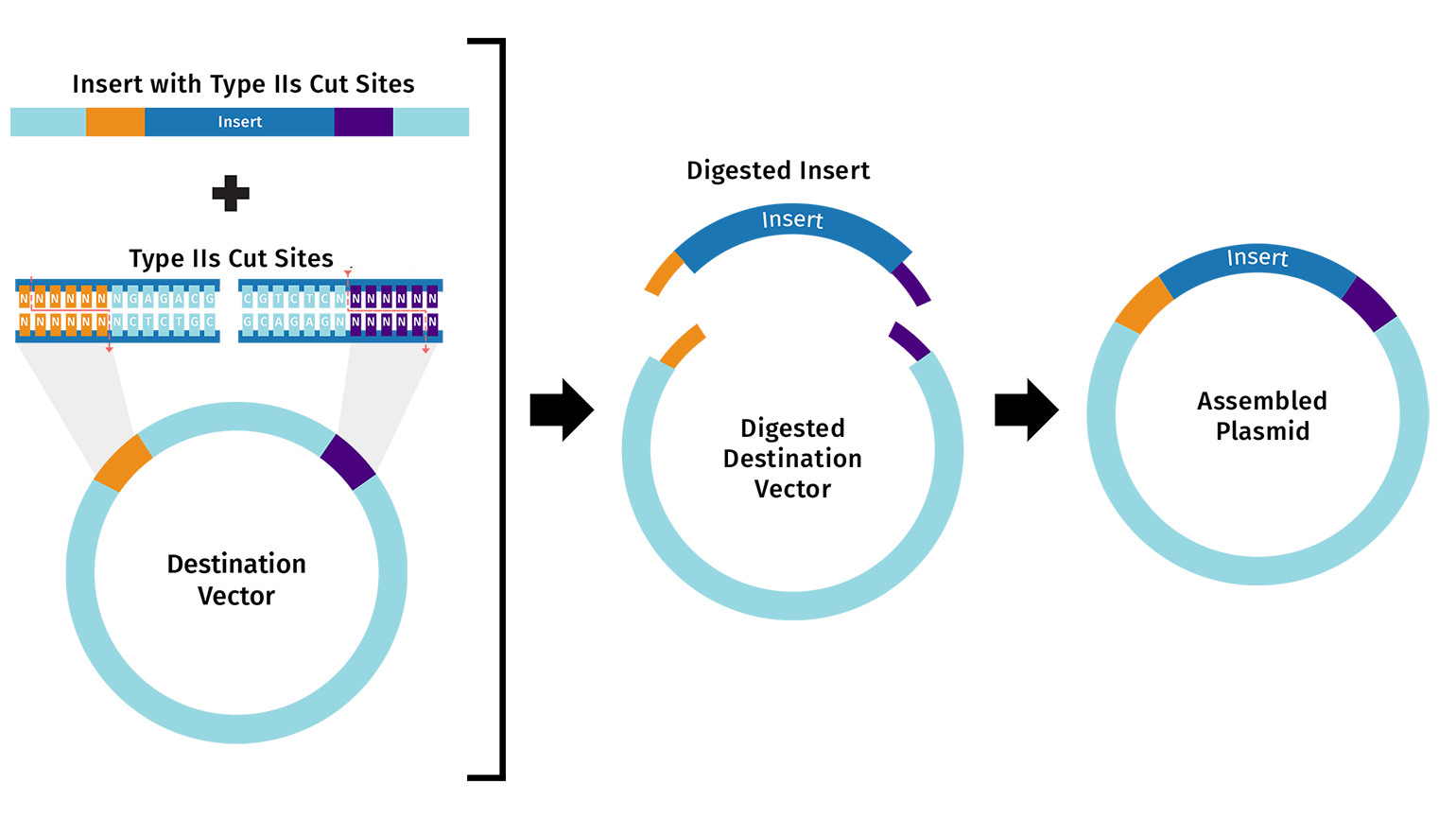This week explores the read–write–edit toolkit: sequencing and synthesis workflows, restriction digests and gel electrophoresis, and early genome-editing frameworks.
This week covers neuromorphic genetic circuits, showing how engineered gene networks can implement neural-network “perceptron”-like computation and learning.
Subsections of Homework
Week 1 - Principles & Practices
This week lays the foundation for ethics, safety, and governance in biotechnology — and we get hands-on with lab basics.
First, describe a biological engineering application or tool you want to develop and why.
So far, one idea I have is to develop an alternative to coffee and tea based on modified bacteria or mushrooms that synthesize caffeine. This would be interesting because it wouldn’t require the extensive transportation, human labor, land use, and resources that coffee and tea currently do. Given the scale of global consumption of these two beverages, their impact is far from negligible. I personally enjoy tea and coffee quite a lot (which is probably what led me to think about this idea). I don’t plan on replacing them entirely, but I think that being able to offer alternatives could only be beneficial.
2. Governance and policy goals
Next, describe one or more governance/policy goals related to ensuring that this application or tool contributes to an “ethical” future, like ensuring non‑malfeasance (preventing harm). Break big goals down into two or more specific sub‑goals.
Safety and security
Equity through open development
Transparency and information sharing
3. Governance actions
Next, describe at least three different potential governance “actions” by considering the four aspects below (Purpose, Design, Assumptions, Risks of Failure & “Success”).
Governance “action”
Actors
Purpose
Design
Assumptions
Risks of Failure & “Success”
Follow established biosafety best practices when working with living organisms
Researchers / academics
Avoid major problems, assess risks in advance, and be prepared if issues arise
Based on existing knowledge and trust in established standards
Error probability is never zero, so being prepared for most plausible scenarios is necessary
Still having biosafety problems; “success” could be not detecting or recognizing that a problem is occurring
Developing a viable solution and undergoing regulatory review
Researchers / academics
Some blends of mushrooms and coffee exist with unproven benefits. Modifying E. coli to synthesize caffeine has been done (e.g. this paper: https://pubs.rsc.org/en/content/articlehtml/2017/ra/c7ra10986e), but an accessible synthetic biology–based alternative for consumers does not yet exist
Requires sufficient knowledge and resources to carry out effective research and development
The main assumption is that it is feasible to develop a viable solution; it might not yet be possible in a sufficiently interesting or practical way
Failure: not being able to create it; or creating it but being unable to certify that it is safe for human consumption
Develop safe and sustainable pathways to scale the technology through hubs/micro‑factories
Company / co‑producers
Make the solution impactful by making it accessible to consumers and replacing a significant share of coffee/tea consumption to reduce environmental impact
Requires international partnerships, building trust, and the ability to share systems while growing the consumer base
Collaborators and consumers will be interested in adopting the solution
Failure: not being able to scale at all or by this model; low interest limiting impact; “success” risk: the solution being appropriated and developed by another actor with harmful or misaligned objectives
4. Scoring of governance options
Next, score (from 1–3, with 1 as the best, or n/a) each of your governance actions against your rubric of policy goals.
Does the option:
Option 1
Option 2
Option 3
Enhance Biosecurity
• By preventing incidents
1
2
2
• By helping respond
1
n/a
n/a
Foster Lab Safety
• By preventing incidents
1
2
3
• By helping respond
1
2
n/a
Protect the environment
• By preventing incidents
1
2
3
• By helping respond
1
n/a
n/a
• By reducing environmental impact
n/a
3
1
Other considerations
• Minimizing costs and burdens to stakeholders
1
1
1
• Feasibility
1
2
2
• Not impeding research
1
1
1
• Promoting constructive applications
1
1
1
• Protecting the consumer
1
1
2
5. Prioritized options and trade‑offs
Last, drawing upon this scoring, describe which governance option, or combination of options, you would prioritize, and why. Outline any trade‑offs you considered as well as assumptions and uncertainties.
The main priority is the strict application of established biosafety and biosecurity practices. This option scores best across safety, feasibility, and environmental protection, and it is essential given that the project involves genetically modified organisms and potential human consumption. Preventing harm, avoiding accidental release, and maintaining public trust are prerequisites for any further development.
The second priority is regulated development and verification. Regulatory oversight is necessary to demonstrate that the technology is safe, transparent, and credible beyond the lab. While this step may slow progress, it protects consumers and ensures that any claims about safety or sustainability are evidence‑based.
