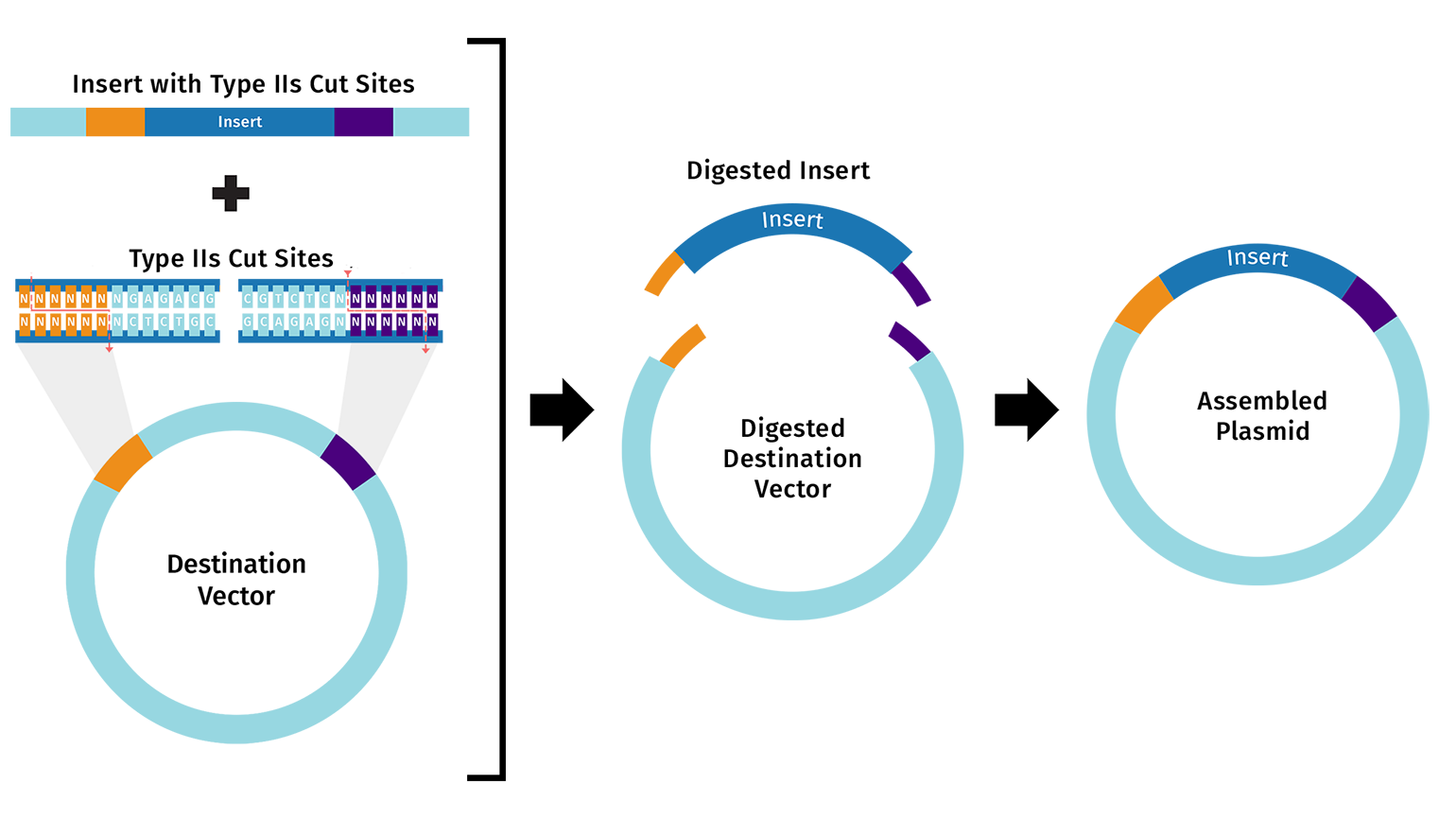I am a Masters of Design Studies candidate in the Ecologies domain at Harvard GSD.
I love cooking, video games, puzzles, animals, martial arts, and movies.
I have previously researched and worked with mycelium as a building material, from cultivation to production, to explore its structural and insulating quality. I am currently researching design strategies that explore architecture’s ability to contribute to marine ecosystems after being submerged by water, such as architectural materials that promote coral colonization after sea levels rise.
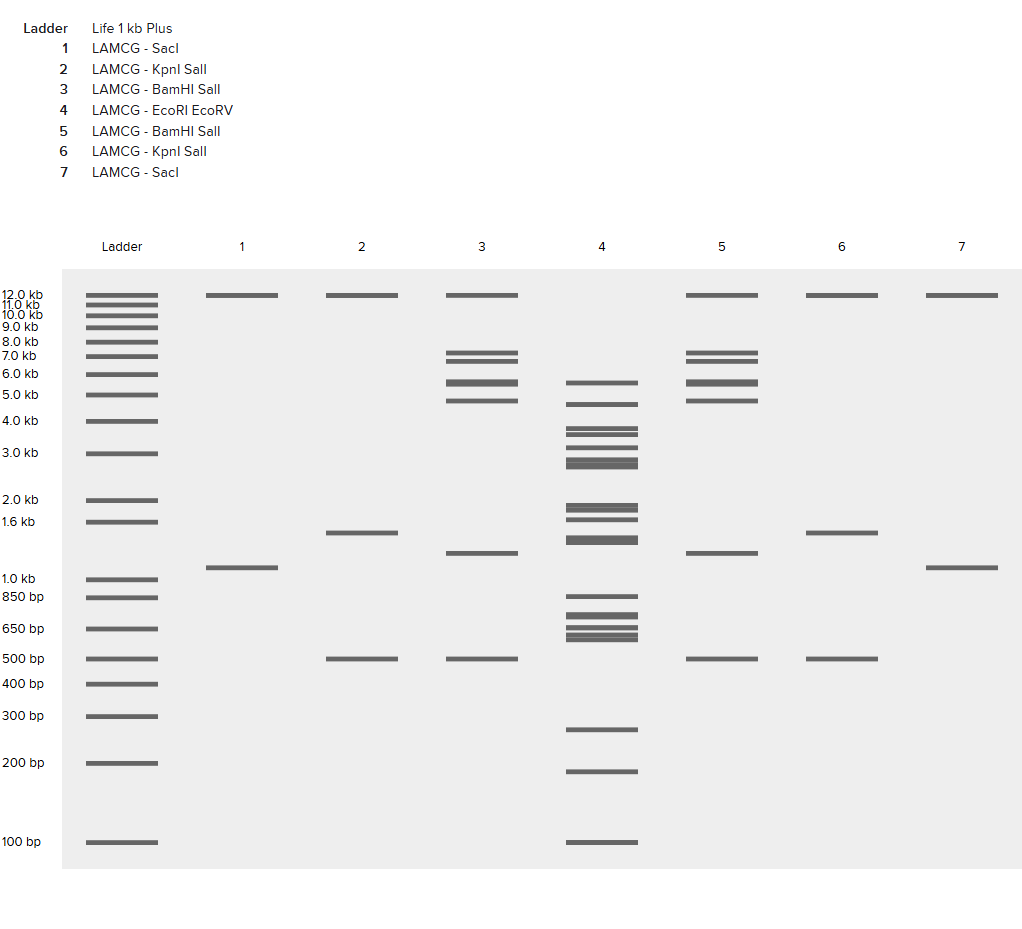
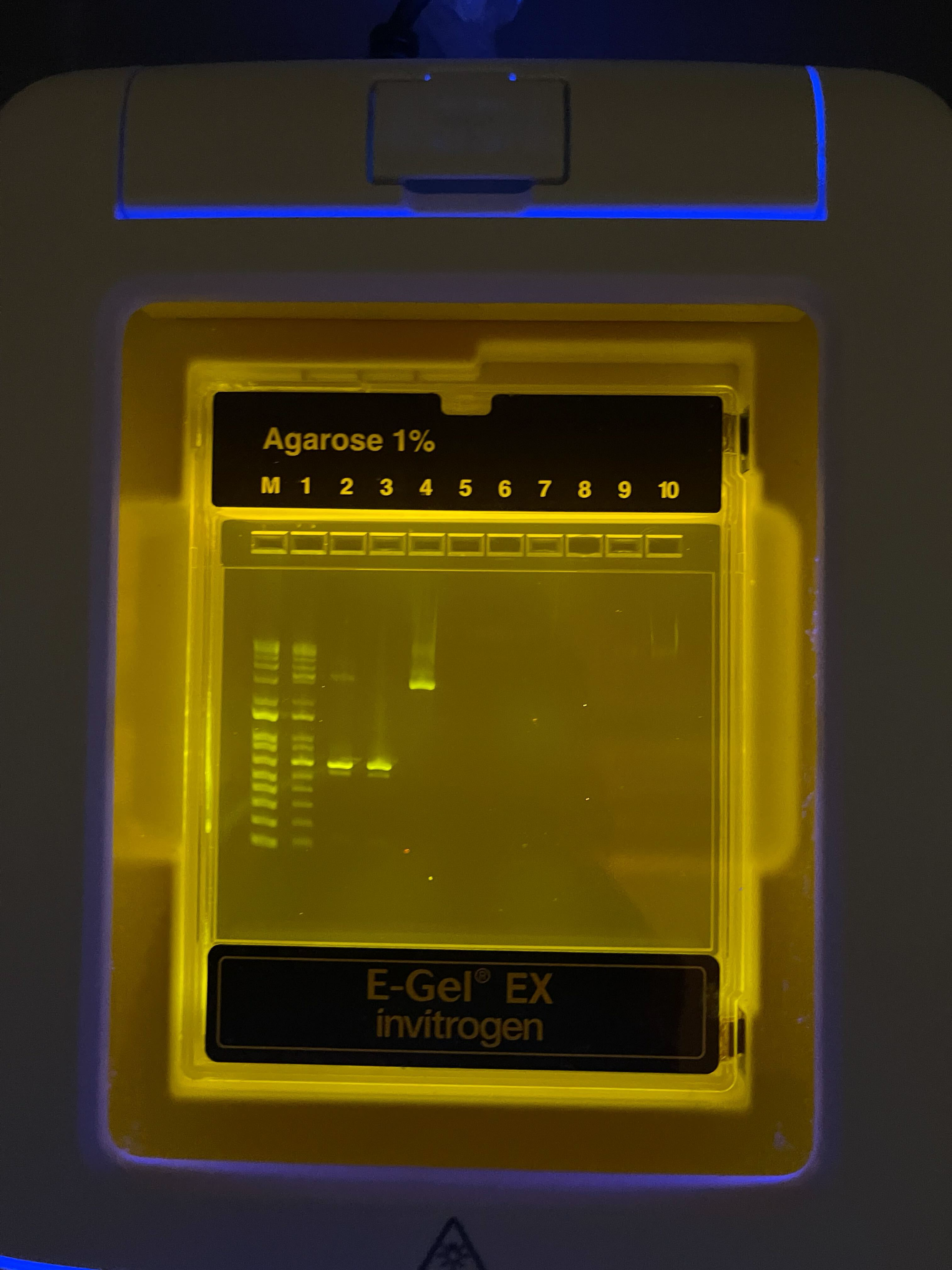
Part 1: Benchling & In-silico Gel Art Part 2: Gel Art - Restriction Digests and Gel Electrophoresis See Week 2 Lab.
Part 3: DNA Design Challenge 3.1. Choose your protein.
Post-Lab Questions 1. Find and describe a published paper that utilizes the Opentrons or an automation tool to achieve novel biological applications.
Dettinger et al. (2022), “Open-source personal pipetting robots with live-cell incubation and microscopy compatibility,” published in Nature Communications.
The authors introduce PHIL (Pipetting Helper Imaging Lid), an open-source, low-cost pipetting robot designed for liquid handling during live-cell experiments and microscopy workflows. PHIL is important because it addresses a real problem in academic labs: many experiments are small-scale, frequently changing, and not well suited to large industrial automation systems, which are often expensive and hard to adapt.
Part A. Conceptual Questions 1. How many molecules of amino acids do you take with a piece of 500 grams of meat? (on average an amino acid is ~100 Daltons)
Assuming 500 g of meat contains about 20–25% protein by weight, that piece has roughly:
100–125 g of protein 100 daltons = 100 g/mol so 100–125 g of amino acid residues = about 1.0–1.25 moles 1 mole = 6.022 × 10^23 molecules So the total is about: 6.0 × 1023 to 7.5 × 1023 amino acid molecules 2. Why do humans eat beef but do not become a cow, eat fish but do not become fish?
Part A: SOD1 Binder Peptide Design (From Pranam) Part 1: Generate Binders with PepMLM
Sequnce:
MATKVVCVLKGDGPVQGIINFEQKESNGPVKVWGSIKGLTEGLHGFHVHEFGDNTAGCTSAGPHFNPLSRKHGGPKDEERHVGDLGNVTADKDGVADVSIEDSVISLSGDHCIIGRTLVVHEKADDLGKGGNEESTKTGNAGSRLACGVIGIAQ Binder Pseudo Perplexity WRYYVAAVALWX 16.2395343215211 WHYYAVAAEWKX 13.6945052943038 WLVPAAAAAHGK 7.93406338297721 WRYGPVAVRHWK 14.3797355975152 FLYRWLPSRRGG 20.6352312728361 Part 2: Evaluate Binders with AlphaFold3
“PepMLM outputs X; substituted X→A for AlphaFold input.”
Binder ipTM Score WRYYVAAVALWA 0.34 WHYYAVAAEWKA 0.23 WLVPAAAAAHGK 0.48 WRYGPVAVRHWK 0.36 FLYRWLPSRRGG 0.38 Across the five AlphaFold3 complex predictions, ipTM values ranged from 0.23 to 0.48. The known binder FLYRWLPSRRGG gave ipTM = 0.38 and appeared weakly defined, remaining largely surface-adjacent/partially detached rather than buried in a clear pocket, with no obvious localization near the N-terminus where A4V sits. Three PepMLM peptides (WHYYAVAAEWKA, 0.23; WRYYVAAVALWA, 0.34; WRYGPVAVRHWK, 0.36) similarly showed low-confidence interfaces, tending to lie loosely on the β-barrel exterior instead of concentrating at the A4V region. In contrast, WLVPAAAAAHGK produced the strongest prediction (ipTM = 0.48) and appeared more plausibly docked along a β-barrel/loop-adjacent surface, making it the only PepMLM-generated peptide that exceeded the known binder’s ipTM in this set. Part 3: Evaluate Properties of Generated Peptides in the PeptiVerse
Part A: DNA Assembly What are some components in the Phusion High-Fidelity PCR Master Mix and what is their purpose?
