Week 6 HW: Genetic Circuits Part 1
Part A: DNA Assembly
What are some components in the Phusion High-Fidelity PCR Master Mix and what is their purpose?
- Phusion High-Fidelity PCR Master Mix is a 2X mix containing Phusion DNA Polymerase, nucleotides, and an optimized reaction buffer including MgCl₂. Functionally, the polymerase synthesizes the new DNA strand, the dNTPs are the nucleotide building blocks it incorporates, the buffer maintains the reaction chemistry, and Mg²⁺ is the essential cofactor that enables polymerase activity and helps stabilize primer-template interactions. Phusion is called “high fidelity” because it has 3′→5′ exonuclease proofreading activity, which lowers the error rate compared with standard Taq-type PCR enzymes.
What are some factors that determine primer annealing temperature during PCR?
- Primer annealing temperature is determined primarily by the melting temperature (Tm) of the primers. A common guideline is to set the annealing temperature about 2–5°C below the lower Tm of the primer pair so that the primers bind efficiently to the intended target without binding too readily to partially matching sites. The Tm itself depends on several factors, especially primer length, GC content, and sequence composition, because GC-rich primers generally bind more strongly than AT-rich primers. Annealing temperature is also influenced by mismatches, which reduce binding stability, and by secondary structures such as hairpins, self-dimers, and heterodimers, which can interfere with normal primer-target annealing. Salt and buffer conditions further affect duplex stability, so annealing temperature is ultimately a function of both primer design and reaction chemistry.
There are two methods from this class that create linear fragments of DNA: PCR, and restriction enzyme digests. Compare and contrast these two methods, both in terms of protocol as well as when one may be preferable to use over the other.
- PCR and restriction enzyme digestion can both produce linear DNA fragments, but they do so through very different mechanisms and are useful in different situations. PCR is an amplification-based method in which a DNA region is copied through repeated cycles of denaturation, primer annealing, and extension using a thermostable polymerase. This makes PCR highly flexible, because the user can selectively amplify a desired region even from a small amount of starting material and can also introduce new sequence features, such as mutations, overlap regions, or added restriction sites, through primer design. By contrast, a restriction enzyme digest does not amplify DNA at all; instead, it cuts existing DNA at specific recognition sequences to release a fragment with defined ends, which may be sticky or blunt depending on the enzyme used. Restriction digestion is usually preferable when the desired fragment is already available in a plasmid or other DNA source and is conveniently flanked by suitable enzyme sites, whereas PCR is preferable when no convenient restriction sites exist or when the fragment must be modified as it is generated. In that sense, PCR is more design-flexible, while restriction digestion is more site-dependent and typically simpler when the necessary cut sites are already present.
How can you ensure that the DNA sequences that you have digested and PCR-ed will be appropriate for Gibson cloning?
- The DNA fragments are appropriate for Gibson cloning when they are designed with the correct overlapping sequences and verified before assembly. The primers should add 20–22 bp overhangs, and neighboring fragments used in Gibson assembly should share about 20–40 bp of sequence identity so they can join correctly. In practice, this means ensuring that the backbone and color fragments have the proper matching overlaps, the correct 5′→3′ orientation, and the expected sequence content before they are combined. After PCR, the products are treated with DpnI to remove the parental plasmid template, then purified, quantified, and checked on a diagnostic gel against the expected fragment sizes in Benchling.
How does the plasmid DNA enter the E. coli cells during transformation?
- Plasmid DNA enters E. coli during transformation by first making the bacterial cells competent, meaning temporarily able to take up external DNA. In chemical transformation, cells are typically treated with salts such as calcium chloride and then subjected to a brief heat shock, which increases membrane permeability and helps the plasmid enter the cell. Heat shock causes the cell membrane to “open up,” allowing the plasmid to enter the cells by diffusion. After recovery and plating, only the bacteria that successfully received the plasmid survive on the antibiotic plate and later express the chromophore color. In electroporation, an electrical pulse creates transient pores in the membrane through which DNA can pass. After either method, the cells are allowed to recover in rich medium so they can repair their membranes and begin expressing the plasmid’s selectable marker, such as an antibiotic-resistance gene, before they are plated on selective agar. Thus, transformation works by temporarily disrupting the membrane barrier long enough for plasmid DNA to cross into the bacterial cell interior.