Scaling the technology through hubs or micro‑factories would only be considered at a later stage. Although this option offers the greatest potential environmental benefits, it also carries higher risks related to misuse, safety, and governance. Scaling should therefore depend on strong biosafety records and regulatory approval.
Overall, prioritizing biosafety first, regulation second, and scaling last offers the most ethically robust path forward. It aligns with non‑malfeasance, protects consumers and the environment, and still leaves room for meaningful impact if the technology proves viable.
6. Ethical concerns and governance ideas
Reflecting on what you learned and did in class this week, outline any ethical concerns that arose, especially any that were new to you. Then propose any governance actions you think might be appropriate to address those issues.
An important ethical concern discussed this week is the risk of monopolies in synthetic biology. When a small number of actors control key technologies or biological systems, access to solutions like treatments can become limited or too expensive, increasing inequalities, as has already been the case with insulin.
To address this, governance actions could include avoiding exclusive control over essential technologies, encouraging open research and knowledge sharing, and ensuring public oversight in the development of synthetic biology–based treatments. Educating the general public about these technologies and their implications is also important to support informed debate and fair access.
7. Use of AI
Can you correct the English mistakes: […]
How to better format the markdown
Week 2 - 1. Lecture Preparation
Next lecture will be around how to Read, Write and Edit DNA.
Homework Questions from Professor Jacobson:
Nature’s machinery for copying DNA is called polymerase. What is the error rate of polymerase? How does this compare to the length of the human genome. How does biology deal with that discrepancy?
Polymerases are enzymes that synthesize long chains of nucleic acids, such as DNA or RNA. Two main types exist: DNA polymerase and RNA polymerase, which assemble DNA and RNA molecules respectively by copying a DNA template strand through base-pairing interactions during semi-conservative replication. The error rate of DNA polymerase is approximately one error per 10⁶ bases.
This error rate may seem low, but considering the length of the human genome (about 3.2 × 10⁹ base pairs), it would result in roughly 3,000 errors per replication cycle, which would make the process unreliable.
To address this issue, biological systems rely on two main error-correction mechanisms. First, many DNA polymerases possess proofreading activity through an associated exonuclease, which detects and removes incorrectly incorporated bases during replication. Second, post-replication mismatch repair proteins, such as MutS, detect and repair mismatches between the parent strand and the daughter strand. Together, these mechanisms significantly reduce the final number of errors, allowing for stable and reliable DNA replication.
How many different ways are there to code (DNA nucleotide code) for an average human protein? In practice what are some of the
reasons that all of these different codes don’t work to code for the protein of interest?
An average human protein is about 1036 base pairs long. Because the genetic code is redundant, each amino acid can be encoded by multiple codons (typically 2–6), so in theory there are an immense number of different DNA sequences that could code for the same protein.
In practice, most of these sequences do not work. Cells show codon bias, preferring certain codons for efficient translation. Some sequences create strong DNA or RNA secondary structures that interfere with transcription or translation. Extreme GC or AT content can reduce stability or cause replication problems. Other sequences accidentally introduce regulatory signals or stop codons. Some are also difficult to synthesize accurately. As a result, only a small fraction of the theoretically possible DNA sequences can reliably produce the intended protein.
Homework Questions from Dr. LeProust:
What’s the most commonly used method for oligo synthesis currently?
The most commonly used method today is solid-phase phosphoramidite chemical synthesis. DNA bases are added one at a time to a growing strand that is attached to a solid support, repeating a cycle of coupling, capping, oxidation, and deprotection
Why is it difficult to make oligos longer than 200nt via direct synthesis?
Each nucleotide addition is not 100% efficient. Even small failure rates compound over many cycles. As the oligo gets longer, the fraction of full-length correct molecules drops rapidly, and truncated or error-containing products dominate. By around 200 nt, yield and accuracy become too low for reliable direct synthesis
Why can’t you make a 2000bp gene via direct oligo synthesis?
A 2000 bp gene would require 2000 sequential chemical coupling steps. With cumulative errors and truncations, the probability of producing a full-length, correct molecule becomes essentially zero. Instead, long genes are made by assembling many shorter oligos (typically 50–200 nt) using enzymatic methods like PCR and ligation, followed by error correction and sequencing
Homework Question from George Church:
[Using Google & Prof. Church’s slide #4]What are the 10 essential amino acids in all animals and how does this affect your view of the “Lysine Contingency”?
The 10 essential amino acids in all animals are:
Histidine, Isoleucine, Leucine, Lysine, Methionine, Phenylalanine, Threonine, Tryptophan, Valine, and Arginine (arginine is strictly essential during growth and development). Animals cannot synthesize these amino acids in sufficient amounts, so they must obtain them from their diet or from other organisms.
Lysine is especially interesting because it is essential, chemically distinct, and metabolically expensive to make. Animals have completely lost the ability to synthesize it, yet lysine is required for protein synthesis, regulation, and many post-translational modifications
In Jurassic Park, InGen claims the dinosaurs were engineered to be lysine-dependent, so they would die if they escaped because lysine would be unavailable in the wild. However, lysine is a common essential amino acid found in many foods, and humans themselves are lysine-dependent. As a medically trained author, Michael Crichton would have known this, and in the story the dinosaurs indeed survive outside containment. Therefor we can imagine that the “lysine contingency” was an intentional deception by InGen, a narrative of safety designed to reassure investors and regulators, rather than a genuine biological containment strategy.
Week 2 - 2. DNA Read, Write & Edit
This week explores the read–write–edit toolkit: sequencing and synthesis workflows, restriction digests and gel electrophoresis, and early genome-editing frameworks.
I’m a commited listener but without acces to a biology lab.
0. Basics of Gel Electrophoresis
I followed the class and take a look at the content around this technique.
1. Benchling & In-silico Gel Art
After creating an account on Benchling and using it to understand better the possibilities it offers.
I made a Digestion Simulation with all the indicated restriction enzymes :
Here is my created image using this same technique :
“Seagull flying in a clear sky”, I acknowleged it is quite simple it’s the first inspiration that came to my mind :)
2. Gel Art - Restriction Digests and Gel Electrophoresis
I don’t have acces to a lab as mentionned ealier
3. DNA Design Challenge
Choose your protein.
I’ve chosen Chitodextrinase, it’s an enzyme that permit the degradation of chitin which a polysaccaride that form structure and rigidity of living orgnisme. It’s found mostly in mushrooms and insect, there for chitodextrinase is produced by animal eating insect or plants to protect themselve from fungi. I found it intersting because i worked on mycelium based composite and chitin is on of the main strucuring compond allowing those object to be resistant and light while being biodegradable. So i thought it would be intersting to understand better the process of it’s decomposition by living organisme.
Here is this protein Sequence :
LOCUS CAM6841167 609 aa linear BCT 18-MAR-2025
DEFINITION Chitodextrinase [Enterobacter rongchengensis]
elipse:
I started my research of an intersting protein thinking about cafeine but it’s an alcaloïde. Then I though that i could be one at the origine of the smell of fresh rain : it come from geosmin a bacterian made metabolite (way smaller than proteines). I also though about melanine but it’s not so simple you have different type and they aren’t protein either. After that I thought about all the things we hears about the protein in our alimentation and remembered that the most protein dense “veggie” in french it’s considered a legumineuse is lentils and the 2nd most important protein in quantity in them is “Albumine” a protein playing an important role in human lymphatic systeme alowing a proper osmotique balance of the plasma.