Phusion High-Fidelity PCR Master Mix is a 2X mix containing Phusion DNA Polymerase, nucleotides, and an optimized reaction buffer including MgCl₂. Functionally, the polymerase synthesizes the new DNA strand, the dNTPs are the nucleotide building blocks it incorporates, the buffer maintains the reaction chemistry, and Mg²⁺ is the essential cofactor that enables polymerase activity and helps stabilize primer-template interactions. Phusion is called “high fidelity” because it has 3′→5′ exonuclease proofreading activity, which lowers the error rate compared with standard Taq-type PCR enzymes. What are some factors that determine primer annealing temperature during PCR?
Assignment Part 1: Intracellular Artificial Neural Networks What advantages do IANNs have over traditional genetic circuits, whose input/output behaviors are Boolean functions?
IANNs have an advantage over traditional Boolean genetic circuits because they can respond to graded, noisy biological signals instead of forcing every input into a simple ON/OFF state. This makes them better for tasks like classification, where a cell needs to weigh several signals together and make a more flexible decision, while Boolean circuits are better for simpler yes/no logic. Describe a useful application for an IANN; include a detailed description of input/output behavior, as well as any limitations an IANN might face to achieve your goal.
General homework questions Explain the main advantages of cell-free protein synthesis over traditional in vivo methods, specifically in terms of flexibility and control over experimental variables. Name at least two cases where cell-free expression is more beneficial than cell production.
Cell-free protein synthesis is more flexible than in vivo expression because you can directly control the reaction conditions without keeping cells alive. It is especially useful for making toxic proteins and proteins that are hard to express in living cells, like some membrane proteins. Describe the main components of a cell-free expression system and explain the role of each component.
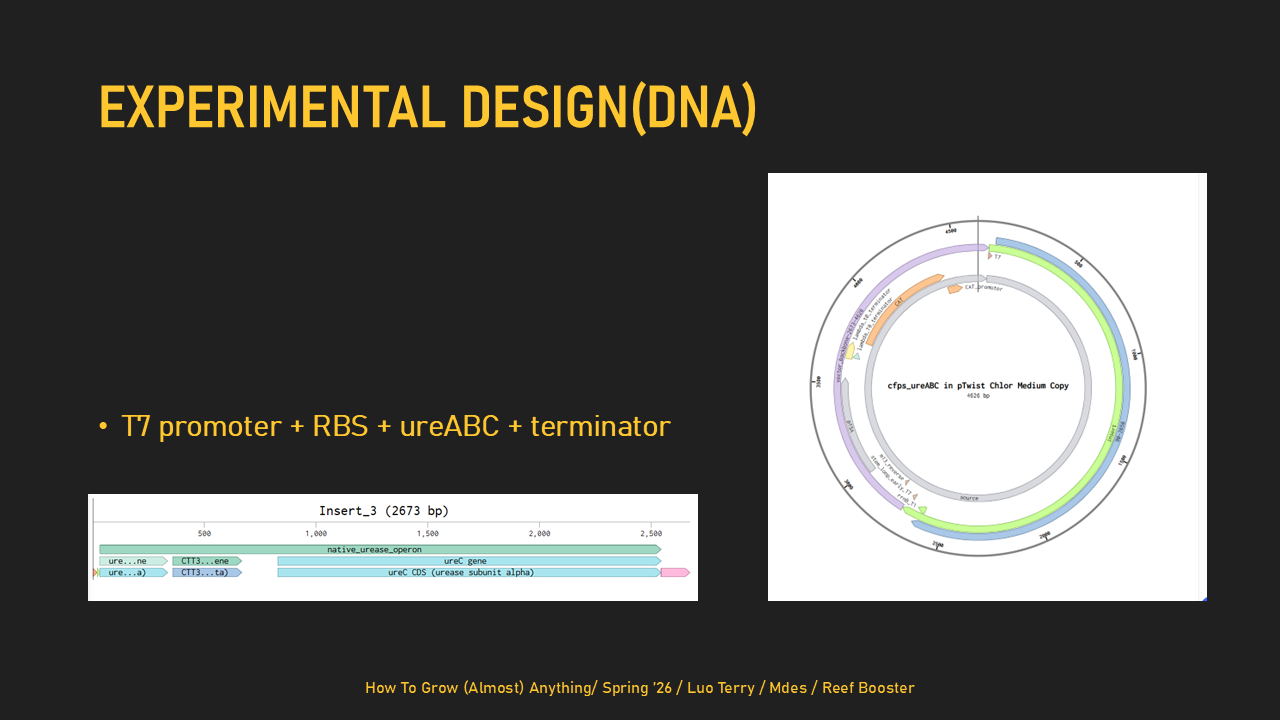
Homework: Final Project For the final project, I will primarily measure whether my designed DNA templates are correct and whether they can produce proteins in a T7 cell-free expression system. I will verify the DNA constructs using DNA sequencing and agarose gel electrophoresis, then test expression using two templates: a minimal ureABC structural urease construct and a separate sfGFP reporter construct. The sfGFP template will serve as a visual positive control, and its expression will be measured by fluorescence, while expression of UreA, UreB, and UreC will be measured using SDS-PAGE. I may also perform an exploratory urease-related activity assay, but this is not the main measurement because the minimal structural construct does not include the full accessory genes required for strong urease activation. Homework: Waters Part I — Molecular Weight Based on the predicted amino acid sequence of eGFP (see below) and any known modifications, what is the calculated molecular weight?
Part B: Cell-Free Protein Synthesis | Cell-Free Reagents Referencing the cell-free protein synthesis reaction composition (the middle box outlined in yellow on the image above, also listed below), provide a 1-2 sentence description of what each component’s role is in the cell-free reaction.
BL21 (DE3) Star lysate (includes T7 RNA polymerase): This provides the core cellular machinery needed for protein synthesis, incl~uding ribosomes, tRNAs, enzymes, and other translation factors. Because it includes T7 RNA polymerase, it can also transcribe DNA templates with a T7 promoter into mRNA.
Subsections of Homework
Week 1 HW: Principles and Practices
Assignment (Class - Ethics)
1. Describe a biological engineering application or tool you want to develop and why.
I want to develop a bioengineered architectural substrate that accelerates coral colonization and marine habitat formation on submerged structures. I call this system Reef-Transition Building Skin (RTBS) - a modular “living interface” that can be attached to coastal buildings, seawalls, piles, and pier foundations, biologically tuned to support coral settlement and marine habitat formation once those structures are periodically or permanently submerged. Sea-level rise guarantees that many coastal structures will eventually enter the water, yet most existing hard infrastructure is ecologically sterile or actively harmful. RTBS responds to this reality by operationalizing the idea that architecture can also serve non-human systems. It is based on the idea that architecture should be designed for more than its period of human occupation: structures that serve people today should be capable of transforming into productive marine habitat in the future, so that when they are submerged they contribute to ecological life rather than becoming inert debris or pollution.
2. Describe one or more governance/policy goals related to ensuring that this application or tool contributes to an “ethical” future, like ensuring non-malfeasance (preventing harm). Break big goals down into two or more specific sub-goals.
Goal A — Prevent harm to ecosystems and people
Avoid invasive impacts: Check local ecological compatibility before deployment to prevent disrupting existing species and habitats.
Avoid toxic pollution: Set strict limits on harmful chemicals and materials that could leach into the water.
Avoid false ecological claims: Require measurable evidence before projects can claim environmental benefits.
Goal B — Ensure real ecological benefit
Prove habitat improvement: Measure ecological conditions before and after installation to confirm actual gains.
Design for local conditions: Adapt structures to each site’s water quality, climate, and wave patterns.
Plan for long-term care: Require maintenance and repair plans if systems fail or cause harm.
Goal C — Support fairness and local control
Benefit local communities: Ensure projects create jobs or ecological benefits for nearby residents.
Include local decision-making: Involve community stakeholders in design and approval.
Share information openly: Publish monitoring results, including failures, not just successes.
3. Describe at least three different potential governance “actions” by considering the four aspects below (Purpose, Design, Assumptions, Risks of Failure & “Success”).
Action 1 - Habitat-based permitting rule
This action would change coastal permits so projects must help marine ecosystems, not just avoid damage. Regulators would require new seawalls, piers, and coastal structures to show measurable ecological benefit, such as increased coral growth or biodiversity, supported by surveys before construction and monitoring for several years after installation. Independent reviewers would verify results, and projects that fail would need to be repaired or redesigned. This assumes ecological performance can be measured fairly and that communities can support monitoring. The risk is weak enforcement, where monitoring becomes symbolic. Even if it works, developers might focus on easy metrics instead of long-term ecosystem health.
Action 2 - Eco-material certification standard
This action creates a certification system to ensure coastal building materials are safe for marine life. Materials would be tested for toxicity, chemical leaching, and durability, and proven in real-world pilot projects before approval. Certified materials would be labeled and traceable, and public infrastructure projects would prioritize their use. This assumes companies are willing to share information and certification bodies stay independent. The risk is greenwashing if standards are weak. Even success could create problems if certification becomes expensive and excludes smaller producers.
Action 3 - Financial incentives for transition-ready architecture
This action makes ecological coastal design financially attractive. Governments and insurers would provide grants, insurance discounts, and performance-based funding to projects that demonstrate real ecological benefits. Funding would depend on monitoring results and require open data and local job training. This assumes insurers accept ecological performance as reducing risk and that ecosystem benefits can be valued financially. The risk is funding projects that don’t actually help ecosystems. Even if successful, incentives could unintentionally encourage more development in vulnerable coastal areas.
4. Score (from 1-3 with, 1 as the best, or n/a) each of your governance actions against your rubric of policy goals. The following is one framework but feel free to make your own:
Does the option:
Habitat-based permitting rule
Eco-material certification standard
Financial incentives for transition-ready architecture
Enhance Biosecurity
• By preventing incidents
2
3
1
• By helping respond
3
2
1
Foster Lab Safety
• By preventing incident
2
1
n/a
• By helping respond
1
2
n/a
Protect the environment
• By preventing incidents
2
3
1
• By helping respond
2
1
3
Other considerations
• Minimizing costs and burdens to stakeholders
n/a
n/a
3
• Feasibility?
3
1
2
• Not impede research
1
n/a
2
• Promote constructive applications
3
1
2
5. Drawing upon this scoring, describe which governance option, or combination of options, you would prioritize, and why. Outline any trade-offs you considered as well as assumptions and uncertainties.
Based on the scoring, I would prioritize the habitat-based permitting rule as the primary governance action, supported by certification and financial incentives. Permitting is the strongest safeguard because it directly prevents ecological harm and sets enforceable minimum standards. Without a regulatory baseline, certification and incentives risk becoming optional or symbolic. A permitting framework ensures that every coastal project must meet habitat-performance thresholds, making environmental protection non-negotiable rather than market-dependent.
Certification and incentives still play important supporting roles. Certification reduces uncertainty about material safety, making compliance easier and more consistent, while incentives help offset costs and encourage adoption. However, relying too heavily on financial incentives could unintentionally encourage risky development in vulnerable coastal areas. Permitting acts as the necessary boundary that keeps ecological goals from being undermined by economic pressures.
The main trade-off is feasibility: strong permitting requires institutional capacity, reliable monitoring, and long-term enforcement. It assumes ecological performance can be measured fairly across diverse sites and that regulators have resources to enforce standards. Uncertainty remains around scaling this system globally and maintaining political support over time. Still, prioritizing permitting creates a stable ethical foundation, with certification and incentives functioning as tools that operate within those ecological limits rather than replacing them.
Assignment (Lab Preparation)
Complete Lab Specific Training in Person.
Complete Safety Training in Atlas
Assignment (Your HTGAA Website)
Personalizing your HTGAA website
Assignment (Week 2 Lecture Prep)
Homework Questions from Professor Jacobson:
Nature’s machinery for copying DNA is called polymerase. What is the error rate of polymerase? How does this compare to the length of the human genome. How does biology deal with that discrepancy?
DNA polymerase makes about one error per 10⁴–10⁵ bases, while the human genome is about 3 × 10⁹ bases long. Cells correct this mismatch through proofreading and mismatch repair, reducing the final mutation rate to roughly one error per genome replication.
How many different ways are there to code (DNA nucleotide code) for an average human protein? In practice what are some of the reasons that all of these different codes don’t work to code for the protein of interest?
An average human protein can be encoded by an astronomically large number of DNA sequences because of codon redundancy, roughly 3ᴸ for a protein of length L. Most of these sequences do not work well in practice due to codon bias, unstable mRNA structure, extreme GC content, hidden regulatory motifs, or disrupted translation kinetics.
Homework Questions from Dr. LeProust:
What’s the most commonly used method for oligo synthesis currently?
The most common oligo synthesis method is solid-phase phosphoramidite chemistry.
Why is it difficult to make oligos longer than 200nt via direct synthesis?
Oligos longer than about 200 nucleotides are difficult because small stepwise synthesis errors accumulate exponentially with length.
Why can’t you make a 2000bp gene via direct oligo synthesis?
A 2000 bp gene cannot be made by direct synthesis because yield collapses and errors dominate, so long genes must be assembled from shorter oligos.
Homework Question from George Church:
What are the 10 essential amino acids in all animals and how does this affect your view of the “Lysine Contingency”?
The 10 essential amino acids in animals are arginine, histidine, isoleucine, leucine, lysine, methionine, phenylalanine, threonine, tryptophan, and valine. Lysine dependence highlights a potential containment strategy because organisms that cannot synthesize lysine cannot grow without an external supply.
Week 2 HW: Read, Write, and Edit DNA
Part 1: Benchling & In-silico Gel Art
Part 2: Gel Art - Restriction Digests and Gel Electrophoresis
See Week 2 Lab.
