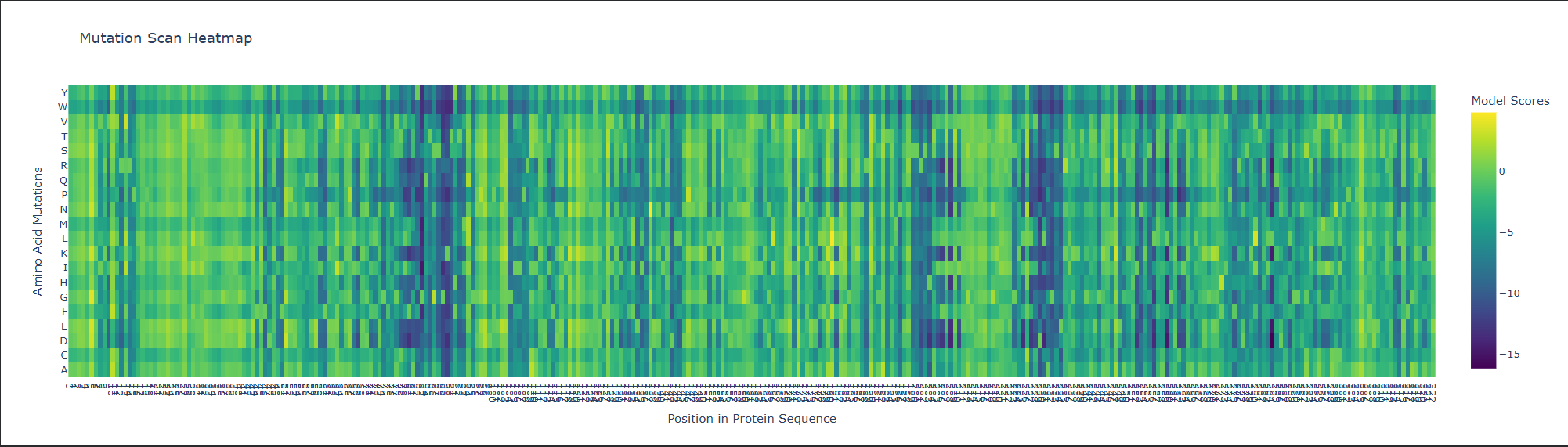
b. Can you explain any particular pattern? (choose a residue and a mutation that stands out)
This is visible as dark vertical columns, where nearly all substitutions are predicted to be unfavorable. One standout region is around residue 240–250, where many mutations score very poorly. This suggests that the position is structurally or functionally important, likely contributing to the stability of the kinesin motor-domain fold or to a conserved functional region. In particular, mutations such as proline are strongly disfavored, which is consistent with disruption of local backbone geometry or secondary structure.
C2. Protein Folding
1. Folding a protein
a. Fold your protein with ESMFold. Do the predicted coordinates match your original structure?
- Yes, it matches.
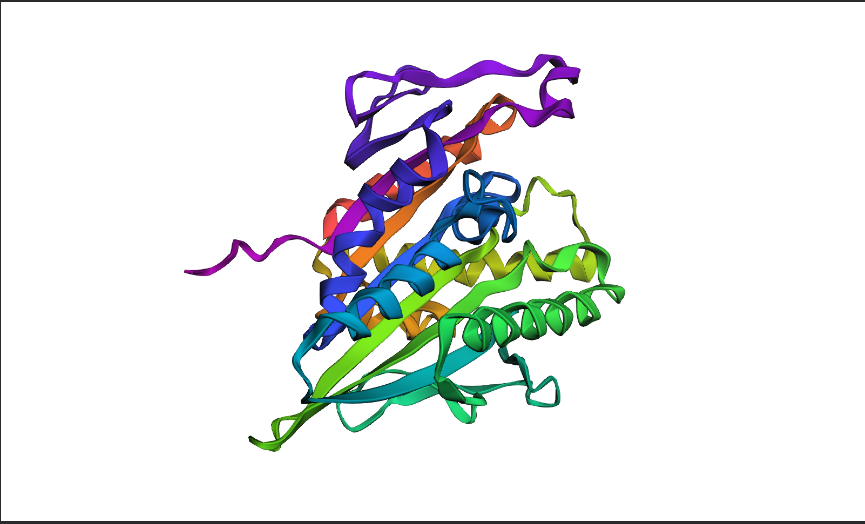
predicted
from pdb
b. Try changing the sequence, first try some mutations, then large segments. Is your protein structure resilient to mutations?
The protein is only partly resilient to mutations. It can tolerate some single conservative mutations, but it is not very resilient to larger sequence changes or to mutations at highly constrained positions.
C3. Protein Generation
Inverse-Folding a protein: Let’s now use the backbone of your chosen PDB to propose sequence candidates via ProteinMPNN
a. Analyze the predicted sequence probabilities and compare the predicted sequence vs the original one.