So for bonus here is an Albumine sequence I founded too :
Reverse Translate: Protein (amino acid) sequence to DNA (nucleotide) sequence.
using : https://www.bioinformatics.org/sms2/rev_trans.html I found the translation of my Chitodextrinase protein amino acid code to nucleotide.
reverse translation of Untitled to a 1827 base sequence of most likely codons.
atgaacaaacgcaccctgctgagcgtgctggtggcgggcgcgtgcgtggcgccgtttatg
gcgcaggcggcgagcctgcaggcgaccaccagcgaaccgtataccattaaagcgagcgat
ctggcgaaaaaagaaaaagaactgaccagctttccgctgatggcgagcgtgaaagaaacc
attcagaccctggataacgcgcaggtggaactgattgaaccgggccgcgcggcgaacccg
gataacgtgaaacgcgtggaaggcattgtgaaagcgagcgattgggaatatctgtttccg
ctgcgcgcgcaggcgtatacctatagcaactttctgaaagcggtgggcaaatttccggcg
ctgtgcaaaacctataacgatggccgcgatagcgatgcgatttgccgcaaagaactggcg
accatgtttgcgcattttgcgcaggaaaccggcggccatgaaagctggcgcccggaagcg
gaatggcgccaggcgctggtgcatgtgcgcgaaatgggctggagcgaaggccagaaaggc
ggctataacggcgaatgcaacccggatgtgtggcagggccagacctggccgtgcggcaaa
gataaagatggcgattttctgagctattttggccgcggcgcgaaacagctgagctataac
tataactatggcccgtttagcgaagcgatgtatggcgatgtgcgcaccctgctggataaa
ccggaactggtggcggatacctggctgaacctggcgagcgcgatttttttttttgcgtat
ccgcagccgccgaaaccgagcatgctgcaggtgattgatggcacctggcagccgaacgat
catgataaagcgaacggcctggtgccgggctttggcgtgaccacccagattattaacggc
ggcgtggaatgcggcggcccgaccgaaattgcgcagagcgaaaaccgcattaaatattat
aaagaatttgcgaactatctgaaagtgccggtgccggcgaacgaagtgctgggctgcgcg
aacatgaaacagtttgatgaaggcggcgcgggcgcgctgaaaatttattgggaacaggat
tggggctggagcgcggataccccgagcggcgcgacctatagctgccagctggtgggctat
cagaccccgtttagcgcgtttaaagaaggcgattatagcaaatgcgtgcagaaatttttt
aacgtgaacattgtgaacgatgatggcagcgcgaccccggatcagaccccggtgaccccg
accccgaccccggcgccgagccaggatgaaaccccggcgccggcgccggtgccggatgaa
accccggcggaaccggcggcggtgaaccatgcgccggtggcggatattgcgggcccgatt
ggcgcggtggatgcgggcgcgcaggtgagcctgagcgcggaaggcagcaccgatgcggat
ggcaacgcgctgacctatacctggcgcagccaggatggccagaccgtgaccggccaggat
aaagcggtggtgacctttaaagcgccggaagcggcgaccgcgcagcagattgaaattagc
ctgaccgtgagcgatggcgaactgagcagcaccaccagctatctgctgaacgtgaaagcg
aaagcggcgccgagccaggatgaaggcaccagcggcaactatgcggcgtggagcgcgaac
agcaaatataaagcgggcgatattgtgaacaaccatggcaaactgtttcagtgcaaaccg
tttccgtatagcggctggtgcaacaacgcgccggcgtattatgaaccgggcgcgggcctg
gcgtgggcggatgcgtggaccgcgctg
Codon optimization.
Different organisms “prefer” different codons for the same amino acid, because their tRNA pools are tuned to their own typical codon usage. If I took a gene from another organism and used it as‑is in a new host, translation could be slow or inefficient, leading to low protein yield or even misfolded, non‑functional protein. Codon optimization rewrites the DNA sequence without changing the amino acid sequence, replacing rare codons with codons that are common in the chosen host to match its tRNA availability and boost expression. I chose to optimize the sequence for E. coli, because it is one of the most widely used and well‑characterized hosts in synthetic biology and industrial protein production, with many tools, protocols, and optimization resources available.
You have a sequence! Now what?
What technologies could be used to produce this protein from your DNA? Describe in your words the DNA sequence can be transcribed and translated into your protein. You may describe either cell-dependent or cell-free methods, or both.
You could produce the protein using recombinant expression in cells (like E. coli, yeast, insect, or mammalian cells) or by cell‑free translation systems that use cell extracts. In both cases, your DNA sequence is first transcribed into messenger RNA by RNA polymerase, using base pairing rules (A–U, C–G) to copy the gene. Then ribosomes translate
the mRNA: every three nucleotides (a codon) are read in order and matched to a tRNA carrying the corresponding amino acid. As the ribosome moves along the mRNA, it links amino acids together into a polypeptide chain that folds into your functional protein
How does it work in nature/biological systems?
Describe how a single gene codes for multiple proteins at the transcriptional level.
A single gene can give multiple proteins mainly through alternative splicing of its pre‑mRNA. During transcription, the whole gene (exons + introns) is copied into a pre‑mRNA. The spliceosome can then join exons together in different combinations (for example skipping an exon, using alternative 5′ or 3′ splice sites, or choosing mutually exclusive exons), producing several distinct mRNA isoforms from the same gene. Each mRNA has a different coding sequence, so translation produces related but different protein isoforms with altered domains or lengths.
4. Prepare a Twist DNA Synthesis Order
Whole process done around the exemple of “Constitutive sfGFP”
.fasta and .gbk file in the folder
5. DNA Read/Write/Edit
DNA Read
(i) What DNA would you want to sequence (e.g., read) and why? This could be DNA related to human health (e.g. genes related to disease research), environmental monitoring (e.g., sewage waste water, biodiversity analysis), and beyond (e.g. DNA data storage, biobank).
I would want to sequence metagenomic DNA extracted from the outer skin/surface of our biolab’s mycelium composite object. This would reveal the microbial communities (bacteria, fungi, etc.) colonizing the surface, helping us understand biofilm formation, contamination risks, and natural protection that mycelium provides against external microbes.
Additionally, I’d sequence internal DNA from the mycelium-wood composite itself (after surface sterilization) to compare the native microbiome within the material versus surface colonizers. This contrast would show whether the mycelium creates a unique internal niche or if external contaminants penetrate.
(ii) In lecture, a variety of sequencing technologies were mentioned. What technology or technologies would you use to perform sequencing on your DNA and why?
I would use Illumina sequencing for the metagenomic DNA from our biolab’s mycelium composite surface and internal samples. Because Illumina provides high accuracy and important throughput (millions of reads), which is adapted for detecting low-abundance microbes on material surfaces and generating statistically robust community profiles. Short reads (150-300 bp) cover the hypervariable regions of 16S rRNA (bacteria/archaea) and ITS (fungi) needed for species identification.
Is your method first-, second- or third-generation or other? How so?
This method is considered second-generation. It sequences millions of DNA fragments simultaneously using PCR-amplified clusters on a flow cell, unlike first-gen Sanger (one fragment at a time) or third-gen single-molecule methods.
What is your input? How do you prepare your input (e.g. fragmentation, adapter ligation, PCR)? List the essential steps.
For the imput and preparation the essentials steps are :
Input: Total DNA (~10-100 ng) extracted from surface swabs and internal core samples. Essential preparation steps:
PCR amplification of 16S V3-V4 (~460 bp) and ITS1 (~250 bp) regions using universal primers
Adapter ligation to add Illumina sequencing handles
Library pooling and normalization
Cluster generation on flow cell via bridge amplification
What are the essential steps of your chosen sequencing technology, how does it decode the bases of your DNA sample (base calling)?
Sequencing by synthesis: DNA polymerase adds one fluorescently labeled nucleotide (with reversible terminator) at a time
Each base (A/T/C/G) emits a unique fluorescent color
Imaging: Camera captures light emission after each incorporation
Cleavage: Fluorescent blocker removed, allowing next base addition
Base calling: Software converts fluorescence intensities → ACGT sequence
What is the output of your chosen sequencing technology?
a FASTQ files containing:
Millions of short reads (150-300 bp) per sample
Phred quality scores per base (Q30 = 99.9% accuracy)
Processed via QIIME2/DADA2 → taxonomic profiles showing bacterial/archaeal/fungal relative abundances on mycelium surface vs. interior
DNA Write
(i) What DNA would you want to synthesize (e.g., write) and why?
To be fully honest i’m not so sure so far, I need to take time to do more research to understand more the possibilities and the outcomes it could bring.
(ii) What technology or technologies would you use to perform this DNA synthesis and why?
(ii) What DNA synthesis technology would I use and why?
I could use phosphoramidite-based chemical DNA synthesis the industry standard employed by companies like Twist Bioscience and IDT. This method offers the highest reliability, scalability, and quality control for custom gene synthesis up to 3 kb, with >99% accuracy at the oligo level.