Part 3: DNA Design Challenge
3.1. Choose your protein.
Skeletal aspartic acid-rich protein 1
Acropora millepora (Staghorn coral)
I chose skeletal aspartic acid rich protein 1 (SAARP1) because it is directly involved in coral calcification and biomineralization, linking a specific protein to the physical formation of the coral skeleton. Skeletal aspartic acid rich protein 1 (SAARP1) is a major component of the skeletal organic matrix (SOM) in the staghorn coral Acropora millepora. It is a highly acidic protein with roughly 20 percent aspartic acid residues and is involved in coral calcification and biomineralization. SAARP1 belongs to a conserved family of acidic proteins that likely helps regulate the formation, structure, and deposition of aragonite crystals in the coral skeleton.
3.2. Reverse Translate: Protein (amino acid) sequence to DNA (nucleotide) sequence.
Even though multiple codons can encode the same amino acid, different organisms prefer different codons. That preference tracks with tRNA abundance and other expression constraints. If you use a DNA sequence with many codons that are “rare” in your expression host, translation can stall, causing lower yield, more misfolding/aggregation, and sometimes premature termination. The codon is optimized for E. Coli because we will be using it in labs as the host.
3.4. You have a sequence! Now what?
Once I have a codon-optimized SAARP1 DNA sequence, I can produce the protein by placing the gene into an E. coli expression plasmid that includes a promoter, ribosome binding site (RBS), transcription terminator, origin of replication, and an antibiotic resistance marker, often plus a purification tag such as 6×His. The plasmid can be introduced into E. coli either via heat shock or electroporation. After the plasmid is inside E. coli, the gene is transcribed into mRNA when the promoter is active (often induced in expression systems). The mRNA is then translated by E. coli ribosomes, which read the mRNA codons and use tRNAs to assemble the SAARP1 amino-acid chain. Codon optimization improves this step by matching E. coli’s preferred codons, reducing ribosome stalling and increasing the likelihood of higher protein yield.
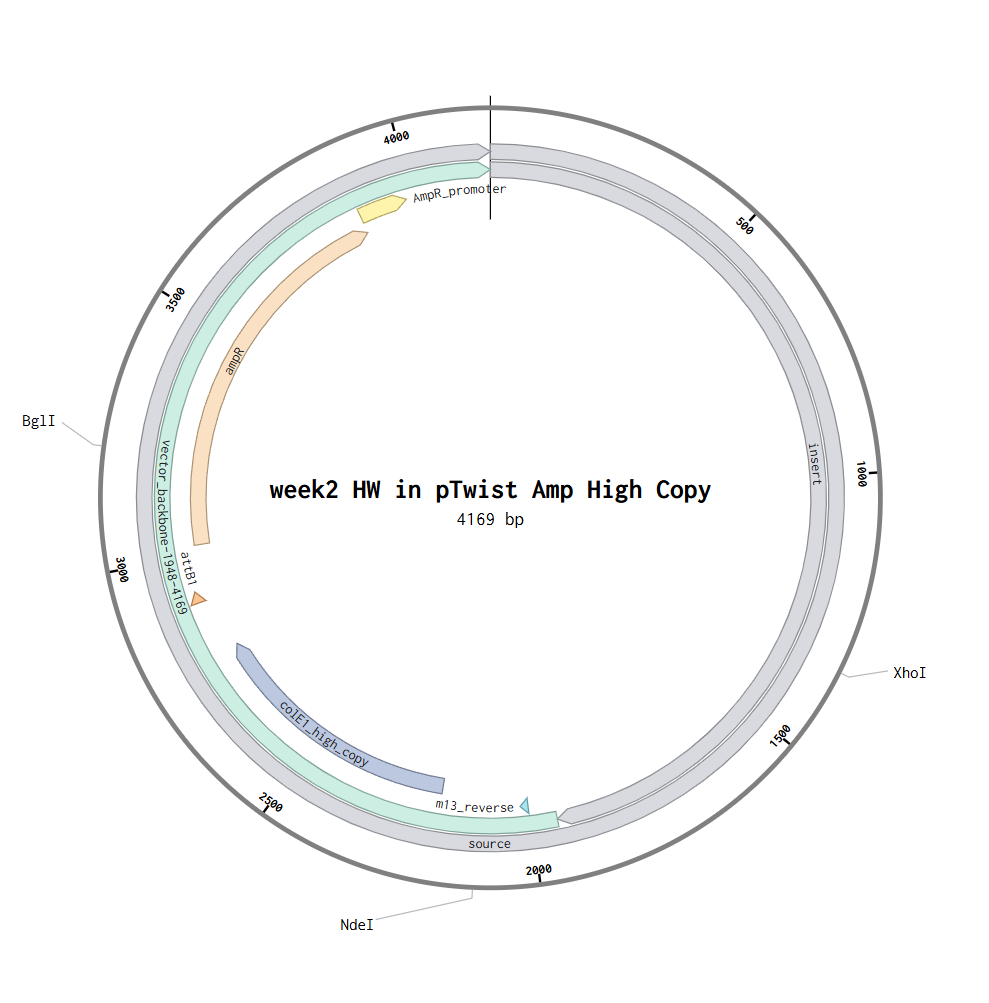
Part 4: Prepare a Twist DNA Synthesis Order
Part 5: DNA Read/Write/Edit
5.1 DNA Read (sequence)
(i) What DNA would you want to sequence and why?
I would sequence some of coral’s DNA like the SAARP1 (skeletal aspartic acid-rich protein 1) gene from Acropora millepora, because I want to explore coral’s potential as architectural material or learn about their calcification performance and stress response under warming and acidification to ensure green future shoreline development.
(ii) What sequencing technology would you use and why?
I would use Sanger sequencing to read the SAARP1 gene because it is accurate, cost-effective, and ideal for sequencing individual gene-length fragments amplified by PCR. PCR and synthetic oligos allow me to isolate the specific gene region, and Sanger sequencing provides precise base-level confirmation of the amplified or edited DNA. Sanger sequencing is considered first-generation sequencing, because it reads one DNA fragment at a time with high accuracy. The input is genomic DNA extracted from coral tissue. The essential preparation steps are: design oligo primers → PCR amplify the SAARP1 locus → purify the PCR product → add sequencing primers → load into the Sanger reaction. Sanger sequencing uses chain-terminating nucleotides labeled with fluorescent dyes. When DNA synthesis stops at each base position, the fragments are separated by size and read by a detector that converts fluorescence into a DNA sequence. The output is a chromatogram and a high-accuracy DNA sequence of the amplified SAARP1 fragment, typically in FASTA format.
5.2 DNA Write (synthesize)
(i) What DNA would you want to synthesize and why?
I would synthesize a codon-optimized SAARP1 coding sequence so I can express it in a chosen host (or produce peptides/biomineralization assays) and test how SAARP1 chemistry affects mineral deposition and coral-like calcification cues.
(ii) What synthesis technology would you use and why?
I would use phosphoramidite oligo synthesis plus assembly (the standard commercial approach) because it is fast, scalable, and ideal for making gene-length constructs via assembly from shorter oligos.
Design sequence in silico → synthesize short oligos → assemble into the full gene (PCR/Gibson-like assembly) → clone into plasmid → sequence-verify.
Long sequences accumulate errors and often need verification and rebuilding. Repetitive/low-complexity regions and extreme GC can reduce synthesis success. Scaling to many variants is feasible, but costs and validation time increase quickly.
5.3 DNA Edit (edit)
(i) What DNA would you want to edit and why?
I would edit the SAARP1 regulatory region (or specific coding residues) in coral to test whether higher or better-timed SAARP1 expression improves calcification under heat/acidification stress.
(ii) What editing technology would you use and why?
I would use CRISPR-based editing, ideally prime editing (or a high-fidelity Cas9 + HDR if prime editing isn’t feasible), because it can make targeted changes without needing large insertions.
1) How does it edit DNA, and what are the essential steps?
CRISPR targets a specific locus with a guide RNA and edits it using an editor enzyme and a repair template or edit-encoding RNA.
Key steps are guide design → delivery into cells/embryos → editing reaction → screening/validation.
2) What prep and inputs are needed?
Inputs include a target sequence, guide RNA, editor protein (Cas9/prime editor), and (if needed) a repair template plus the recipient cells/embryos and delivery method (microinjection/electroporation/viral vectors depending on system).
3) Limitations (efficiency/precision)?
Delivery and survival in coral embryos can be difficult and can cause mosaic edits.
Off-target edits and unintended repair outcomes can occur.
Even precise edits may have ecological and ethical constraints if used outside controlled research settings.
Week 3 HW: Lab Automation
Post-Lab Questions
1. Find and describe a published paper that utilizes the Opentrons or an automation tool to achieve novel biological applications.
Dettinger et al. (2022), “Open-source personal pipetting robots with live-cell incubation and microscopy compatibility,” published in Nature Communications.
The authors introduce PHIL (Pipetting Helper Imaging Lid), an open-source, low-cost pipetting robot designed for liquid handling during live-cell experiments and microscopy workflows. PHIL is important because it addresses a real problem in academic labs: many experiments are small-scale, frequently changing, and not well suited to large industrial automation systems, which are often expensive and hard to adapt.
The paper shows that PHIL can automate tasks such as media exchange, stimulation, and immunostaining while remaining compatible with time-lapse microscopy. This makes it possible to run dynamic live-cell experiments with less manual intervention and better reproducibility. Another key strength is accessibility: the system is built from 3D-printable parts and low-cost components, making advanced lab automation more realistic for smaller or resource-limited research labs.
2. Write a description about what you intend to do with automation tools for your final project.
Automate a screening workflow for coral-related biomineralization conditions
I want to use an automation tool (such as Opentrons) to set up and run a small screening experiment using coral proteins. The robot would prepare multiple reaction conditions (different buffers, salts, and controls) in a consistent and repeatable way.
Compare material or condition combinations in a plate-based format
I want to test which conditions may better support coral-relevant mineralization behavior (for example, comparing protein vs control conditions across different solution chemistries). Automation helps because it can precisely mix and dispense many combinations with less manual error.
Use a custom holder / insert setup for non-standard samples
If needed, I may design a simple 3D-printed holder to keep small material samples or test coupons in a fixed position during pipetting. This would make the workflow more reproducible and easier to scale across replicates.
Final Project Ideas
See slide deck
Week 4 HW: Protein Design Part 1
Part A. Conceptual Questions
1. How many molecules of amino acids do you take with a piece of 500 grams of meat? (on average an amino acid is ~100 Daltons)
Assuming 500 g of meat contains about 20–25% protein by weight, that piece has roughly:
100–125 g of protein
100 daltons = 100 g/mol
so 100–125 g of amino acid residues = about 1.0–1.25 moles
1 mole = 6.022 × 10^23 molecules
So the total is about:
6.0 × 1023 to 7.5 × 1023 amino acid molecules
2. Why do humans eat beef but do not become a cow, eat fish but do not become fish?
Because human body digests and obsorbs. It breaks food down into raw materials and then rebuilds those materials into human body. After absorption, cells use those pieces according to human DNA, human enzymes, and human metabolism. So even if the amino acids came from cow muscle, human body reassembles them into muscle, enzymes, skin, and so on.
Proteins are digested into amino acids
Fats are digested into fatty acids and glycerol
Carbohydrates are digested into simple sugars
3. Why are there only 20 natural amino acids?
There are 20 canonical amino acids in the standard genetic code because evolution seems to have settled on a set that is chemically versatile enough to build functional proteins while still being simple enough for reliable biosynthesis and translation.
5. Where did amino acids come from before enzymes that make them, and before life started?
The main idea is that early Earth and space already had simple carbon-containing molecules like water, carbon dioxide, methane, ammonia, hydrogen cyanide, and related compounds. With energy sources such as lightning, UV radiation, heat, impacts, and mineral surfaces, those molecules could react abiotically and make amino acids. That is the basic lesson of the classic Miller-type experiments and newer prebiotic chemistry work.
6. If you make an α-helix using D-amino acids, what handedness (right or left) would you expect?
A helix made from D-amino acids would generally be left-handed.
7. Can you discover additional helices in proteins?
Yes, but unlikely.
8. Why are most molecular helices right-handed?
In biology, most proteins are built from L-amino acids. That stereochemistry makes a right-handed α-helix lower in energy than a left-handed one, mainly - -because it gives:
better backbone bond geometry
better hydrogen-bond alignment
fewer steric clashes between backbone and side chains
So for ordinary proteins, right-handed helices are favored because they fit the geometry of L-amino acids better.
9. Why do β-sheets tend to aggregate?
What is the driving force for β-sheet aggregation?
Beta-sheet aggregation happens because exposed beta-strands can strongly bond to each other through backbone hydrogen bonds, while also hiding hydrophobic parts from water, making the growing clump very stable.
10. Why do many amyloid diseases form β-sheets?
Can you use amyloid β-sheets as materials?
Amyloid diseases often involve β-sheets because β-sheet-like cross-β fibrils are a generic low-energy aggregation state available to many proteins, not just one special sequence.
Amyloid β-sheets can be used as materials. Researchers use amyloid or amyloid-like fibrils as biomaterials, hydrogels, coatings, scaffolds, filtration media, and nanostructured building blocks because they are stiff, high-aspect-ratio, self-assembling, chemically robust, and sequence-programmable.
Part B: Protein Analysis and Visualization
1. Briefly describe the protein you selected and why you selected it.
Skeletal aspartic acid-rich protein 1
Acropora millepora (Staghorn coral)
2. Identify the amino acid sequence of your protein.
How long is it? What is the most frequent amino acid? You can use this Colab notebook to count the frequency of amino acids.
The length of the protein is: 392 aminoacids.
The most common amino acid is: D, which appears 74 times.
How many protein sequence homologs are there for your protein? Hint: Use Uniprot’s BLAST tool to search for homologs.
250
Does your protein belong to any protein family?
SAARP1 belongs to the coral acid-rich skeletal organic matrix protein group (SAARP/CARP-related SOMPs), rather than to a large universal protein family with a standard conserved domain architecture.