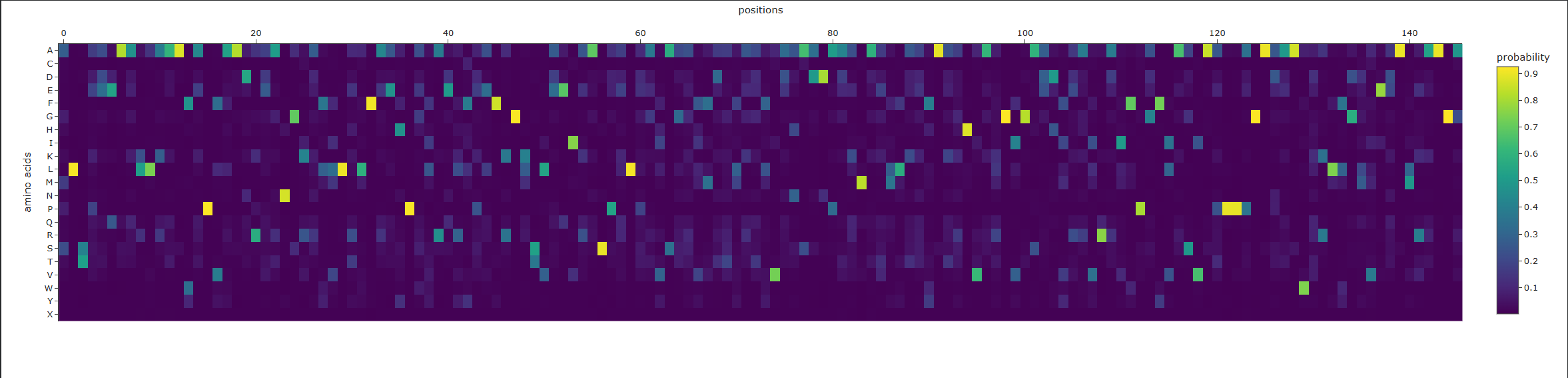
Original:
MADLAECNIKVMCRFRPLNESEVNRGDKYIAKFQGEDTVVIASKPYAFDRVFQSSTSQEQVYNDCAKKIVKDVLEGYNGTIFAYGQTSSGKTHTMEGKLHDPEGMGIIPRIVQDIFNYIYSMDENLEFHIKVSYFEIYLDKIRDLLDVSKTNLSVHEDKNRVPYVKGCTERFVCSPDEVMDTIDEGKSNRHVAVTNMNEHSSRSHSIFLINVKQENTQTEQKLSGKLYLVDLAGSEKVSKTGAEGAVLDEAKNINKSLSALGNVISALAEGSTYVPYRDSKMTRILQDSLGGNCRTTIVICCSPSSYNESETKSTLLFGQRAKTI
New:
ASTGSNIKVFCRVRPLSEEEIAAGDEDVIEFEGDNTVILDGEPYSFDKVYRPTTTQKEIYEDAAKEIVDNVLAGKNGTIFAIGPTGSGKTTTMLGDLDDPEKCGILPRIIERIFEKIEEEKGDVEYEITMSYFRIHNEKITDLLDPSKRNLKVKKDENGKPYVEGLTEVKVNSVEEAKALIKKGLENSKIGVEDVTKFDATSTRVIVITVKSKNKKTGEEKEGKLYLVDLASTDWRGRTGAGGAGASGGAPLHPDIAALEACIKALSAGSATVPYDLSPLTKLLQDALGGDSDNTIIGCIRPSSSRREEAKRVLEFLSLAKRS
b. Input this sequence into ESMFold and compare the predicted structure to your original.
Part D. Group Brainstorm on Bacteriophage Engineering
Group member: Sean Murphy, Alayah Hines, Terry Luo
- Drafted by Sean
Background
- The MS2 L protein is a 75-amino-acid polypeptide that lyses E. coli by an incompletely understood mechanism. Its C-terminal transmembrane (TM) domain inserts into the cytoplasmic membrane and oligomerizes, causing depolarization that triggers host autolytic enzymes to degrade the murein layer. Recessive, conservative missense mutations clustered around a conserved LS dipeptide strongly implies L engages an unidentified host protein target rather than simply disrupting the bilayer. The dispensable N-terminal domain binds chaperone DnaJ (with solved PDB structures), modulating lysis timing. Its removal causes lysis ~20 min earlier. No experimental structure of L exists.
Goals
- (1) Stabilize L for more robust membrane accumulation. (2) Accelerate lysis by bypassing DnaJ-dependent regulatory timing and improving delivery of functional L to the membrane. Because the downstream lytic target is unknown, we do not attempt to enhance per-molecule toxicity at the point of target engagement; we focus on removing regulatory brakes and increasing the supply of functional protein.