Essential steps:
Solid-phase phosphoramidite synthesis of short oligonucleotides (50-200 bp)
Gibson assembly or Golden Gate to stitch oligos into full-length genes
Error correction via enzymatic selection or hybridization capture
Cloning into a plasmid vector
Sanger sequencing verification
Key limitations:
Error rates accumulate with length (>3 kb requires multi-fragment assembly)
GC-rich, repetitive, or hairpin-forming sequences reduce yield
Cost scales linearly with length (~$0.10-0.20/base)
Why it fits: Despite limitations, gene-scale synthesis (1-3 kb) is now routine and robust enough for most synthetic biology applications. For a caffeine pathway, multiple short genes can be synthesized separately then assembled.
Scaling reality check: Unlike chemical reactors, biological systems face oxygen gradients, metabolic stress, and contamination at scale. Lab success doesn’t guarantee industrial performance, so small-scale mastery comes first.
DNA Edit
(i) What DNA would you want to edit and why?
Same as for the Write,i’m not sure yet. I don’t think I have a big enough understanding.
(ii) What technology or technologies would you use to perform these DNA edits and why?
I would use *CRISPR-Cas9 genome editing because it enables precise insertion or modification of DNA at specific genomic locations using programmable guide RNAs, making it far more flexible and efficient than older methods like TALENs or ZFNs.
How CRISPR edits DNA:
Design guide RNA (gRNA) to target specific 20-bp genomic sequence (adjacent to PAM site)
Deliver Cas9 protein + gRNA as RNP complex or plasmids into cells
Cas9 creates double-strand break at target site
Provide donor DNA template with desired gene flanked by homology arms
Homology-directed repair (HDR) integrates new sequence during break repair
Potential off-target cuts (mitigated by high-fidelity Cas9 variants)
Delivery challenges in some cell types
Large donors (>2 kb) reduce integration efficiency
Why CRISPR fits: Despite limitations, it remains the most precise, flexible, and accessible editing tool available, with continuous improvements (Cas12a, base editing, prime editing) addressing early shortcomings. For engineering caffeine pathways into bacteria or fungi, CRISPR provides the necessary precision at multiple loci.
Week 3 - Lab Automation
This week we get hands-on (or at least code-on) with pipetting robots
1. Python Script for Opentrons Artwork
LifeFab node at London only have 3 colors : Purple, Pink and Blue. Therefore I try making imaging working with just those ones.
I got and adapted the code to operate the opentrons to produce my image. I’ll stay listening to my node to know what are the possibilities, adapt my work and maybe make it.
2. Post-Lab Questions
Find and describe a published paper that utilizes the Opentrons or an automation tool to achieve novel biological applications.
Slowpoke: An Automated Golden Gate Cloning Workflow for Opentrons OT-2 and Flex, Malcı Koray Malcı et al. American Chemical Society, https://doi.org/10.1021/acssynbio.5c00629
This paper presents Slowpoke, an open-source, automated workflow for Golden Gate DNA assembly in synthetic biology. It is designed to run on affordable liquid-handling robots such as the Opentrons OT-2 and Opentrons Flex, making high-throughput cloning accessible to standard laboratories rather than only large biofoundries.
Slowpoke automates the full cloning pipeline: Golden Gate assembly of modular DNA parts, transformation into Escherichia coli, plating, and colony PCR screening. By precisely pipetting and handling dozens of DNA combinations in parallel, the robot enables rapid construction of large combinatorial genetic libraries with high accuracy and minimal human intervention. This automation allows researchers to quickly build and test multi-gene pathways, regulatory circuits, and standardized toolkit constructs, accelerating the design–build–test cycle.
Write a description about what you intend to do with automation tools for your final project.
I’m not really sure yet but I think I might be able to run experiment from a cloud lab.
3. Final Project Ideas
Synthetic Biology Based Solution for Caffeine Production
Caffeine has become one of the most widely consumed psychoactive substances in the world. The estimated global coffee consumption in 2023/2024 was 10.6 million tonnes (ICO). This level of production generates significant impacts on human labor, land use, water consumption, transportation, and other environmental and socio-economic factors.
Caffeine naturally occurs in plants such as coffee, tea, and guarana, and it can be chemically extracted from these sources or produced synthetically through chemical processes starting from compounds such as ammonia. It could be interesting to explore synthetic biology approaches to develop alternative production solutions that could be deployed closer to areas of consumption and scaled to reduce these environmental impacts.
Some research has already been conducted in this area and could be starting points:
Could Synth Bio Improve Concrete: Reduce C02 FootPrint and/or Self repairability ?
No need to present concret its usages and impacts.
Quite self explanatory title. Exploration of the actual possibilities, research on the strain of bacteria used, how and what are key limitations. Could lead into trying to improve them and doing concrete experiments
Some research has already been conducted in this area and could be starting points:
Synthetic Biology for Fermentation and Probiotics Improvements
Fermentation as been a technique to keep for for longer and even get more nutrients out of them though almost symbiotic relationship with bacterias and yeast. It could be interesting to take a closer looks on it and maybe try to characterise best levain colony and evaluate the value they brings.
It could also be done around other type of fermentation processes, we could optimize the good nutrients and probiotics creation of lactic acid based fermentation or pickling of different vegetables and seed as: cabbage, peppers, mustard and many more
Some research has already been conducted in this area and could be starting points:
Genomics and synthetic community experiments uncover the key metabolic roles of acetic acid bacteria in sourdough starter microbiomes - https://pubmed.ncbi.nlm.nih.gov/39287380/
This week focuses on how sequence, structure, and energetics can be modeled and manipulated to create or optimize proteins with specified functions.
Part A. Conceptual Questions
Answer any NINE of the following questions from Shuguang Zhang:
How many molecules of amino acids do you take with a piece of 500 grams of meat? (on average an amino acid is ~100 Daltons)
This paper : “Samicho, Z. , Mutalib, S. R. A. and Abdullah, N. (2013) Amino acid composition of droughtmaster beef at various beef cuts. Agricultural Sciences, 4, 61-64. doi: 10.4236/as.2013.45B012. " We get the information that the different cut of beef meat contains from 10 to 20 grams of amino acis per 100g of meat.
From that we can approximate the number of molecules of amino acids in 500 grams of meat.
Knowing that on average: There is ~15g of amino acid in 100g of meat, amino acid is ~100 Daltons, 1 Dalton is 1,67x10e-24
15 x 5 = 75 (g of amnio acids in 500 g of meat)
75 / 100 x 1,67x10e-24 = 4,49x10e23
So there is approximately 4,5x10e23 amino acid in 500g of meats
Why do humans eat beef but do not become a cow, eat fish but do not become fish?
We don’t became the animals (or any other aliments) we eat because the proces of digestion break down the protein, cells, fat and other compound into amino acids that are absorbed and then used to create our human cells.
Why are there only 20 natural amino acids?
There is now 22 and there isn’t an absolute answer from the knowledge we have today. They have been know to appeared a long time ago (The 20 standard amino acids encoded by the Genetic Code were adopted during the RNA World, around 4 billion years ago) and life keep this configuration even if there little expection here and there (Pyrolysine is only found in some extremophiles) and human are know to use 21 since Pyrolysine was discovered in 2002. Some clues I understood were the limited genetic space to encode and decode those amino acids, that this number is likely a large enough panel to have a diversity of properties and possibilities but small enough to be practical to create. And the cost of creating the living pathway to create and use new ones would be really important for the benefice it would provide.
Can you make other non-natural amino acids? Design some new amino acids.
yes definitly. an amino acid is a compound that contain both amino and carboxylic acid functional groups. There for by adding a C between the CR and COOH I just created a new amino acids that is non-natural because all the natural amino acid are alpha ones so like so: H2N - CR- COOH (R is the amino acid side chain)
Where did amino acids come from before enzymes that make them, and before life started?
Amino acids likely formed naturally on early Earth before life existed through chemical reactions like: Primitive atmosphere reactions, Gases like methane, ammonia, water vapor, and hydrogen were hit by lightning or UV light, forming amino acids. This was shown in the Miller-Urey experiment. Deep-sea vents: Hot underwater vents provided energy and minerals that helped simple molecules combine into amino acids. From space: Amino acids have been found in meteorites, meaning they can form in space and may have been delivered to Earth. So, amino acids did not need enzymes or life to form they came first through natural chemistry, and life later appeared with a way to use them.
If you make an α-helix using D-amino acids, what handedness (right or left) would you expect?
In natural proteins, α-helices are formed almost exclusively from L-amino acids. The typical α-helix is a right-handed helix, and this geometry is favored by L-amino acids. If you built a helix from D-amino acids, it would form a left-handed helix instead (mirror image).