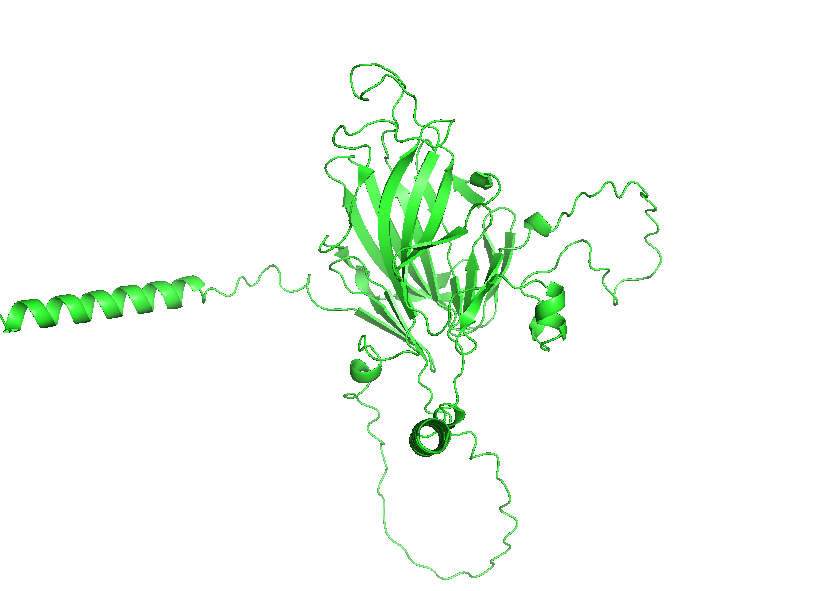
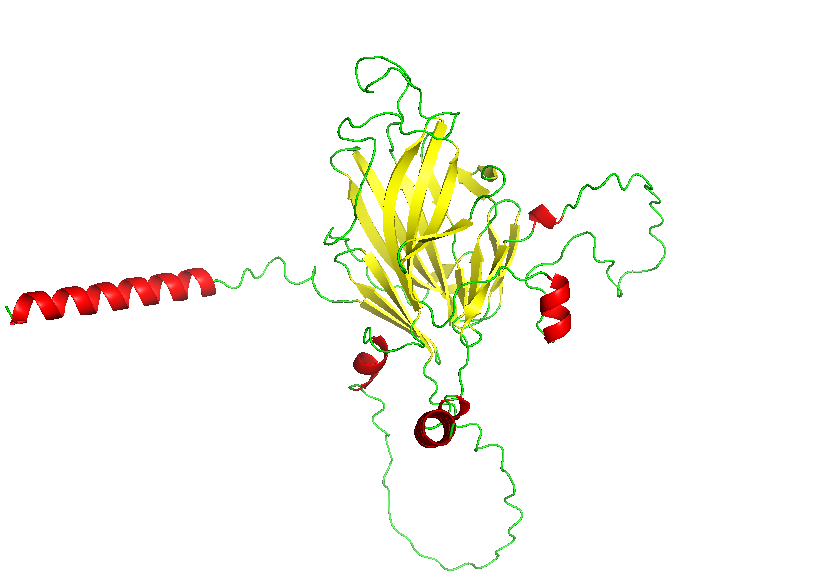
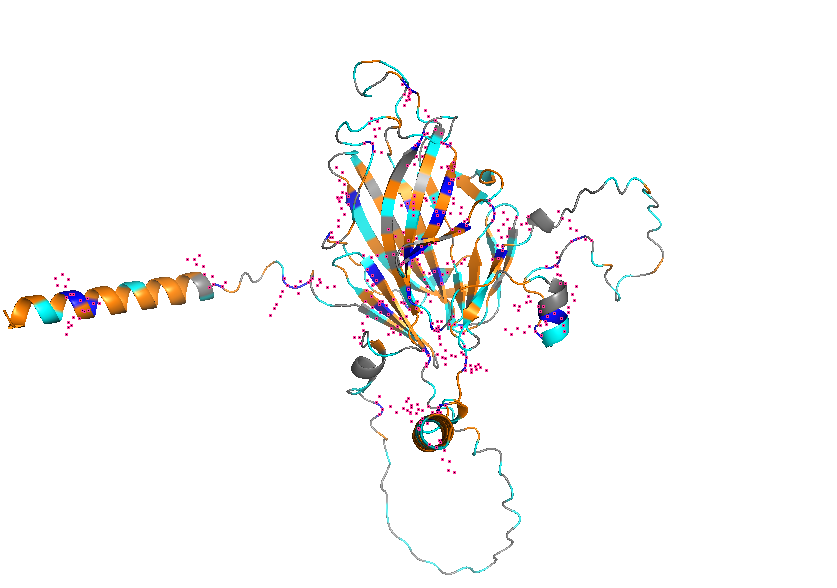
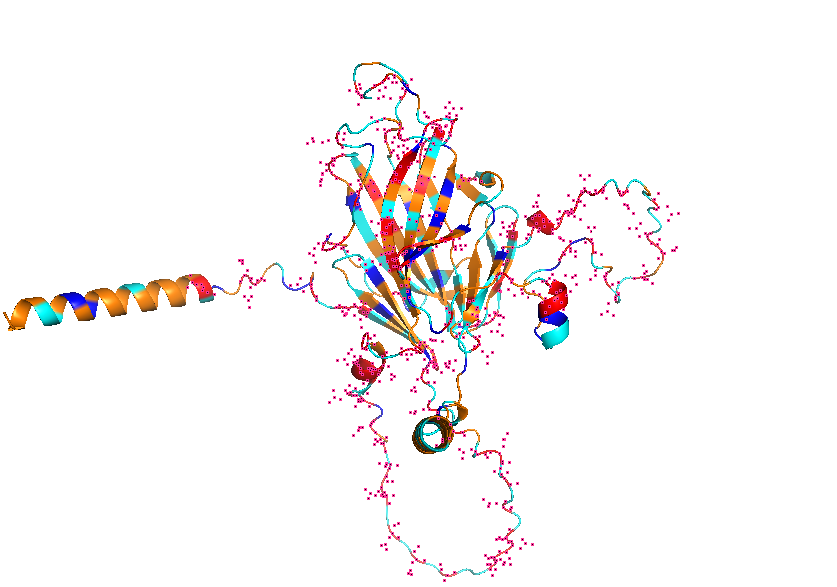
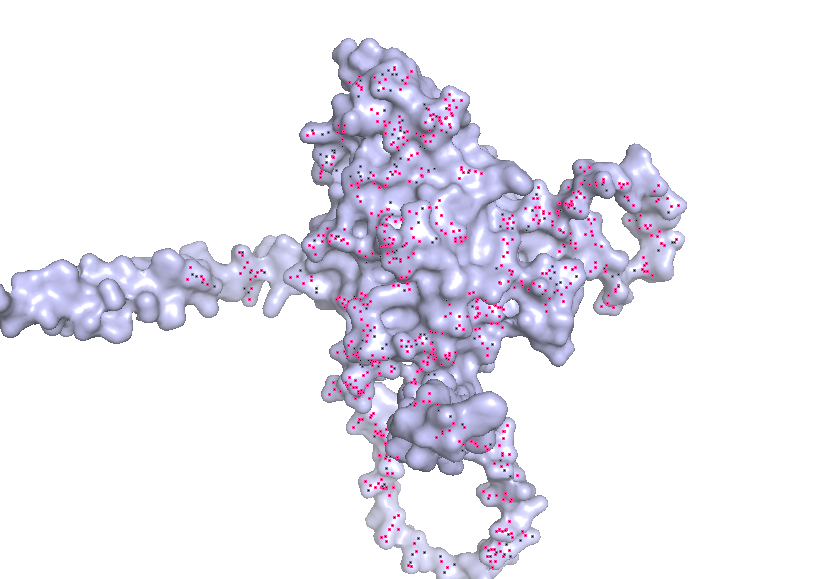
3. Identify the structure page of your protein in RCSB
AF_AFB3EWY6F1 is the RCSB AlphaFold structure page for skeletal aspartic acid-rich protein 1 from Acropora millepora. This is not an experimentally solved structure but a computed AlphaFold model, so it has no experimental resolution. Its overall confidence is moderate (global pLDDT about 77.99), with a more reliable structured core and several lower-confidence, likely disordered regions. The model contains only a single protein chain (A) and no other molecules. No clear standard structural classification family is listed on the page; the protein is more appropriately described as a coral skeletal acid-rich biomineralization protein.
4. Open the structure of your protein in any 3D molecule visualization software:
Visualize the protein as “cartoon”, “ribbon” and “ball and stick”.
Color the protein by secondary structure. Does it have more helices or sheets?
Color the protein by residue type. What can you tell about the distribution of hydrophobic vs hydrophilic residues?
Visualize the surface of the protein. Does it have any “holes” (aka binding pockets)?
Part C. Using ML-Based Protein Design Tools
1BG2 | pdb_00001bg2, HUMAN UBIQUITOUS KINESIN MOTOR DOMAIN
C1. Protein Language Modeling
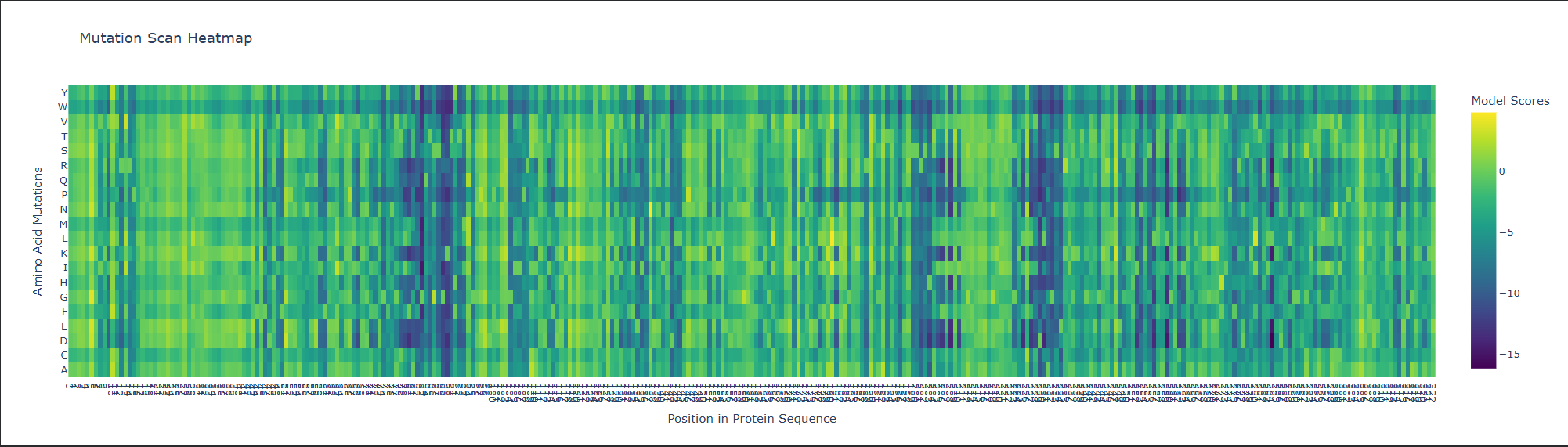
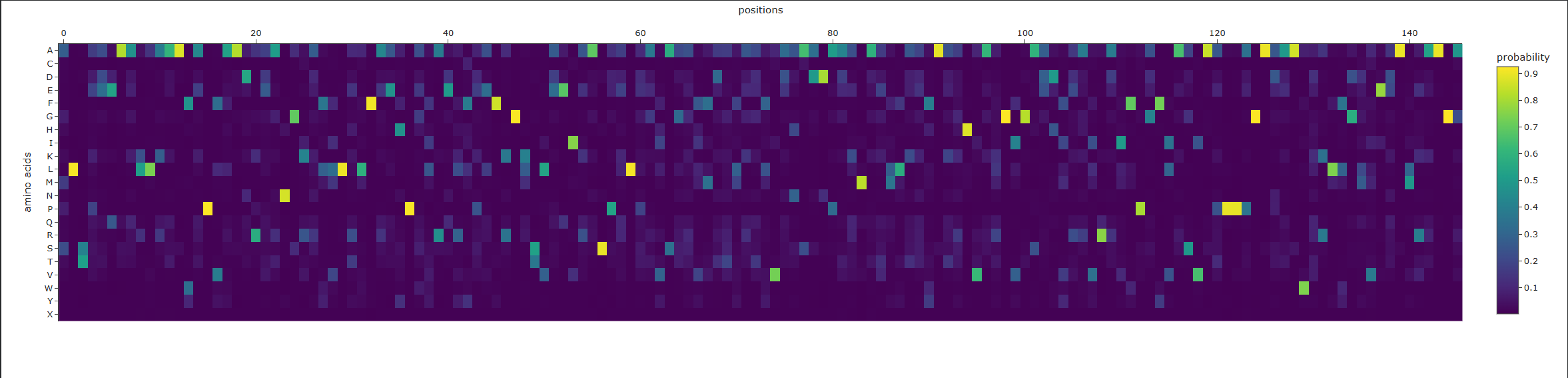
1. Deep Mutational Scans
a. Use ESM2 to generate an unsupervised deep mutational scan of your protein based on language model likelihoods.
b. Can you explain any particular pattern? (choose a residue and a mutation that stands out)
This is visible as dark vertical columns, where nearly all substitutions are predicted to be unfavorable. One standout region is around residue 240–250, where many mutations score very poorly. This suggests that the position is structurally or functionally important, likely contributing to the stability of the kinesin motor-domain fold or to a conserved functional region. In particular, mutations such as proline are strongly disfavored, which is consistent with disruption of local backbone geometry or secondary structure.
C2. Protein Folding
1. Folding a protein
a. Fold your protein with ESMFold. Do the predicted coordinates match your original structure?
Yes, it matches.
predicted
from pdb
b. Try changing the sequence, first try some mutations, then large segments. Is your protein structure resilient to mutations?
The protein is only partly resilient to mutations. It can tolerate some single conservative mutations, but it is not very resilient to larger sequence changes or to mutations at highly constrained positions.
C3. Protein Generation
Inverse-Folding a protein: Let’s now use the backbone of your chosen PDB to propose sequence candidates via ProteinMPNN
a. Analyze the predicted sequence probabilities and compare the predicted sequence vs the original one.
b. Input this sequence into ESMFold and compare the predicted structure to your original.