Describe another assembly method in detail (such as Golden Gate Assembly)
- One alternative to Gibson Assembly is Golden Gate Assembly, a one-pot cloning method that uses a Type IIS restriction enzyme such as BsaI or BsmBI together with T4 DNA ligase. Unlike standard restriction enzymes, Type IIS enzymes cut outside of their recognition sequences, which allows the user to design custom overhangs that determine exactly how the DNA fragments will join together. During the reaction, the enzyme cuts the fragments to expose these programmed overhangs, and ligase then seals the matching ends together in the intended order. Because the recognition sites are usually removed during assembly, the final product is often considered scarless or seamless. Golden Gate is especially useful for modular and multi-fragment cloning because many parts can be assembled in a single reaction, but it requires careful design to ensure that the DNA parts do not contain unwanted internal sites for the chosen Type IIS enzyme.
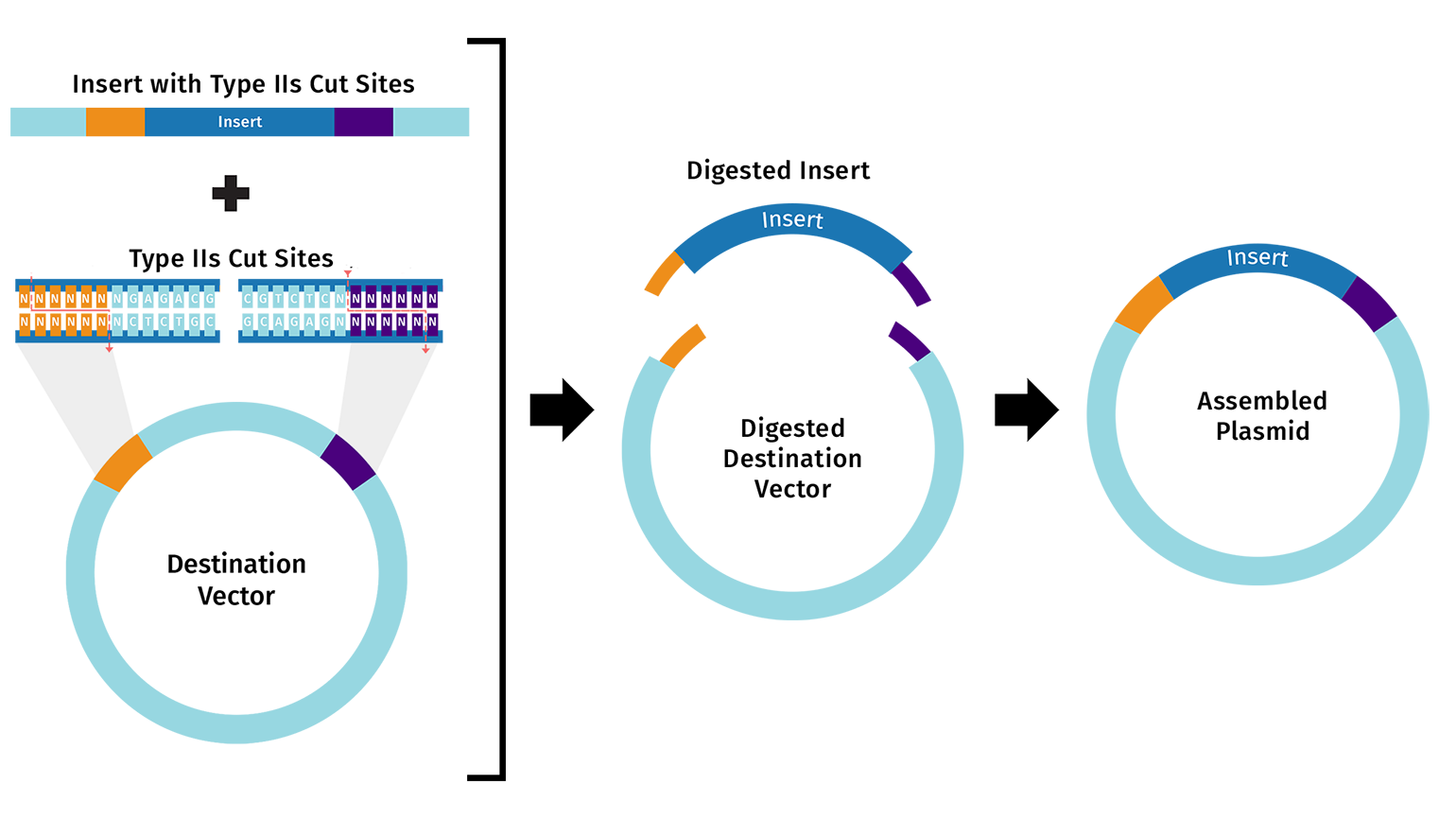
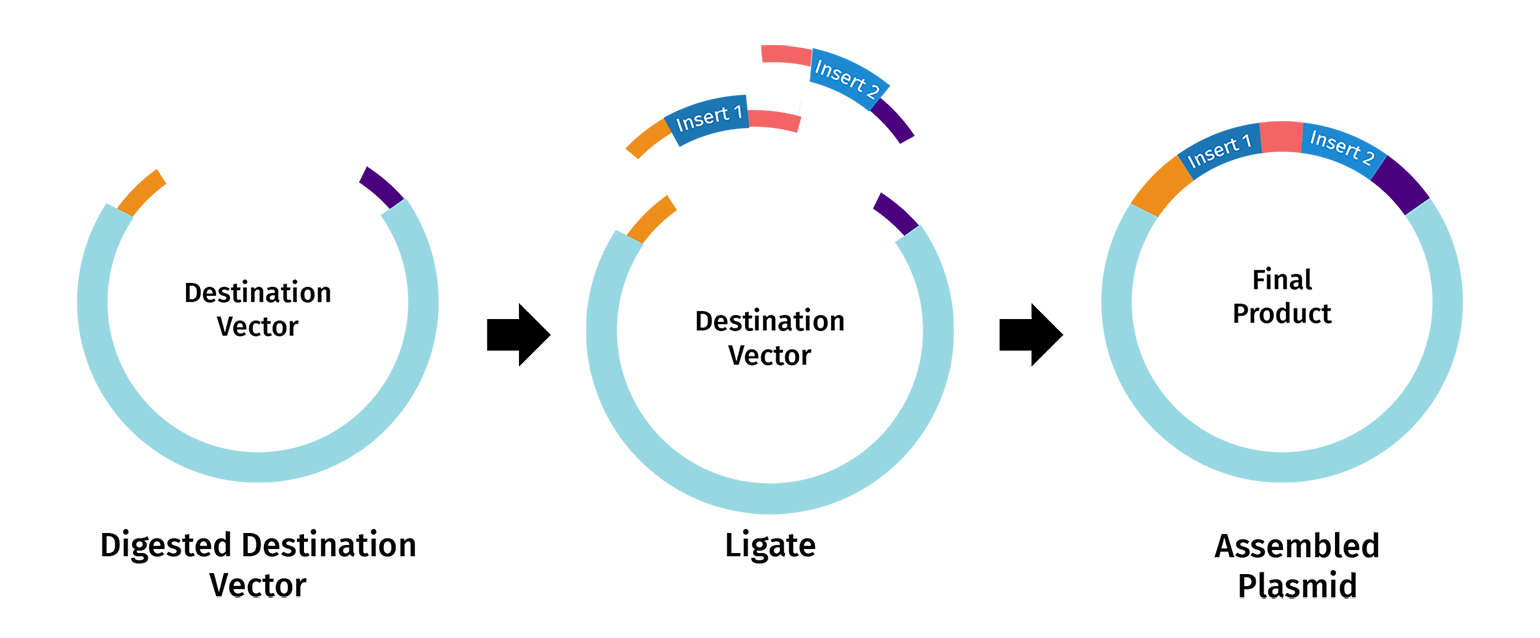
Images are from snapgene.com
Part B: Asimov Kernel
- Construct:0. Repressilator

pTetR → A1 RBS → LacI → L3S2P24 Bacterial Terminator → pLacI → A1 RBS → LambdaCI → L3S2P24 Bacterial Terminator → pLambdaCI → A1 RBS → TetR → L3S2P24 Bacterial Terminator → pUC-SpecR V1 backbone → (back to pTetR)
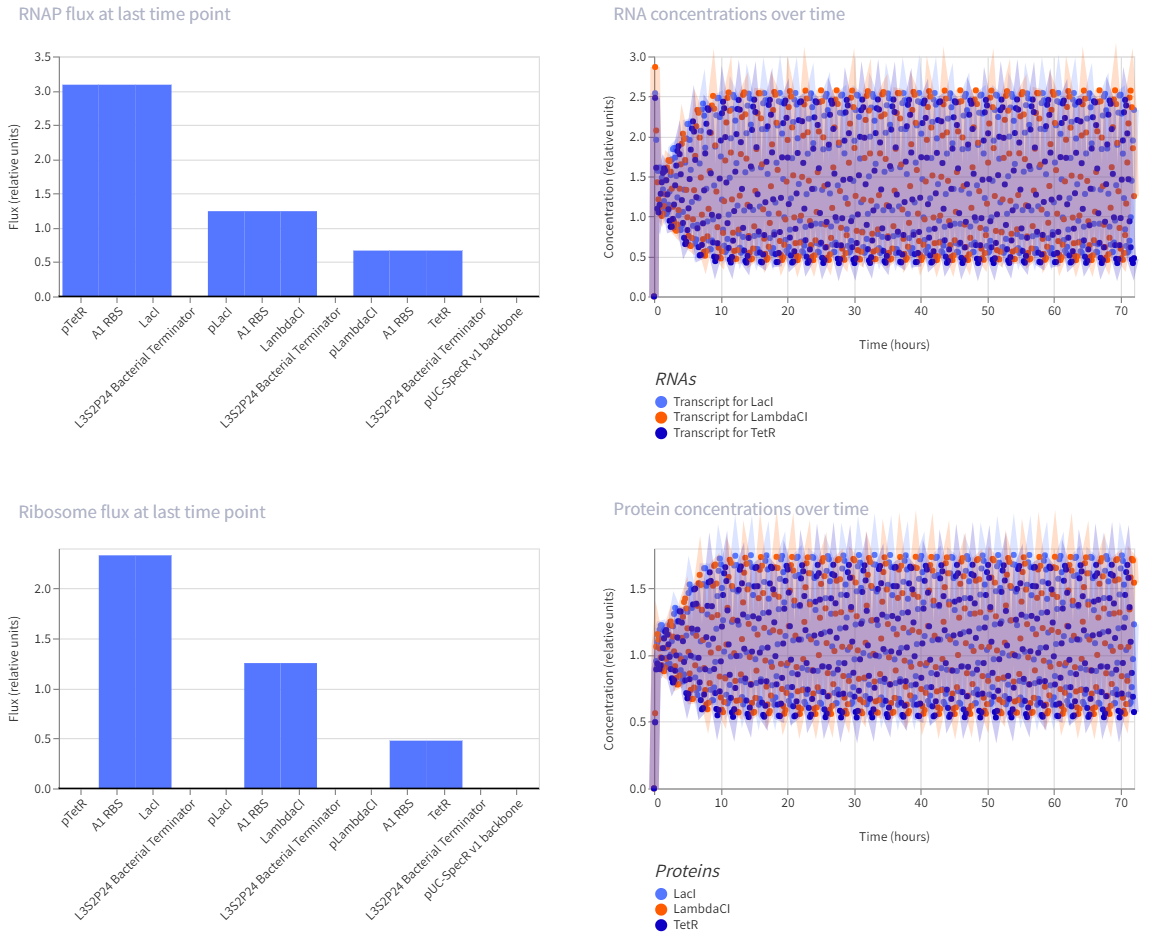
- The repressilator is a synthetic genetic oscillator made from three repressors arranged in a circular negative-feedback loop. In the construct, pTetR drives LacI, pLacI drives LambdaCI, and pLambdaCI drives TetR. Each protein represses the promoter of the next gene: LacI represses pLacI, LambdaCI represses pLambdaCI, and TetR represses pTetR. In simple terms, this means each gene takes a turn shutting off the next one, causing expression to rotate through the circuit instead of remaining constant. This creates oscillations in both RNA and protein levels over time, which is visible in the simulation graphs. The RNA and protein plots show repeating up-and-down patterns, indicating that the circuit is functioning as an oscillator rather than a simple on/off switch.