L and D for amino acids refer to the spatial arrangement (chirality) of the atoms around the α carbon. This shift between right-handed helif to a left-handed one will make the properties of that protein.
Can you discover additional helices in proteins?
Yes, proteins already have multiple helix types beyond α-helices and in principles new ones can be discovered or designed using physical tools or computational tools like alpha fold to analyse the structure of the protein and find uncomun ones but they’re usually variants rather than entirely new structural families.
Why are most molecular helices right-handed?
Mort molecular helices are right-handed because the amino acids used by life are L-amino acids, and their geometry naturally packs into right-handed helices with the lowest energy. It’s likely a “choice” made by life at some point, that became the norm.
Why do β-sheets tend to aggregate?
β-sheets are protein structures where strands lie side by side and are held together by hydrogen bonds, They tend to aggregate because their edges have exposed hydrogen bonds that easily attach to other strands, and their flat shape plus hydrophobic side chains makes them stack and stick together easily.
What is the driving force for β-sheet aggregation?
The driving force for their aggregation is mainly the formation of more hydrogen bonds between strands and the hydrophobic effect, where nonpolar side chains cluster together to avoid water, making the structure more stable.
10 and 11 skiped
Part B: Protein Analysis and Visualization
Briefly describe the protein you selected and why you selected it.
I’ve chosen Chitodextrinase, it’s an enzyme that permit the degradation of chitin which a polysaccaride that form structure and rigidity of living orgnisme. It’s found mostly in mushrooms and insect, there for chitodextrinase is produced by animal eating insect or plants to protect themselve from fungi. I found it intersting because i worked on mycelium based composite and chitin is on of the main strucuring compond allowing those object to be resistant and light while being biodegradable. So i thought it would be intersting to understand better the process of it’s decomposition by living organisme.