Part D. Group Brainstorm on Bacteriophage Engineering
Group member: Sean Murphy, Alayah Hines, Terry Luo
Drafted by Sean
Background
The MS2 L protein is a 75-amino-acid polypeptide that lyses E. coli by an incompletely understood mechanism. Its C-terminal transmembrane (TM) domain inserts into the cytoplasmic membrane and oligomerizes, causing depolarization that triggers host autolytic enzymes to degrade the murein layer. Recessive, conservative missense mutations clustered around a conserved LS dipeptide strongly implies L engages an unidentified host protein target rather than simply disrupting the bilayer. The dispensable N-terminal domain binds chaperone DnaJ (with solved PDB structures), modulating lysis timing. Its removal causes lysis ~20 min earlier. No experimental structure of L exists.
Goals
(1) Stabilize L for more robust membrane accumulation. (2) Accelerate lysis by bypassing DnaJ-dependent regulatory timing and improving delivery of functional L to the membrane. Because the downstream lytic target is unknown, we do not attempt to enhance per-molecule toxicity at the point of target engagement; we focus on removing regulatory brakes and increasing the supply of functional protein.
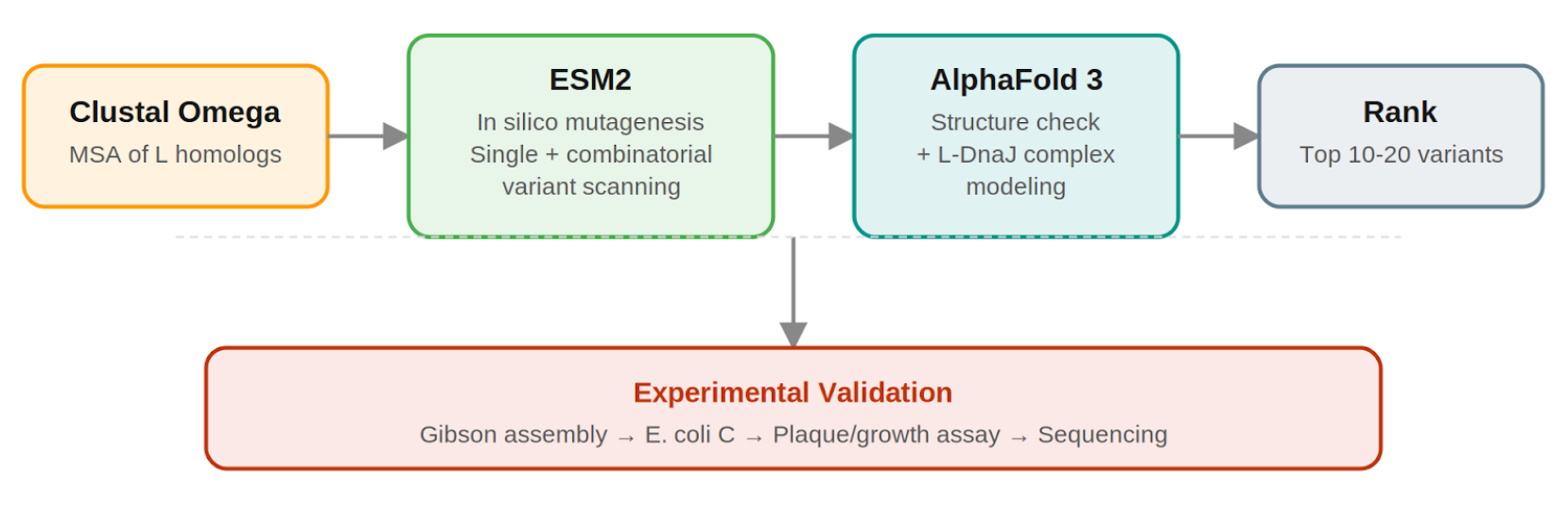
Pipeline: Three Tools, Each Non-Redundant
Clustal Omega (Conservation Map). Align L homologs across Leviviridae (MS2, f2, R17, GA, PP7, AP205, PRR1, M12, KU1, JP34). Conserved C-terminal residues, especially the LS motif, are presumed to mediate the unknown heterotypic interaction and are excluded from mutation. This map constrains all downstream design.
ESM2 + Deep Combinatorial Scanning (Fitness Oracle). Score every single-point mutation by log-likelihood change: increases at mutable positions indicate stabilizing substitutions (Goal 1). N-terminal scanning identifies mutations that disrupt DnaJ binding (Goal 2). A strict preservation rule applies near the LS motif: mutations are evaluated for maintenance of wild-type fitness, not improvement. The genetics show even conservative changes there cause recessive loss of function. Pairwise combinatorial scanning (about ~2M pairs) captures epistatic synergies at mutable positions. This could be potentially pushed further with enough compute.
AlphaFold 3 (Structural Filter + Complex Model). Predicts variant structures as a sanity check (does the TM helix survive?) and models the L–DnaJ complex to verify that N-terminal truncations/mutations disrupt the regulatory interface. Used as a filter, not a design engine. PAE matrix identifies confident interface contacts.
Ranking.
Composite score: ESM2 log-likelihood gain (stability) + conservation preservation (all essential residues intact) + AF3-predicted DnaJ-binding disruption (for timing bypass). Top 10–20 variants advance to experimental validation.
Pipeline Schematic
Why Not More Tools?
ProteinMPNN is excluded because it is trained on crystallized globular PDB proteins, not predicted structures of disordered membrane peptides. Compute is invested in combinatorial ESM2 depth.
Pitfalls
No experimental structure: All structural reasoning rests on AF3 predictions for a challenging target; mitigated by treating AF3 as a filter and cross-referencing against the conservation map.
Unknown lytic target: The central limitation. We cannot optimize target-binding affinity for an unidentified partner; engineering is restricted to upstream properties (stability, membrane delivery, DnaJ bypass).
Autolysin bottleneck: If lysis rate is limited by host autolytic enzyme activity rather than L accumulation, stabilization gains may show diminishing returns; the plaque assay will reveal this.
“PepMLM outputs X; substituted X→A for AlphaFold input.”
Binder
ipTM Score
WRYYVAAVALWA
0.34
WHYYAVAAEWKA
0.23
WLVPAAAAAHGK
0.48
WRYGPVAVRHWK
0.36
FLYRWLPSRRGG
0.38
Across the five AlphaFold3 complex predictions, ipTM values ranged from 0.23 to 0.48. The known binder FLYRWLPSRRGG gave ipTM = 0.38 and appeared weakly defined, remaining largely surface-adjacent/partially detached rather than buried in a clear pocket, with no obvious localization near the N-terminus where A4V sits. Three PepMLM peptides (WHYYAVAAEWKA, 0.23; WRYYVAAVALWA, 0.34; WRYGPVAVRHWK, 0.36) similarly showed low-confidence interfaces, tending to lie loosely on the β-barrel exterior instead of concentrating at the A4V region. In contrast, WLVPAAAAAHGK produced the strongest prediction (ipTM = 0.48) and appeared more plausibly docked along a β-barrel/loop-adjacent surface, making it the only PepMLM-generated peptide that exceeded the known binder’s ipTM in this set.
Part 3: Evaluate Properties of Generated Peptides in the PeptiVerse
Peptide
Predicted binding affinity
Solubility
Hemolysis probability
Net charge (pH 7)
Molecular weight
WRYYVAAVALWA
Medium binding(8.472)
Soluble(0.999)
Non-hemolytic(0.178)
0.76
1468.7
WHYYAVAAEWKA
Weak binding(5.407)
Soluble(1.000)
Non-hemolytic(0.042)
-0.15
1494.6
WLVPAAAAAHGK
Weak binding(5.328)
Soluble(1.000)
Non-hemolytic(0.019)
0.85
1191.4
WRYGPVAVRHWK
Weak binding(5.615)
Soluble(1.000)
Non-hemolytic(0.018)
2.85
1554.8
FLYRWLPSRRGG
Weak binding(5.968)
Soluble(1.000)
Non-hemolytic(0.047)
2.76
1507.7
Comparing PeptiVerse to the AlphaFold3 complexes, the two signals only partially agree. Structurally, WLVPAAAAAHGK had the highest interface confidence (ipTM = 0.48), but PeptiVerse predicts only weak binding (5.328)—so higher ipTM did not map cleanly to higher predicted affinity in this set. Instead, the strongest PeptiVerse binder is WRYYVAAVALWA with medium binding (8.472) even though its AF3 interface confidence was lower (ipTM = 0.34) and its pose looked more loosely surface-associated. Encouragingly, all peptides are predicted soluble (~1.0) and non-hemolytic (hemolysis probabilities 0.018–0.178), so there’s no obvious developability red flag from solubility or hemolysis. Balancing predicted binding and therapeutic properties, WRYYVAAVALWA stands out as the best overall: it has the strongest predicted affinity, remains soluble, and is non-hemolytic with a moderate net charge (+0.76).
Peptide to advance: WRYYVAAVALWA — it offers the best binding/developability tradeoff (highest predicted affinity while still soluble and non-hemolytic), and its AF3 pose is at least compatible with a surface-binding interaction even if the interface confidence is modest.
Part 4: Generate Optimized Peptides with moPPIt
Peptide
Hemolysis
Non-Fouling
Solubility
Affinity
Motif
Specifity
RFEEEERRRRRA
0.9574970
0.9754648
0.8333333
5.5774255
0.0013071
0.9743590
GKCTLNNSQCQV
0.8824576
0.6816904
0.8333333
6.0879078
0.8724478
0.6346154
PSPKKKRRKRCL
0.9656924
0.9467938
0.7500000
6.5786953
0.0183732
0.9743590
DEKDDDHTCHEK
0.9114821
0.8636953
1.0000000
5.7854242
0.7418348
0.7628205
moPPIt produced more “designed” peptides than PepMLM: instead of mostly natural-looking, Ala-rich 12-mers, it generated strongly biased sequences (very cationic like PSPKKKRRKRCL/RFEEEERRRRRA or more acidic like DEKDDDHTCHEK) because it was optimizing multiple objectives at once. With hemolysis, non-fouling, solubility, affinity, motif, and specificity all enabled (weights = 1; motif positions 1–4), the peptides generally looked developable: non-fouling scores were high (0.68–0.98) and solubility was good (0.75–1.00). Predicted affinity ranked highest for PSPKKKRRKRCL (6.58), then GKCTLNNSQCQV (6.09), DEKDDDHTCHEK (5.79), and RFEEEERRRRRA (5.58), while motif matching varied a lot (very low for RFEEEERRRRRA, higher for GKCTLNNSQCQV and DEKDDDHTCHEK). Before any real advancement, I would next run AlphaFold3 to confirm the peptides actually dock to SOD1 (ipTM + binding site), then validate with basic binding experiments (SPR/BLI/MST), plus solubility, hemolysis/cytotoxicity, and stability tests, since model scores don’t guarantee true binding in biology.
Part C: Final Project: L-Protein Mutants
Soluble (1–40):
P13L — LLR +0.100
S15A — LLR +0.036
R30L — LLR −0.130
Transmembrane (41–75):
A45P — LLR +0.038
I46F — LLR −0.096
Every mutation is supported by lab evidence (lysis happens and the protein is detectable).
LLR scores are mostly near-neutral to mildly positive; even the slightly negative ones are not extremely deleterious by the model, which is a reasonable sanity check.
I prioritized variants that were lysis-positive in the experimental mutant dataset, and further preferred those with detectable protein levels, since the project goal includes improving stability/expression in addition to lysis. I then cross-referenced each candidate with the ESM mutational LLR scan as a plausibility check, avoiding strongly disfavored substitutions; however, when directly compared across the set of tested mutants, LLR showed no meaningful correlation with lysis outcomes (AUC ≈ 0.48), so the experimental phenotype was treated as the primary selection signal. To satisfy the assignment constraints, I selected three soluble-domain mutants and two transmembrane mutants.
Week 6 HW: Genetic Circuits Part 1
Part A: DNA Assembly
What are some components in the Phusion High-Fidelity PCR Master Mix and what is their purpose?
Phusion High-Fidelity PCR Master Mix is a 2X mix containing Phusion DNA Polymerase, nucleotides, and an optimized reaction buffer including MgCl₂. Functionally, the polymerase synthesizes the new DNA strand, the dNTPs are the nucleotide building blocks it incorporates, the buffer maintains the reaction chemistry, and Mg²⁺ is the essential cofactor that enables polymerase activity and helps stabilize primer-template interactions. Phusion is called “high fidelity” because it has 3′→5′ exonuclease proofreading activity, which lowers the error rate compared with standard Taq-type PCR enzymes.
What are some factors that determine primer annealing temperature during PCR?
Primer annealing temperature is determined primarily by the melting temperature (Tm) of the primers. A common guideline is to set the annealing temperature about 2–5°C below the lower Tm of the primer pair so that the primers bind efficiently to the intended target without binding too readily to partially matching sites. The Tm itself depends on several factors, especially primer length, GC content, and sequence composition, because GC-rich primers generally bind more strongly than AT-rich primers. Annealing temperature is also influenced by mismatches, which reduce binding stability, and by secondary structures such as hairpins, self-dimers, and heterodimers, which can interfere with normal primer-target annealing. Salt and buffer conditions further affect duplex stability, so annealing temperature is ultimately a function of both primer design and reaction chemistry.
There are two methods from this class that create linear fragments of DNA: PCR, and restriction enzyme digests. Compare and contrast these two methods, both in terms of protocol as well as when one may be preferable to use over the other.
PCR and restriction enzyme digestion can both produce linear DNA fragments, but they do so through very different mechanisms and are useful in different situations. PCR is an amplification-based method in which a DNA region is copied through repeated cycles of denaturation, primer annealing, and extension using a thermostable polymerase. This makes PCR highly flexible, because the user can selectively amplify a desired region even from a small amount of starting material and can also introduce new sequence features, such as mutations, overlap regions, or added restriction sites, through primer design. By contrast, a restriction enzyme digest does not amplify DNA at all; instead, it cuts existing DNA at specific recognition sequences to release a fragment with defined ends, which may be sticky or blunt depending on the enzyme used. Restriction digestion is usually preferable when the desired fragment is already available in a plasmid or other DNA source and is conveniently flanked by suitable enzyme sites, whereas PCR is preferable when no convenient restriction sites exist or when the fragment must be modified as it is generated. In that sense, PCR is more design-flexible, while restriction digestion is more site-dependent and typically simpler when the necessary cut sites are already present.