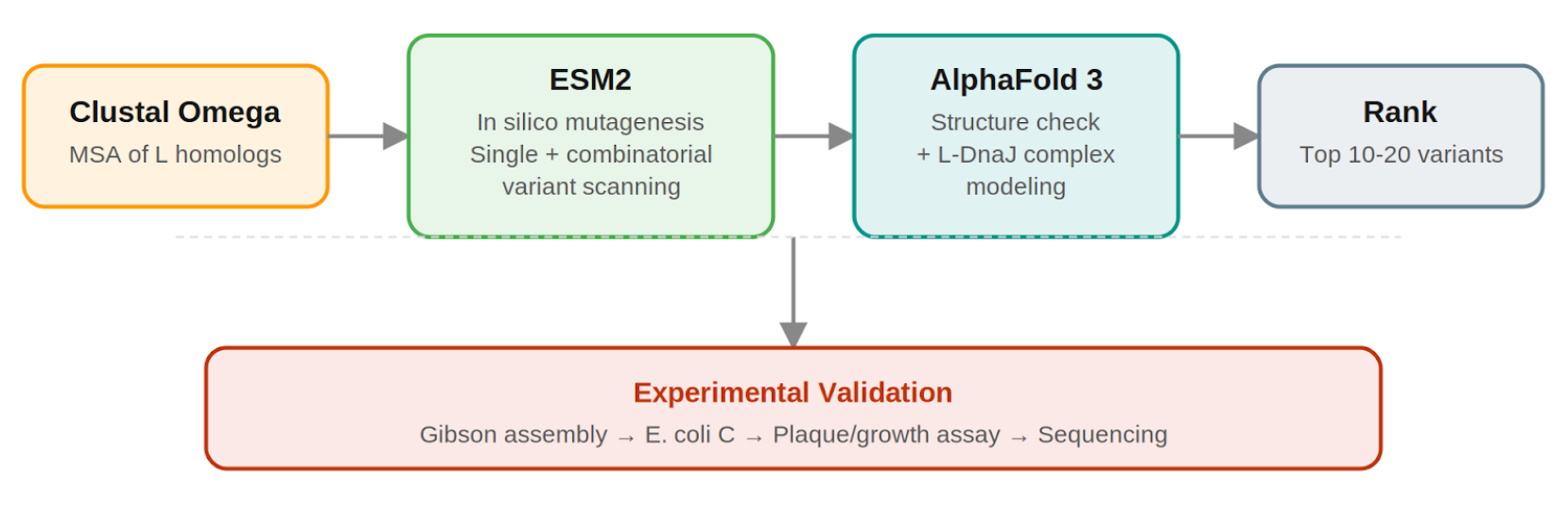
Pipeline: Three Tools, Each Non-Redundant
- Clustal Omega (Conservation Map). Align L homologs across Leviviridae (MS2, f2, R17, GA, PP7, AP205, PRR1, M12, KU1, JP34). Conserved C-terminal residues, especially the LS motif, are presumed to mediate the unknown heterotypic interaction and are excluded from mutation. This map constrains all downstream design.
- ESM2 + Deep Combinatorial Scanning (Fitness Oracle). Score every single-point mutation by log-likelihood change: increases at mutable positions indicate stabilizing substitutions (Goal 1). N-terminal scanning identifies mutations that disrupt DnaJ binding (Goal 2). A strict preservation rule applies near the LS motif: mutations are evaluated for maintenance of wild-type fitness, not improvement. The genetics show even conservative changes there cause recessive loss of function. Pairwise combinatorial scanning (about ~2M pairs) captures epistatic synergies at mutable positions. This could be potentially pushed further with enough compute.
- AlphaFold 3 (Structural Filter + Complex Model). Predicts variant structures as a sanity check (does the TM helix survive?) and models the L–DnaJ complex to verify that N-terminal truncations/mutations disrupt the regulatory interface. Used as a filter, not a design engine. PAE matrix identifies confident interface contacts.
Ranking.
- Composite score: ESM2 log-likelihood gain (stability) + conservation preservation (all essential residues intact) + AF3-predicted DnaJ-binding disruption (for timing bypass). Top 10–20 variants advance to experimental validation.
Pipeline Schematic
Why Not More Tools?
- ProteinMPNN is excluded because it is trained on crystallized globular PDB proteins, not predicted structures of disordered membrane peptides. Compute is invested in combinatorial ESM2 depth.
Pitfalls
- No experimental structure: All structural reasoning rests on AF3 predictions for a challenging target; mitigated by treating AF3 as a filter and cross-referencing against the conservation map.
- Unknown lytic target: The central limitation. We cannot optimize target-binding affinity for an unidentified partner; engineering is restricted to upstream properties (stability, membrane delivery, DnaJ bypass).
- Autolysin bottleneck: If lysis rate is limited by host autolytic enzyme activity rather than L accumulation, stabilization gains may show diminishing returns; the plaque assay will reveal this.