How long is it? What is the most frequent amino acid?
The lenght of the sequence is 611, the most frequent amino acid is Asparagine (N) with 51 apeareance.
How many protein sequence homologs are there for your protein?
yes there are many homologues from differents many differents bacterias
Does your protein belong to any protein family?
This protein belongs to the glycoside hydrolase family 18 (GH18) and contains a conserved chitinase catalytic domain along with carbohydrate-binding modules (CBMs), consistent with chitin-degrading enzymes.
Identify the structure page of your protein in RCBS
When was the structure solved? Is it a good quality structure? Good quality structure is the one with good resolution. Smaller the better (Resolution: 2.70 Å)
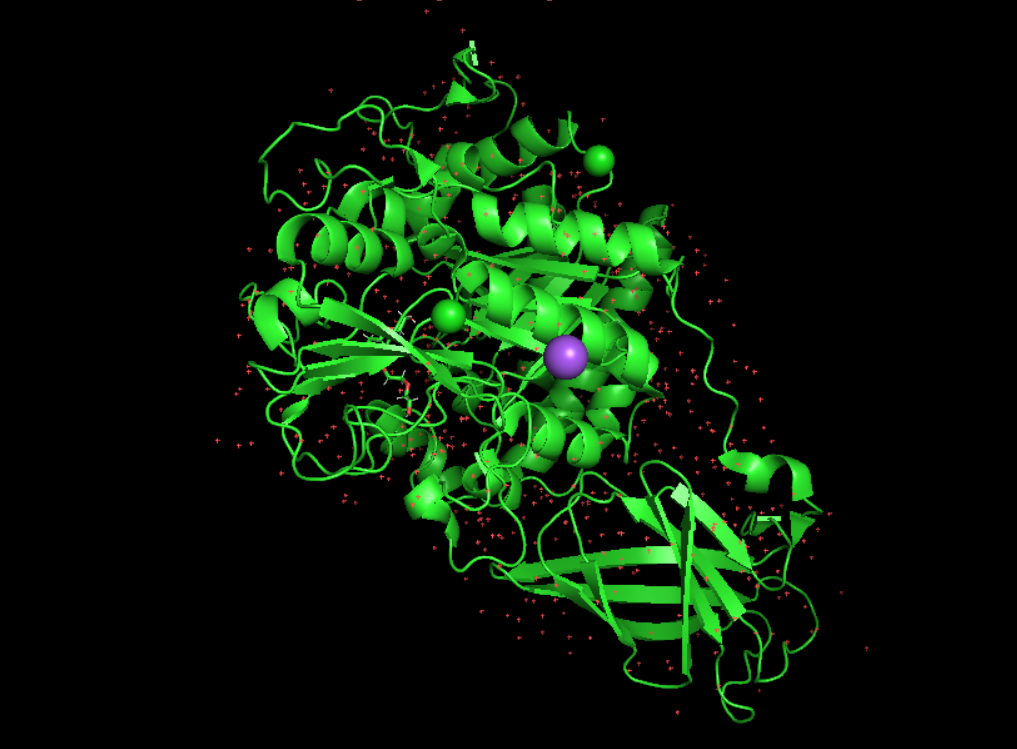
it was solve in 2001 and in 2023 for the specific chitodextrinase from the Clostridium perfringens. the methodology used is X-RAY DIFFRACTION 1.5 Å so yes the quality structure is good. - https://www.rcsb.org/structure/8OTB
Are there any other molecules in the solved structure apart from protein?
No it’s just the protein. But there is other solved structure with the protein and other molecules like : inhibitor bisdionin C, chitin, chitosan for example.
it seems not to belong to any structure classification family because i don’t have any return from SCOP, on RCSB PDB i have those information : Global Symmetry : Asymmetric - C1 Global Stoichiometry: Monomer - A1
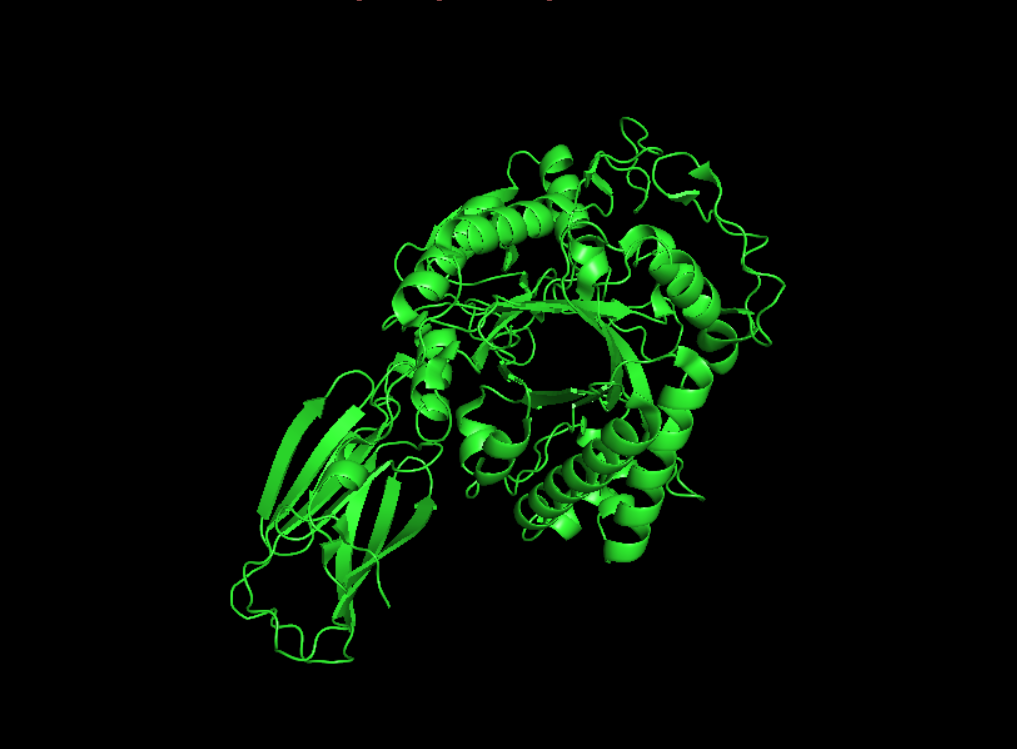
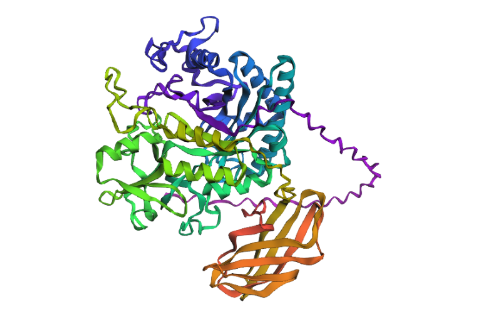
Open the structure of your protein in any 3D molecule visualization software:
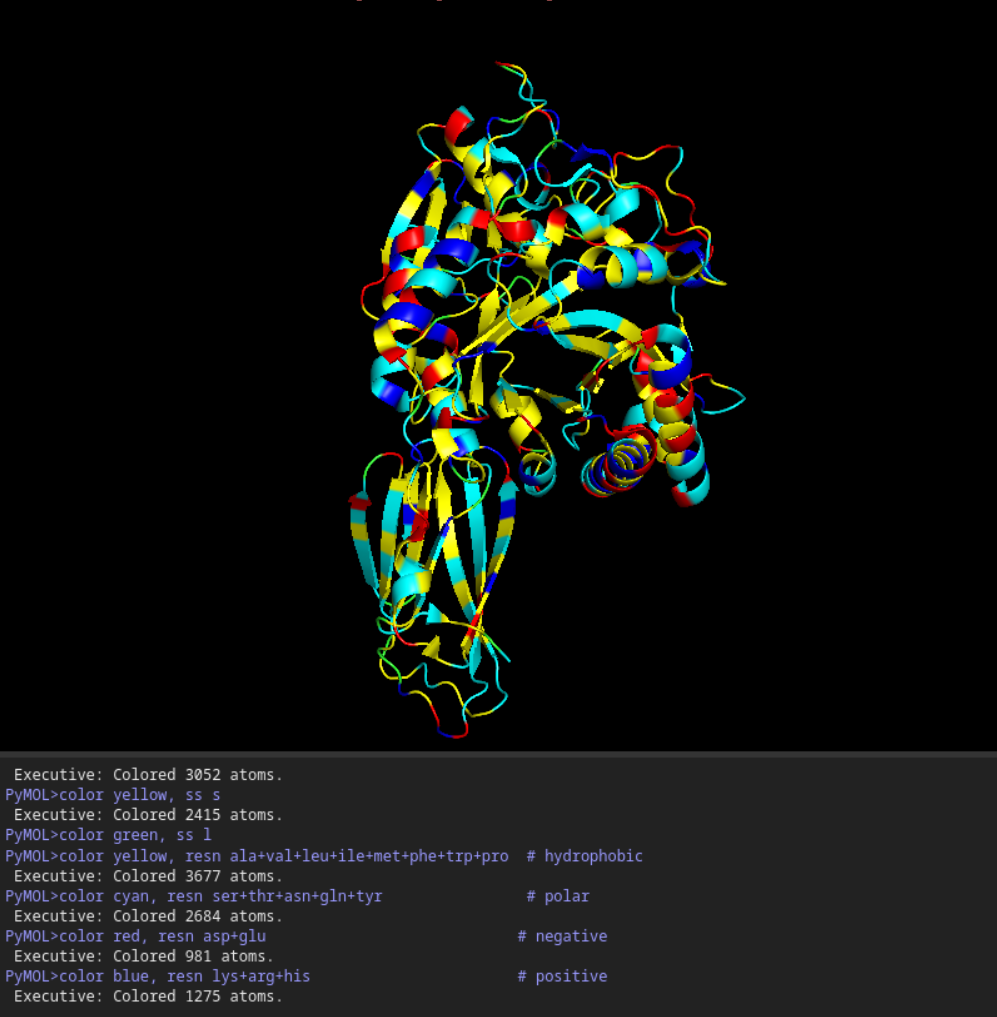
1st Visualisation
Visualisation as cartoon, riboon and ball and stick
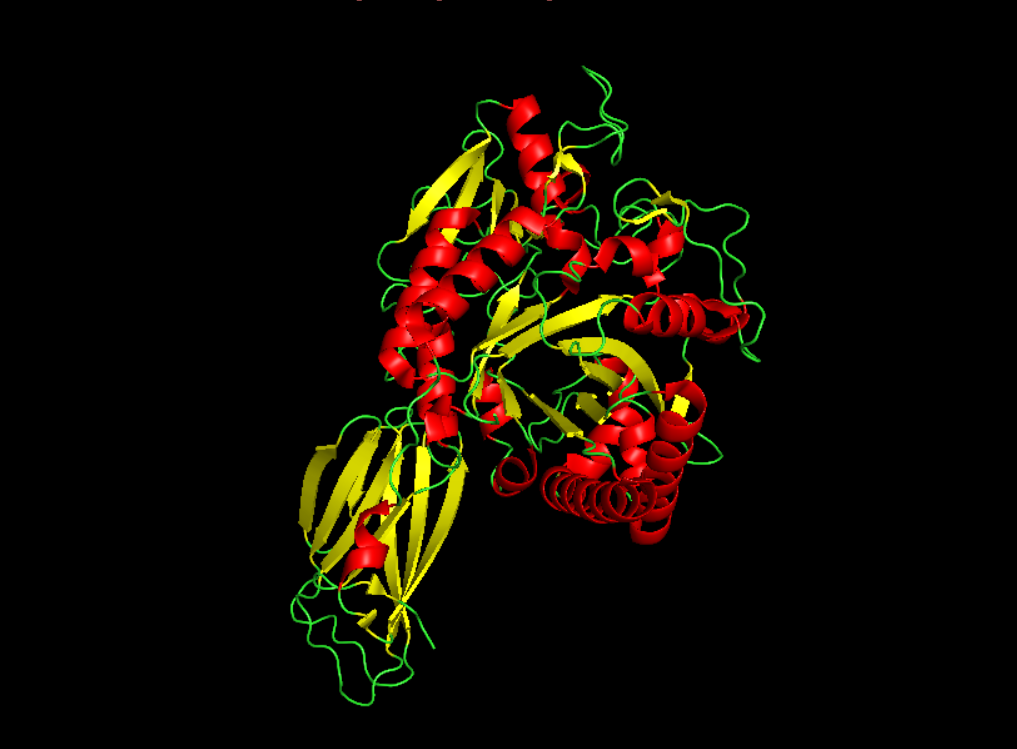
Color the protein by secondary structure. Does it have more helices or sheets?
It seems to have more few more helices than sheets.
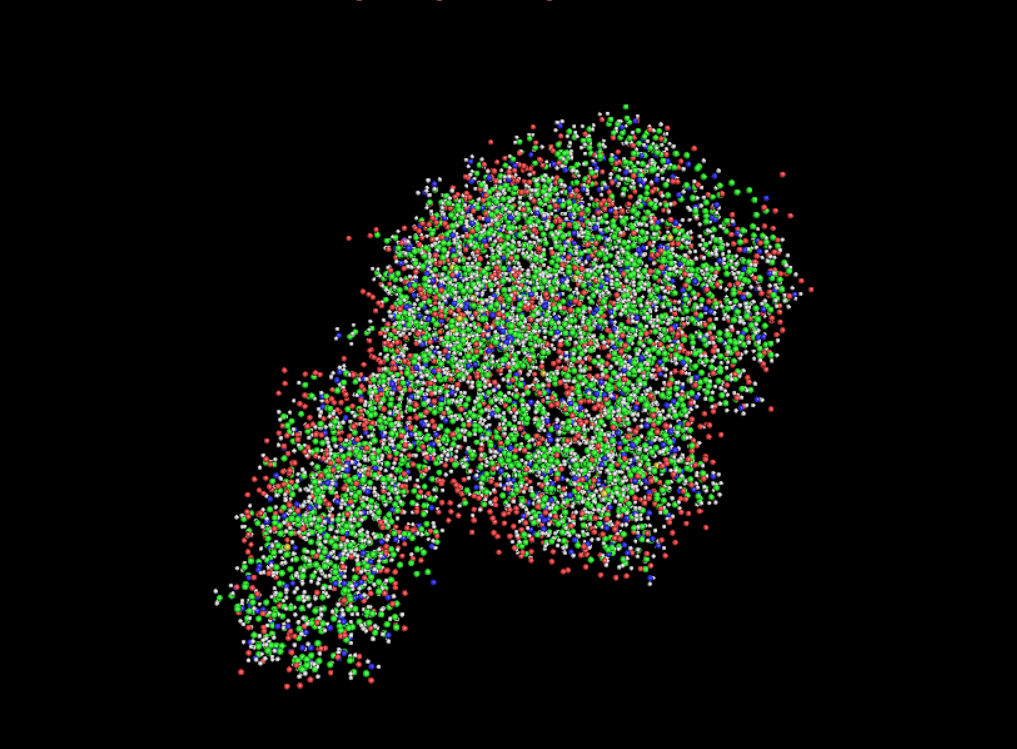
Color the protein by residue type. What can you tell about the distribution of hydrophobic vs hydrophilic residues?