How can you ensure that the DNA sequences that you have digested and PCR-ed will be appropriate for Gibson cloning?
The DNA fragments are appropriate for Gibson cloning when they are designed with the correct overlapping sequences and verified before assembly. The primers should add 20–22 bp overhangs, and neighboring fragments used in Gibson assembly should share about 20–40 bp of sequence identity so they can join correctly. In practice, this means ensuring that the backbone and color fragments have the proper matching overlaps, the correct 5′→3′ orientation, and the expected sequence content before they are combined. After PCR, the products are treated with DpnI to remove the parental plasmid template, then purified, quantified, and checked on a diagnostic gel against the expected fragment sizes in Benchling.
How does the plasmid DNA enter the E. coli cells during transformation?
Plasmid DNA enters E. coli during transformation by first making the bacterial cells competent, meaning temporarily able to take up external DNA. In chemical transformation, cells are typically treated with salts such as calcium chloride and then subjected to a brief heat shock, which increases membrane permeability and helps the plasmid enter the cell. Heat shock causes the cell membrane to “open up,” allowing the plasmid to enter the cells by diffusion. After recovery and plating, only the bacteria that successfully received the plasmid survive on the antibiotic plate and later express the chromophore color. In electroporation, an electrical pulse creates transient pores in the membrane through which DNA can pass. After either method, the cells are allowed to recover in rich medium so they can repair their membranes and begin expressing the plasmid’s selectable marker, such as an antibiotic-resistance gene, before they are plated on selective agar. Thus, transformation works by temporarily disrupting the membrane barrier long enough for plasmid DNA to cross into the bacterial cell interior.
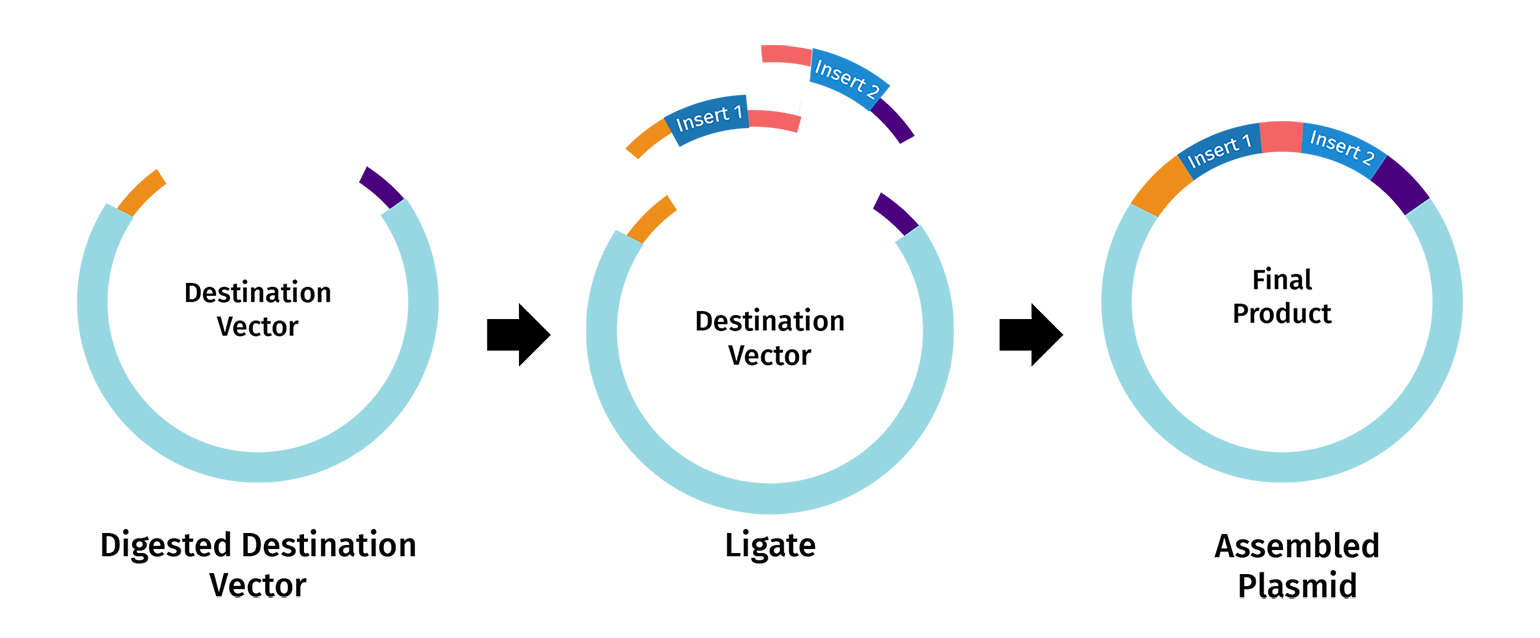
Describe another assembly method in detail (such as Golden Gate Assembly)
One alternative to Gibson Assembly is Golden Gate Assembly, a one-pot cloning method that uses a Type IIS restriction enzyme such as BsaI or BsmBI together with T4 DNA ligase. Unlike standard restriction enzymes, Type IIS enzymes cut outside of their recognition sequences, which allows the user to design custom overhangs that determine exactly how the DNA fragments will join together. During the reaction, the enzyme cuts the fragments to expose these programmed overhangs, and ligase then seals the matching ends together in the intended order. Because the recognition sites are usually removed during assembly, the final product is often considered scarless or seamless. Golden Gate is especially useful for modular and multi-fragment cloning because many parts can be assembled in a single reaction, but it requires careful design to ensure that the DNA parts do not contain unwanted internal sites for the chosen Type IIS enzyme.
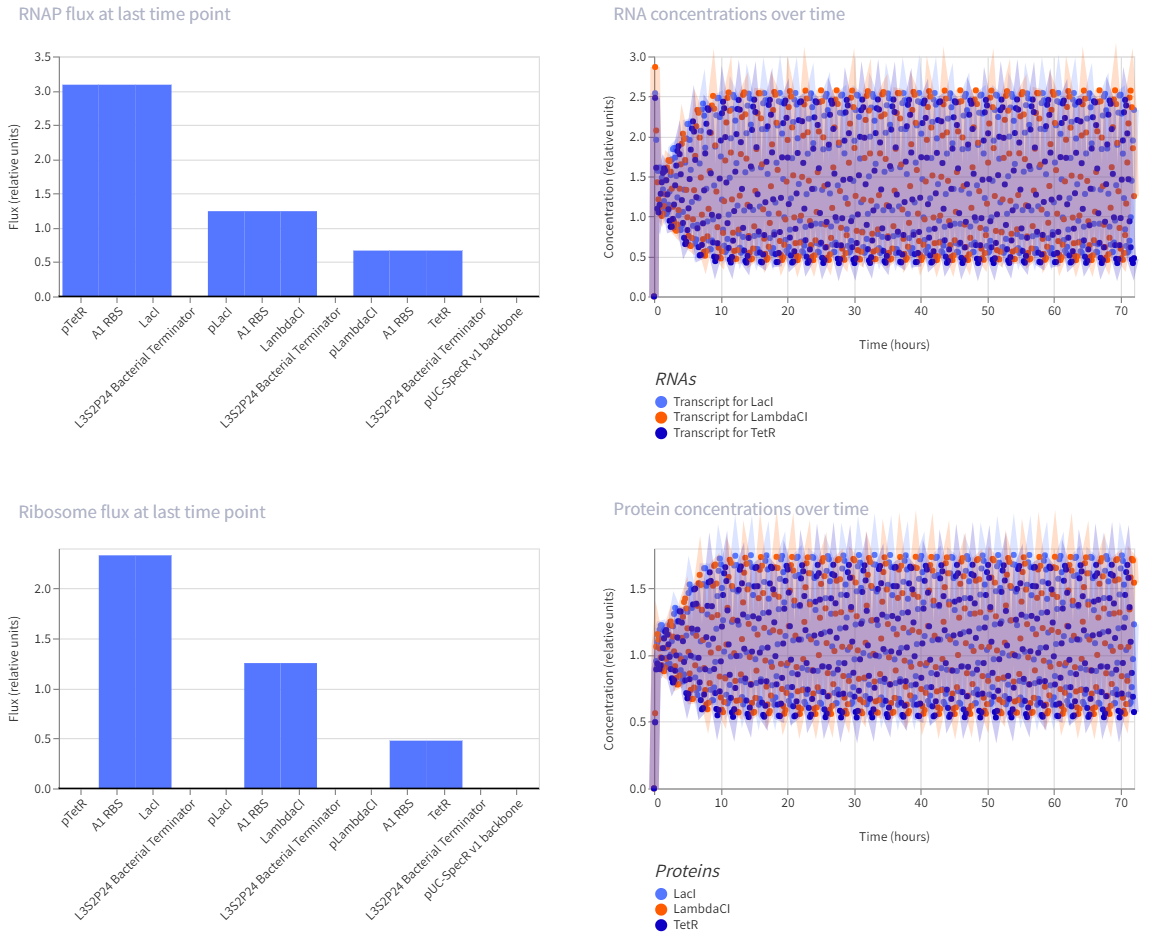
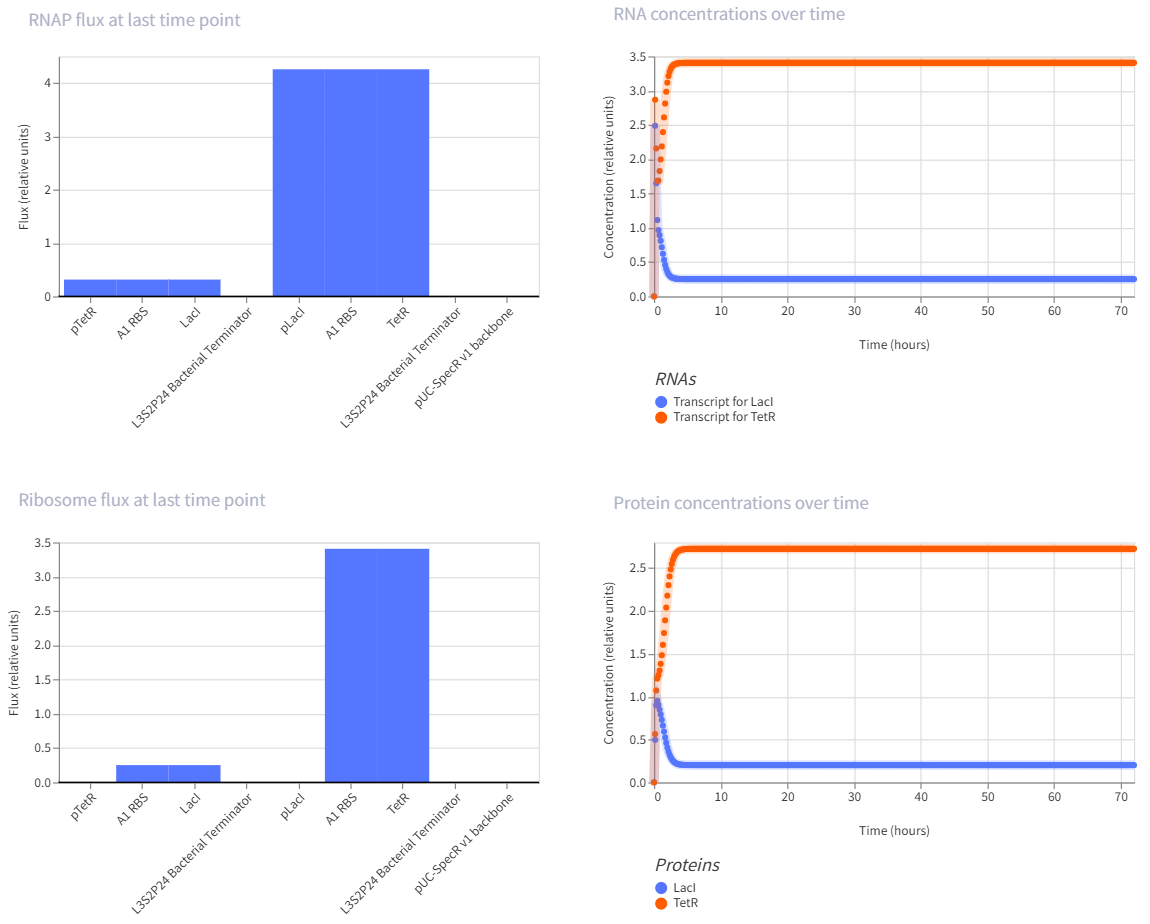
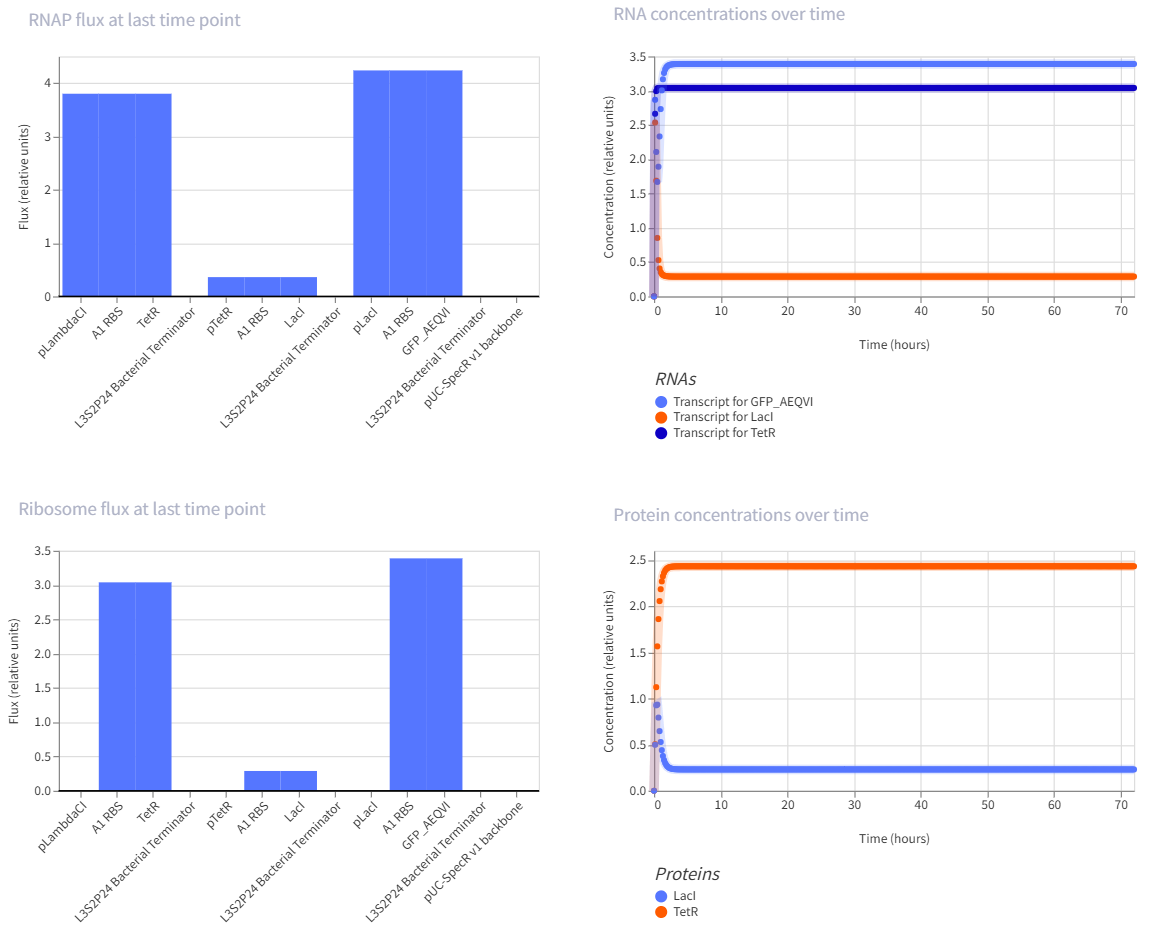
The repressilator is a synthetic genetic oscillator made from three repressors arranged in a circular negative-feedback loop. In the construct, pTetR drives LacI, pLacI drives LambdaCI, and pLambdaCI drives TetR. Each protein represses the promoter of the next gene: LacI represses pLacI, LambdaCI represses pLambdaCI, and TetR represses pTetR. In simple terms, this means each gene takes a turn shutting off the next one, causing expression to rotate through the circuit instead of remaining constant. This creates oscillations in both RNA and protein levels over time, which is visible in the simulation graphs. The RNA and protein plots show repeating up-and-down patterns, indicating that the circuit is functioning as an oscillator rather than a simple on/off switch.