- Construct:1. Negative autoregulation circuit

pLacI → A1 RBS → LacI → L3S2P24 Bacterial Terminator → pUC-SpecR V1 backbone → (back to pLacI)
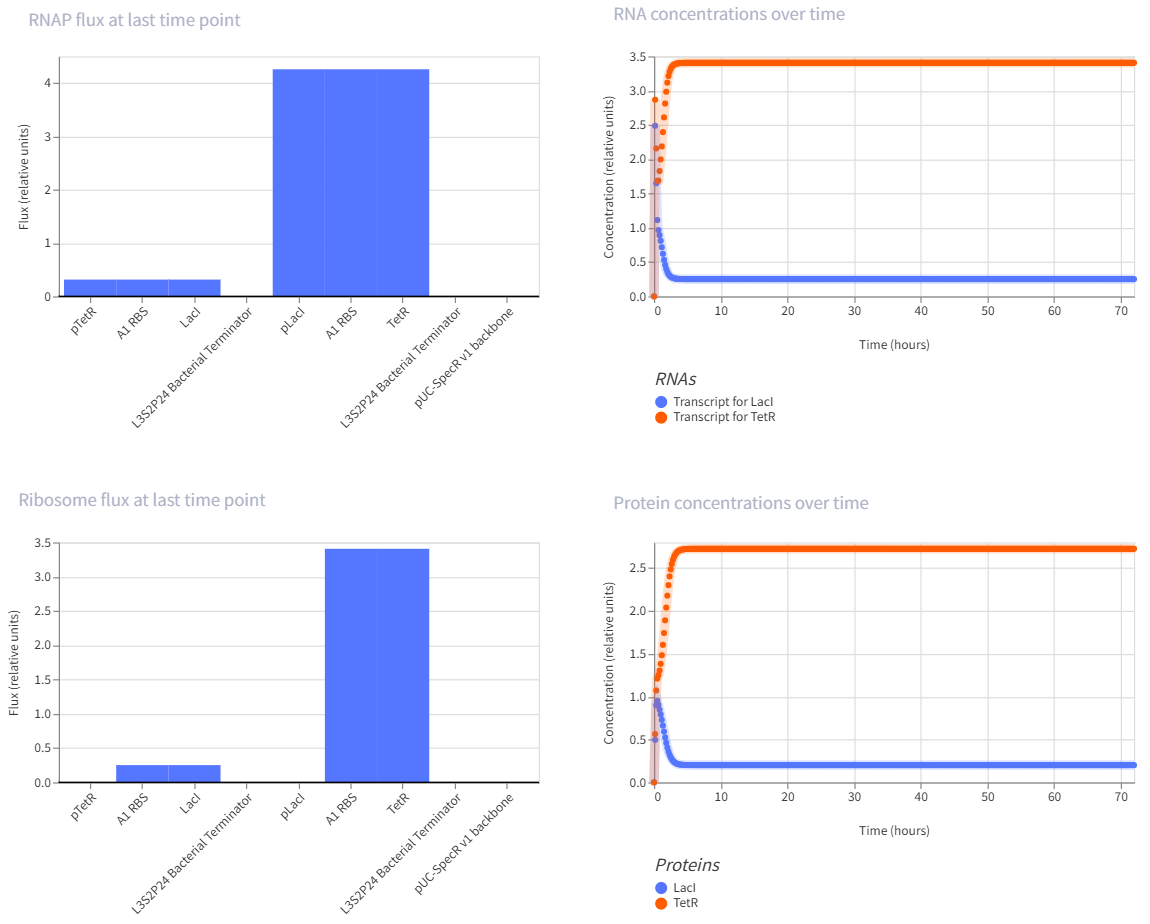
- This is a self-repressing gene. It is one of the classic simple synthetic circuits, and negative autoregulation in E. coli is known to speed response time compared with a non-self-regulated unit.
- Construct:2. Genetic toggle switch
pTetR → A1 RBS → LacI → L3S2P24 Bacterial Terminator → pLacI → A1 RBS → TetR → L3S2P24 Bacterial Terminator → pUC-SpecR V1 backbone → (back to pTetR)
- The toggle switch is one of the landmark synthetic gene circuits in E. coli. It uses two repressors that inhibit each other, which can create two stable states rather than one oscillating pattern.