Once again it is quite balanced but it seems to have more hydrophilic residues.
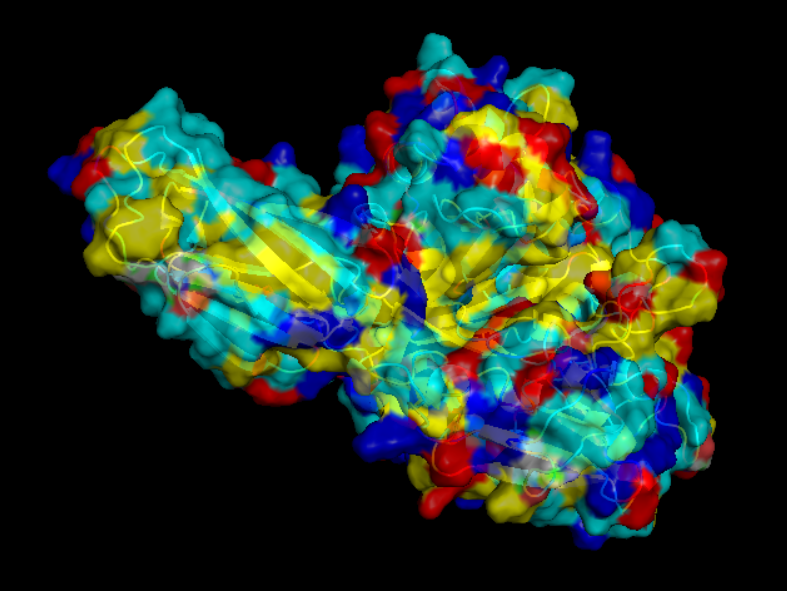
Visualize the surface of the protein. Does it have any “holes” (aka binding pockets)?
yes it look like it does have binding pockets, I don’t know if it is many. I can notice that some are quite deep.
Part C. Using ML-Based Protein Design Tools
To continue, I choosed to keep working with the Chitodextrinase.
Deep Mutational Scans
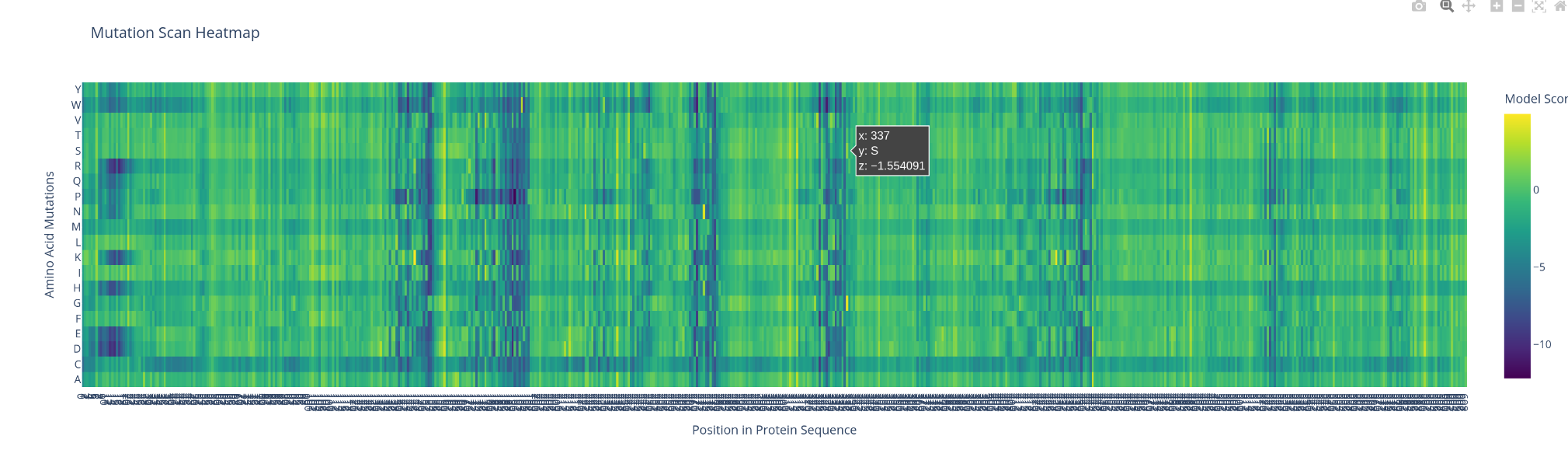
Use ESM2 to generate an unsupervised deep mutational scan of your protein based on language model likelihoods.
Can you explain any particular pattern? (choose a residue and a mutation that stands out)
what catch my sigh first is the apparition of line and cluster of vertical line, the bigger ones are negative impact ones. There is also a horitonzal negative line for multiple amino acids like C W R and H. I could spot few bright spot were the switch might be positive.
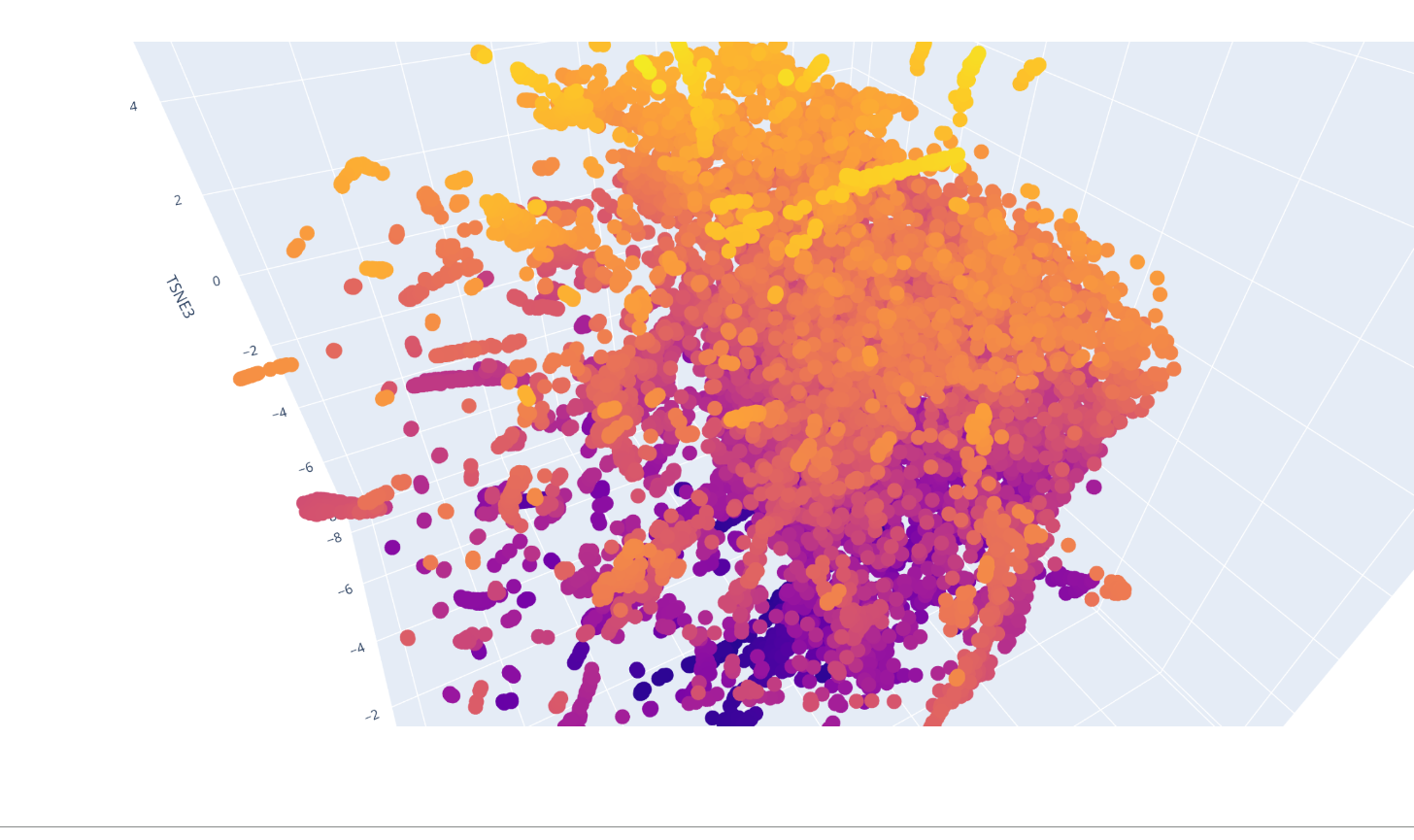
Latent Space Analysis
I was able to make the calculus of the space but not to explore it and understand which were the protein forming clusters.