This is a self-repressing gene. It is one of the classic simple synthetic circuits, and negative autoregulation in E. coli is known to speed response time compared with a non-self-regulated unit.
The toggle switch is one of the landmark synthetic gene circuits in E. coli. It uses two repressors that inhibit each other, which can create two stable states rather than one oscillating pattern.
This is a multi-step repression chain, which is easier than the repressilator but still interesting because it adds delay and layered regulation. Reviews of synthetic circuits in E. coli discuss repression cascades built by inserting modules like LacI and cI between an input layer and a reporter, and note that longer cascades tend to increase response time and noise.
Week 7 HW: Genetic Circuits Part 2
Assignment Part 1: Intracellular Artificial Neural Networks
What advantages do IANNs have over traditional genetic circuits, whose input/output behaviors are Boolean functions?
IANNs have an advantage over traditional Boolean genetic circuits because they can respond to graded, noisy biological signals instead of forcing every input into a simple ON/OFF state. This makes them better for tasks like classification, where a cell needs to weigh several signals together and make a more flexible decision, while Boolean circuits are better for simpler yes/no logic.
Describe a useful application for an IANN; include a detailed description of input/output behavior, as well as any limitations an IANN might face to achieve your goal.
One useful application would be an engineered therapeutic cell that detects a tumor-like signal pattern by integrating several biomarkers, such as hypoxia, inflammation, and growth signaling, and then turns on a treatment response only when the overall pattern matches cancer. A limitation is that real cells are noisy and variable, so it can be hard to tune the network precisely and avoid false positives, crosstalk, or unreliable behavior in different cells.
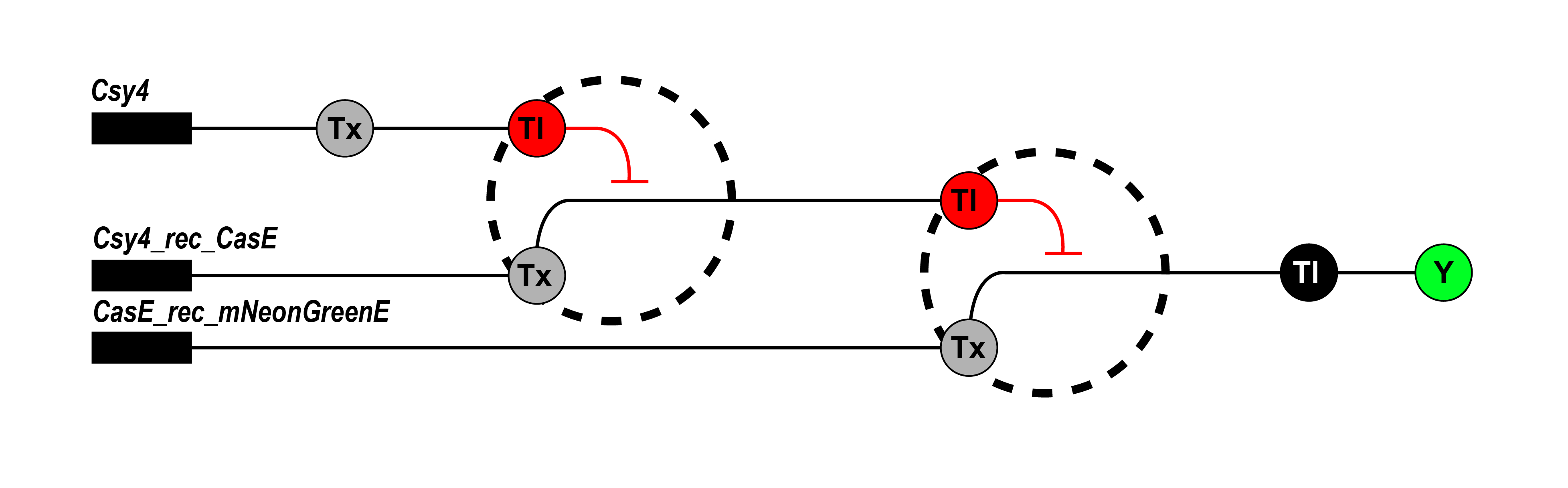
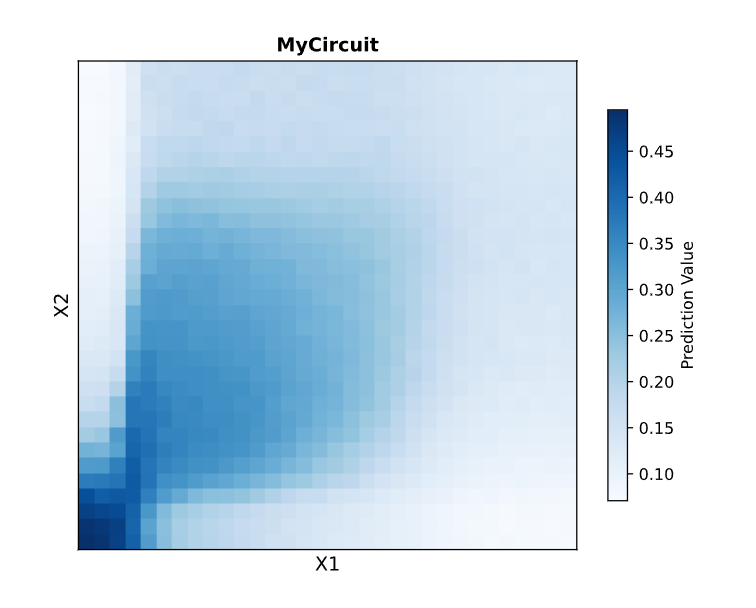
Draw a diagram for an intracellular multilayer perceptron where layer 1 outputs an endoribonuclease that regulates a fluorescent protein output in layer 2.
Assignment Part 2: Fungal Materials
What are some examples of existing fungal materials and what are they used for? What are their advantages and disadvantages over traditional counterparts?
Existing fungal materials include mycelium-based packaging, insulation/building panels, leather-like materials, and fungal chitin/chitosan for wound dressings. Their main advantages are that they can be biodegradable, grown on agricultural waste, and can offer good thermal insulation, acoustic absorption, and fire resistance; their disadvantages are that they often have lower mechanical strength, high water absorption, and less standardized performance than plastics, foams, or conventional leather.
What might you want to genetically engineer fungi to do and why? What are the advantages of doing synthetic biology in fungi as opposed to bacteria?
If I could genetically engineer fungi, I would want them to grow faster and make materials that are stronger, more water-resistant, or more functional for uses like packaging, insulation, or leather alternatives. Fungi have some advantages over bacteria for synthetic biology because they naturally form mycelial networks, can perform eukaryotic protein processing, and are often very good at secreting proteins, which makes them more suitable for material production and some complex biomolecules than bacteria.
Assignment Part 3: First DNA Twist Order
Week 9 HW: Cell Free Systems
General homework questions
Explain the main advantages of cell-free protein synthesis over traditional in vivo methods, specifically in terms of flexibility and control over experimental variables. Name at least two cases where cell-free expression is more beneficial than cell production.
Cell-free protein synthesis is more flexible than in vivo expression because you can directly control the reaction conditions without keeping cells alive. It is especially useful for making toxic proteins and proteins that are hard to express in living cells, like some membrane proteins.
Describe the main components of a cell-free expression system and explain the role of each component.
A cell-free system includes the extract or machinery for transcription and translation, the DNA template, amino acids, nucleotides, salts, cofactors, and an energy source. Together, these parts let the system read the gene and build the protein outside of a living cell.
Why is energy provision regeneration critical in cell-free systems? Describe a method you could use to ensure continuous ATP supply in your cell-free experiment.
Energy regeneration is important because protein synthesis uses a lot of ATP and GTP, and the reaction will stop if energy runs out. One way to maintain ATP supply is to include an energy regeneration system such as phosphoenolpyruvate or a slower source like maltodextrin.
Compare prokaryotic versus eukaryotic cell-free expression systems. Choose a protein to produce in each system and explain why.
Prokaryotic systems are faster, cheaper, and good for simple proteins, while eukaryotic systems are better for proteins that need more complex folding or modifications. For example, GFP could be made in an E. coli system, while a human receptor protein would be better in a eukaryotic system.
How would you design a cell-free experiment to optimize the expression of a membrane protein? Discuss the challenges and how you would address them in your setup.
To express a membrane protein, I would add liposomes, nanodiscs, or mild detergents so the protein has a membrane-like environment while it is being made. The main problems are aggregation and misfolding, so I would test different additives and reaction conditions to improve stability and function.
Imagine you observe a low yield of your target protein in a cell-free system. Describe three possible reasons for this and suggest a troubleshooting strategy for each.
Low yield could come from poor DNA design, non-optimal reaction conditions, or the protein being unstable or aggregating. I would troubleshoot by redesigning the construct, adjusting salts and temperature, and adding folding helpers or membrane mimics if needed.
Homework question from Kate Adamala
Pick a function and describe it.
a. What would your synthetic cell do? What is the input and what is the output?
My synthetic minimal cell would support coral mineralization by producing urease activity in a controlled, cell-like system. The input would be urea, and the output would be ammonia and inorganic carbon, which can help create conditions that support calcification.
b. Could this function be realized by cell-free Tx/Tl alone, without encapsulation?
Yes, the basic function could be done with cell-free Tx/Tl alone because the system only needs to express urease. But encapsulation would make it more useful as a synthetic cell by localizing the reaction and making it easier to apply as a material.
c. Could this function be realized by genetically modified natural cell?
Yes, a genetically modified microbe could also express urease and help change the local chemical environment. Still, a synthetic minimal cell may be safer and easier to control because it is non-living and cannot grow or spread.
d. Describe the desired outcome of your synthetic cell operation.
The desired outcome is that the synthetic cell produces urease when activated and helps shift the surrounding chemistry toward conditions that support coral calcification. In the long term, this could help probiotic materials support reef building on coastal or architectural surfaces.
Design all components that would need to be part of your synthetic cell.
a. What would the membrane be made of?
The membrane would be made of phospholipids, possibly with cholesterol for extra stability. This would create a vesicle that can hold the cell-free system and function like a minimal synthetic cell.
b. What would you encapsulate inside?
Inside, I would encapsulate the cell-free transcription-translation system, DNA for urease expression, amino acids, nucleotides, salts, cofactors, and an energy source. I would also include any helper components needed for urease folding and activity.
c. Which organism would your Tx/Tl system come from?
I would use a bacterial cell-free system, since urease expression can be handled in a bacterial context and the design does not require mammalian regulation. A bacterial system is simpler and more practical for a first prototype.
d. How will your synthetic cell communicate with the environment?
The system would communicate with the environment by taking in urea from the surrounding water and converting it through urease activity. The products of that reaction would diffuse out and change the local chemical conditions around coral or the material surface.
Experimental details
a. List all lipids and genes.
The main lipids would be phospholipids such as POPC, with cholesterol if needed for stability. The genes would include the urease gene cluster and any accessory genes needed for proper urease assembly and activity.
b. How will you measure the function of your system?
I would measure function by checking whether urease is successfully expressed and whether urea is broken down. I would also measure changes in pH, ammonia production, or other chemical signs that the system is creating conditions favorable for mineralization.
Homework question from Peter Nguyen
Write a one-sentence summary pitch sentence describing your concept.
I propose a freeze-dried probiotic-inspired material that uses cell-free urease expression to create local chemical conditions that support coral mineralization on coastal and architectural surfaces.
How will the idea work, in more detail? Write 3-4 sentences or more.