- Construct:3. Repression cascade with GFP output

pLambdaCI → A1 RBS → TetR → L3S2P24 Bacterial Terminator → pTetR → A1 RBS → LacI → L3S2P24 Bacterial Terminator → pLacI → A1 RBS → GFP → L3S2P24 Bacterial Terminator → pUC-SpecR V1 backbone → (back to pLambdaCI)
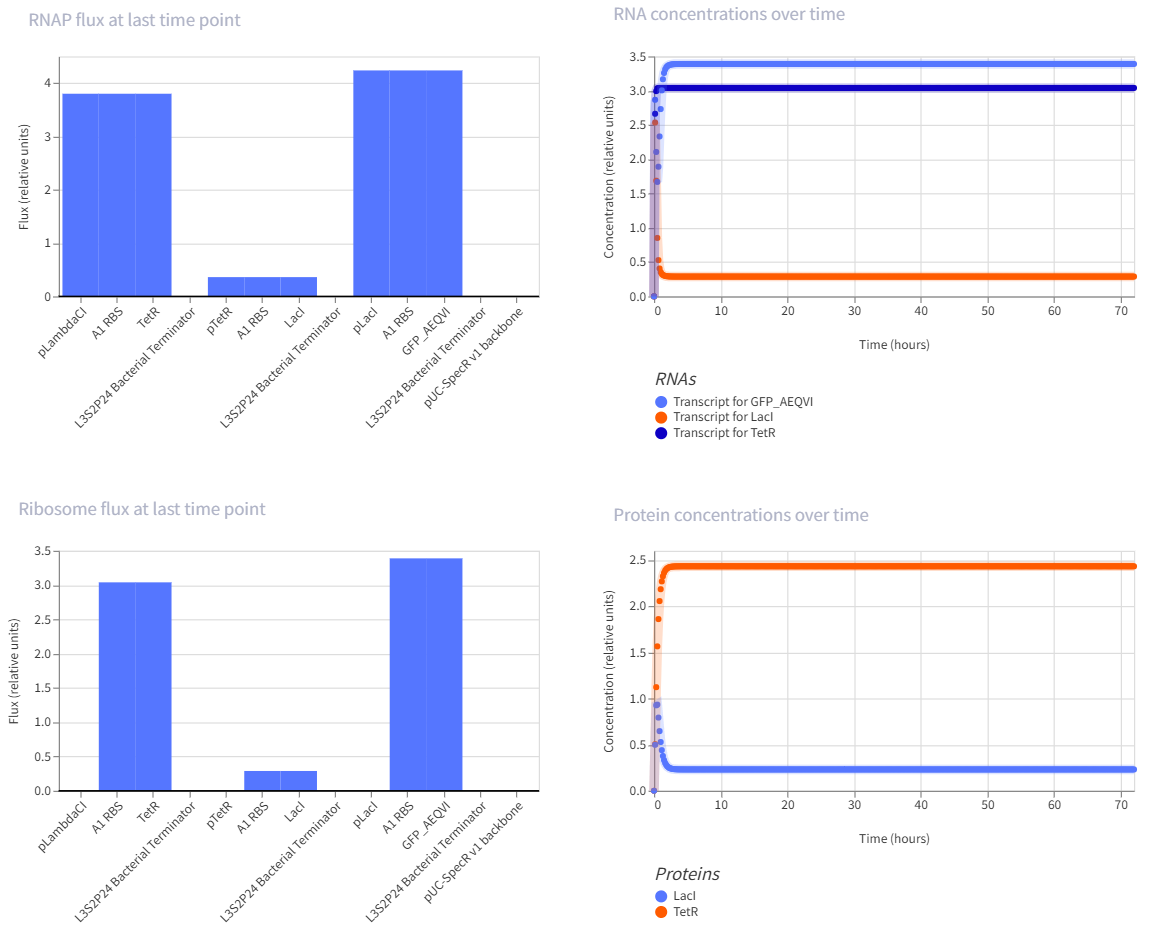
- This is a multi-step repression chain, which is easier than the repressilator but still interesting because it adds delay and layered regulation. Reviews of synthetic circuits in E. coli discuss repression cascades built by inserting modules like LacI and cI between an input layer and a reporter, and note that longer cascades tend to increase response time and noise.