The material would contain freeze-dried cell-free components programmed to express urease when activated by water and supplied with urea. Once active, the system would break down urea into products that may help create conditions favorable for coral calcification. This would let a surface act not just as passive infrastructure, but as a bioactive partner in reef-building processes.
What societal challenge or market need will this address?
This idea addresses the need for new ways to support coral reef restoration and climate-adaptive coastal design. It also responds to the growing need for architectural materials that do more than resist environmental change and instead actively contribute to ecological recovery.
How do you envision addressing the limitation of cell-free reactions (e.g., activation with water, stability, one-time use)?
Freeze-drying helps the system stay stable until it is needed, and water activation is useful because the material would only turn on in wet coastal conditions. Since cell-free reactions are temporary, the material could be designed as a replaceable or renewable layer rather than a permanent system.
Homework question from Ally Huang
Provide background information that describes the space biology question or challenge you propose to address. Explain why this topic is significant for humanity, relevant for space exploration, and scientifically interesting. (Maximum 100 words)
Water safety is a major challenge in space because contamination can threaten astronaut health. A freeze-dried cell-free biosensor would be useful because it is lightweight, stable, and easy to activate when needed.
Name the molecular or genetic target that you propose to study. Examples of molecular targets include individual genes and proteins, DNA and RNA sequences, or broader -omics approaches. (Maximum 30 words)
My target is a reporter circuit that responds to a contamination-related molecule and produces GFP. This makes hidden contamination easier to detect.
Describe how your molecular or genetic target relates to the space biology question or challenge your proposal addresses. (Maximum 100 words)
The target matters because it converts possible water contamination into a visible fluorescent signal. That would make routine testing easier in a resource-limited space environment.
Clearly state your hypothesis or research goal and explain the reasoning behind it. (Maximum 150 words)
My hypothesis is that a freeze-dried cell-free biosensor can act as a simple first-pass test for water contamination in space. If it gives a reliable fluorescent response, it could become a useful lightweight diagnostic tool.
Outline your experimental plan - identify the sample(s) you will test in your experiment, including any necessary controls, the type of data or measurements that will be collected, etc. (Maximum 100 words)
My hypothesis is that a freeze-dried cell-free biosensor can act as a simple first-pass test for water contamination in space. If it gives a reliable fluorescent response, it could become a useful lightweight diagnostic tool.
Week 10 HW: Imaging and Measurement
Homework: Final Project
For the final project, I will primarily measure whether my designed DNA templates are correct and whether they can produce proteins in a T7 cell-free expression system. I will verify the DNA constructs using DNA sequencing and agarose gel electrophoresis, then test expression using two templates: a minimal ureABC structural urease construct and a separate sfGFP reporter construct. The sfGFP template will serve as a visual positive control, and its expression will be measured by fluorescence, while expression of UreA, UreB, and UreC will be measured using SDS-PAGE. I may also perform an exploratory urease-related activity assay, but this is not the main measurement because the minimal structural construct does not include the full accessory genes required for strong urease activation.
Homework: Waters Part I — Molecular Weight
Based on the predicted amino acid sequence of eGFP (see below) and any known modifications, what is the calculated molecular weight?
28006.60.
Calculate the molecular weight of the eGFP using the adjacent charge state approach described in the recitation.
Z= 933.7349/(933.7349-903.7148)
the charge state of the 933.7349 peak is about 30+, so the 903.7148 peak is 31+. The protein mass is about 27982.88 Da.
The accuracy error is about 0.085%.
Can you observe the charge state for the zoomed-in peak in the mass spectrum for the intact eGFP? If yes, what is it? If no, why not.
No, you cannot confidently determine the charge state from the zoomed-in peak alone. The isotopic peaks are not clearly resolved enough, so the spacing needed to identify z is not visible; the charge state is better assigned from the full charge-state distribution in the main spectrum.
Homework: Waters Part II — Secondary/Tertiary structure
Based on learnings in the lab, please explain the difference between native and denatured protein conformations. For example, what happens when a protein unfolds? How is that determined with a mass spectrometer? What changes do you see in the mass spectrum between the native and denatured protein analyses?
In the native state, eGFP stays folded and compact, so it has fewer exposed protonation sites and usually carries fewer charges, which gives peaks at higher m/z. In the denatured state, the protein unfolds, exposes more basic sites, and picks up more charges, so the spectrum shifts to a broader charge-state distribution at lower m/z.
Zooming into the native mass spectrum of eGFP from the Waters Xevo G3 QTof MS, can you discern the charge state of the peak at ~2800? What is the charge state? How can you tell?
Yes, the charge state of the peak at about 2800 m/z can be estimated from the isotope spacing in the zoomed-in spectrum. The peaks are spaced by about 0.5 m/z, and since isotope spacing is approximately 1/z, that means the charge state is about 2+.
Homework: Waters Part III — Peptide Mapping - primary structure
How many Lysines (K) and Arginines (R) are in eGFP? Please circle or highlight them in the eGFP sequence given in Waters Part I question 1 above.
eGFP contains 20 lysines (K) and 7 arginines (R), for a total of 27 trypsin cleavage residues.
How many peptides will be generated from tryptic digestion of eGFP?
19
Based on the LC-MS data for the Peptide Map data generated in lab (please use Figure 5a as a reference) how many chromatographic peaks do you see in the eGFP peptide map between 0.5 and 6 minutes? You may count all peaks that are >10% relative abundance.
Between 0.5 and 6 minutes, there are about 19 chromatographic peaks above 10% relative abundance in the peptide map.
Assuming all the peaks are peptides, does the number of peaks match the number of peptides predicted from question 2 above? Are there more peaks in the chromatogram or fewer
Yes, the number of chromatographic peaks does match the number of predicted peptides.
Identify the mass-to-charge of the peptide shown in Figure 5b. What is the charge of the most abundant charge state of the peptide (use the separation of the isotopes to determine the charge state). Calculate the mass of the singly charged form of the peptide.
The main peptide peak in Figure 5b is at m/z 525.76712. The isotope spacing is about 0.5 m/z, so the most abundant charge state is 2+. The singly charged mass is therefore about 1050.52 Da
Identify the peptide based on comparison to expected masses in the PeptideMass tool. What is mass accuracy of measurement? Please calculate the error in ppm.
The peptide is most likely FEGDTLVNR, which has a theoretical mass of about 1050.52145 Da. Using the experimental mass 1050.52438 Da, the error is about 2.8 ppm
What is the percentage of the sequence that is confirmed by peptide mapping?
The peptide map confirms about 88% sequence coverage of eGFP.
Homework: Waters Part II — Secondary/Tertiary structure
The 7FU decamer is expected at 3.4 MDa, the 8FU didecamer at 8.0 MDa, the 8FU 3-decamer at 12.0 MDa, and the 8FU 4-decamer at 16.0 MDa. In Figure 7, these correspond approximately to the peaks at 3.4, 8.33, 12.67, and the broad weak signal around 16–17 MDa, respectively.
Week 11 HW: Building Genomes
Part B: Cell-Free Protein Synthesis | Cell-Free Reagents
Referencing the cell-free protein synthesis reaction composition (the middle box outlined in yellow on the image above, also listed below), provide a 1-2 sentence description of what each component’s role is in the cell-free reaction.
BL21 (DE3) Star lysate (includes T7 RNA polymerase):
This provides the core cellular machinery needed for protein synthesis, incl~uding ribosomes, tRNAs, enzymes, and other translation factors. Because it includes T7 RNA polymerase, it can also transcribe DNA templates with a T7 promoter into mRNA.
Potassium glutamate:
This helps maintain the ionic environment needed for ribosomes and enzymes to work properly. It supports protein synthesis by mimicking intracellular salt conditions.
HEPES-KOH pH 7.5:
This acts as a buffer to keep the reaction at a stable pH. A stable pH is important because transcription and translation enzymes are sensitive to pH changes.
Magnesium glutamate:
Magnesium is essential for ribosome function, RNA stability, and many enzyme reactions in transcription and translation. If magnesium is too low or too high, protein production drops.
Potassium phosphate monobasic:
This helps buffer the reaction and contributes phosphate needed in the overall chemical environment. It also helps maintain proper ionic strength.
Potassium phosphate dibasic:
This works together with monobasic phosphate to maintain the phosphate buffer system. Together they help keep the reaction chemically stable.
Ribose:
Ribose serves as part of the energy regeneration system and can help support longer-lasting reactions. It provides carbon that can feed metabolic pathways in the lysate.
Glucose:
Glucose is another energy source that helps regenerate ATP through metabolism in the extract. This is important for sustaining transcription and translation over time.
AMP:
AMP is part of the nucleotide and energy balance in the reaction. It can be recycled through the lysate’s metabolic pathways to help support ATP regeneration.
CMP:
CMP is a pyrimidine nucleotide precursor that supports RNA synthesis and nucleotide recycling. It helps maintain the pool of RNA building blocks.
GMP:
GMP is a guanine nucleotide precursor that supports RNA synthesis and nucleotide recycling. It contributes to making the GTP needed for transcription and translation.
UMP:
UMP is another RNA nucleotide precursor that supports transcription and nucleotide balance. It helps maintain enough pyrimidine nucleotides in the reaction.
Guanine:
Guanine can be salvaged by enzymes in the lysate and converted into guanine nucleotides such as GMP and GTP. This helps support transcription even if GMP is not added directly in high amounts.
17 amino acid mix:
This provides most of the amino acids needed to build proteins during translation. They are the basic building blocks for the new protein.
Tyrosine:
Tyrosine is added separately because it may be less stable or less soluble in the standard mix. It is still required as one of the amino acids for protein synthesis.
Cysteine:
Cysteine is also added separately because it is more chemically sensitive than many other amino acids. It is needed for protein synthesis and for forming disulfide-containing proteins if relevant.
Nicotinamide:
Nicotinamide supports metabolic enzyme activity by helping maintain redox cofactor pools such as NAD-related systems. This can improve energy regeneration and reaction longevity.
Nuclease-free water:
This is used to bring the reaction to the correct final volume and concentration. It is nuclease-free so the DNA and RNA are not degraded.
Describe the main differences between the 1-hour optimized PEP-NTP master mix and the 20-hour NMP-Ribose-Glucose master mix shown in the Google Slide above. (2-3 sentences)
The main difference is cost and how the system generates usable nucleotides. The 1-hour PEP-NTP mix directly adds the more expensive NTPs, while the 20-hour NMP-ribose mix uses cheaper NMPs plus ribose/glucose and a longer incubation time so the reaction can regenerate the needed NTPs inside the mix itself.
Bonus question: How can transcription occur if GMP is not included but Guanine is?
Transcription can still occur because guanine can be converted by nucleotide salvage enzymes in the lysate into GMP, and then further into GDP and GTP. In other words, the extract can recycle guanine into the usable nucleotide form needed by RNA polymerase.
Part C: Planning the Global Experiment | Cell-Free Master Mix Design
Given the 6 fluorescent proteins we used for our collaborative painting, identify and explain at least one biophysical or functional property of each protein that affects expression or readout in cell-free systems. (Hint: options include maturation time, acid sensitivity, folding, oxygen dependence, etc) (1-2 sentences each)
sfGFP
sfGFP is a good cell-free reporter because it was engineered to fold very robustly, so it is less likely to lose signal from misfolding than standard GFPs. It also matures relatively fast, with FPbase listing a maturation time of about 13.6 min, so fluorescence appears fairly quickly after translation.
mRFP1
A key limitation of mRFP1 in cell-free systems is its slow maturation: FPbase lists it at about 60 min, so the protein can be made before the red signal is fully visible. It also has a relatively low quantum yield (0.25) and brightness compared with newer red proteins, so its readout is weaker and slower.
mKO2
For mKO2, two important issues are slow maturation and moderate acid sensitivity. FPbase lists a maturation time of about 108 min and a pKa of 5.5, so if the reaction is acidic or you measure too early, the orange signal may look weaker than the actual expression level.
mTurquoise2
mTurquoise2 is strong in cell-free systems because it has a very high quantum yield (0.93), which means it gives a bright signal even at moderate expression levels. It also has very low acid sensitivity (pKa 3.1) and a fairly fast maturation time of about 33.5 min, so its fluorescence is usually reliable across a wider pH range than many other FPs.
mScarlet_I
mScarlet-I is useful because it was engineered for faster maturation than many older red fluorescent proteins; FPbase lists it at about 36 min. It is also very bright, but its moderate acid sensitivity (pKa 5.4) means the signal can still drop if the cell-free reaction becomes too acidic.
Electra2
Electra2 stands out because it is a bright blue fluorescent protein with FPbase brightness of about 61.48, which helps a lot when blue channels are usually dimmer. It also shows strong reported photostability in FPbase, so its signal should hold up better during repeated imaging of the same cell-free artwork.
Create a hypothesis for how adjusting one or more reagents in the cell-free mastermix could improve a specific biophysical or functional property you identified above, in order to maximize fluorescence over a 36-hour incubation. Clearly state the protein, the reagent(s), and the expected effect.
Hypothesis: To maximize mKO2 fluorescence over a 36-hour incubation, I would use the 2 µL reagent-addition window to add extra HEPES-KOH (pH 7.5) plus a concentrated GMP stock, or, if I had to stay strictly within the existing reagent list, extra guanine + ribose. I am targeting mKO2 because it has a relatively slow maturation time (~108 min) and moderate acid sensitivity (pKa ~5.5), so its final orange signal is especially vulnerable to both delayed chromophore development and pH drift during a long cell-free reaction.
Expected effect: Extra HEPES should improve buffering and keep the reaction closer to neutral over 36 hours, which should preserve fluorescence for an acid-sensitive orange FP like mKO2, while extra GMP should reduce the burden on the lysate’s purine-salvage pathway and help sustain the pool of GTP/NTPs needed for transcription later in the reaction. That matters because E. coli extracts can convert NMPs to NTPs in glucose-powered CFPS, but in your current mix the guanine side is starting from guanine with 0 GMP, so supplementing that branch should help maintain mRNA production for longer and ultimately increase total mature mKO2 fluorescence at the 36-hour endpoint.
**The second phase of this lab will be to define the precise reagent concentrations for your cell-free experiment. You will be assigned artwork wells with specific fluorescent proteins and receive an email with instructions this week (by April 24). **