Folding a protein
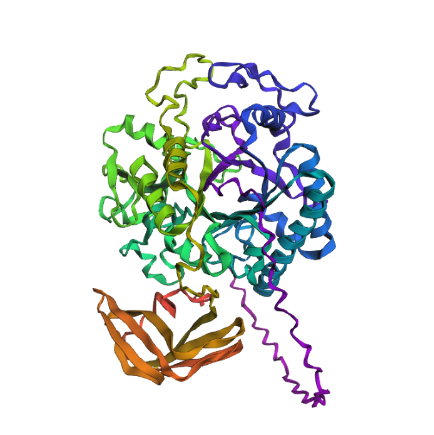
Fold your protein with ESMFold. Do the predicted coordinates match your original structure?
Yes, it does match quite closely to the original structure.
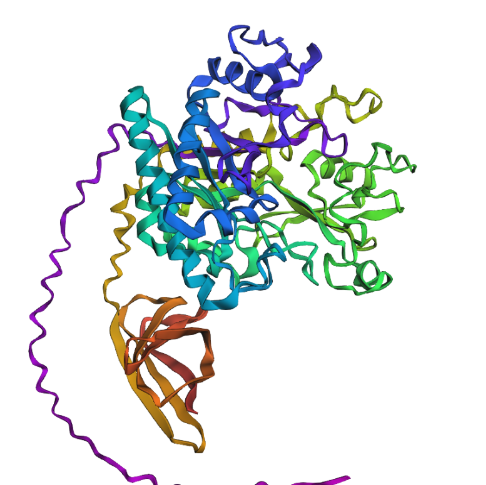
Try changing the sequence, first try some mutations, then large segments. Is your protein structure resilient to mutations?
First I just tried with some mutations: switching 2 D to E and 2 P to A, it gave me thise result :
Which seems to have barely change the estimated structure
Then I changed an relativaly important chunk of amino acid to a random one, from : QIMPTLCMDWAYRYYRGVLPAEKILMGIAYYT to AADRYGVPYMIITWCYQPCFSKSFCEDRGAG in the middle of the protein’s amino acid code. It gave me this result :
Which seems also quite similare to what we would get before execpt few helix turning flat.
From my thin experience I have the impression that this protein structure would be quite resilient.
Inverse-Folding a protein:
Didn’t worked out.
Part D. Group Brainstorm on Bacteriophage Engineering
i was late and didn’t organise properly so I’ve done it alone.
Goal: Increase stability of the MS2 L protein, as this is the most computationally tractable and directly linked to reliable folding and function.
Approach:
Use BLAST + Clustal Omega to identify conserved vs. mutable residues → avoid destabilizing core regions.
Predict structure with ESMFold, then analyze functional/interaction sites with PeSTo.
Apply ProteinMPNN / ESM-IF to redesign sequences that better fit the backbone and improve stability.
Use ESM2/ESM-3 for in silico mutagenesis and ranking of mutations.
Re-validate top variants with ESMFold / AlphaFold to ensure proper folding.
Why this works: Stability is well-modeled by current AI tools, and inverse folding methods are specifically designed to optimize sequences without altering overall structure.
Limits: Predictions may be inaccurate, stability ≠ function, and models don’t capture full cellular context (e.g., chaperones, membrane effects), so experimental validation is essential.
Week 5 - Protein Design Part II
This week we learn how cutting-edge AI and protein language models are used to design functional proteins and peptides “in silico”.
Part A: SOD1 Binder Peptide Design
Your challenge:
Design short peptides that bind mutant SOD1.
Then decide which ones are worth advancing toward therapy.
You will use three models developed in our lab:
PepMLM: target sequence-conditioned peptide generation via masked language modeling
PeptiVerse: therapeutic property prediction
moPPIt: motif-specific multi-objective peptide design using Multi-Objective Guided Discrete Flow Matching (MOG-DFM)
Part 1: Generate Binders with PepMLM
Begin by retrieving the human SOD1 sequence from UniProt (P00441) and introducing the A4V mutation.
Generate four peptides of length 12 amino acids conditioned on the mutant SOD1 sequence.
Binder
Pseudo Perplexity
WRSYAYXLRLGE
16,61684100000000
WRVGAYAARWKK
11,673187000000000
WHYPVAAVAHKK
12,46979000000000
WHVPVAAVAWKE
12,689345000000000
FLYRWLPSRRGG
n/a - known one
for the 1st one X likely means wichever amino acid so I defined one to run the alphafold compute
Part 2: Evaluate Binders with AlphaFold3
Navigate to the AlphaFold Server: alphafoldserver.com
For each peptide, submit the mutant SOD1 sequence followed by the peptide sequence as separate chains to model the protein-peptide complex.
Record the ipTM score and briefly describe where the peptide appears to bind. Does it localize near the N-terminus where A4V sits? Does it engage the β-barrel region or approach the dimer interface? Does it appear surface-bound or partially buried?
In a short paragraph, describe the ipTM values you observe and whether any PepMLM-generated peptide matches or exceeds the known binder.
Binder
ipTM
Descriptions
WRSYAYXLRLGE
0.33
on the side of the B barrel, but on the other side than the one with the V
WRVGAYAARWKK
0.41
also on the barrel but close the N-terminus with the V4, cf image
WHYPVAAVAHKK
0.33
along the barrel but horizontaly and not close the the V4
WHVPVAAVAWKE
0.31
at the other side of the protein than the N-terminus
FLYRWLPSRRGG
0.39
at the other side of the protein than the N-terminus but
Part 3: Evaluate Properties of Generated Peptides in the PeptiVerse
-> To finish, I’ve have a bit accumulated lateness the part on computing took me quite some time to make it work but i’m catching up.
Structural confidence alone is insufficient for therapeutic development. Using PeptiVerse, let’s evaluate the therapeutic properties of your peptide! For each PepMLM-generated peptide:
Paste the peptide sequence.
Paste the A4V mutant SOD1 sequence in the target field.
Check the boxes
Predicted binding affinity
Solubility
Hemolysis probability
Net charge (pH 7)
Molecular weight
Compare these predictions to what you observed structurally with AlphaFold3. In a short paragraph, describe what you see. Do peptides with higher ipTM also show stronger predicted affinity? Are any strong binders predicted to be hemolytic or poorly soluble? Which peptide best balances predicted binding and therapeutic properties?
Choose one peptide you would advance and justify your decision briefly.
Part 4: Generate Optimized Peptides with moPPIt
Now, move from sampling to controlled design. moPPIt uses Multi-Objective Guided Discrete Flow Matching (MOG-DFM) to steer peptide generation toward specific residues and optimize binding and therapeutic properties simultaneously. Unlike PepMLM, which samples plausible binders conditioned on just the target sequence, moPPIt lets you choose where you want to bind and optimize multiple objectives at once.
Open the moPPit Colab linked from the HuggingFace moPPIt model card
Make a copy and switch to a GPU runtime.
In the notebook:
Paste your A4V mutant SOD1 sequence.
Choose specific residue indices on SOD1 that you want your peptide to bind (for example, residues near position 4, the dimer interface, or another surface patch).
Set peptide length to 12 amino acids.
Enable motif and affinity guidance (and solubility/hemolysis guidance if available). Generate peptides.
After generation, briefly describe how these moPPit peptides differ from your PepMLM peptides. How would you evaluate these peptides before advancing them to clinical studies?
Part B: BRD4 Drug Discovery Platform Tutorial
skipped so far.
Part C: Final Project: L-Protein Mutants
cf doc
Week 6 - Genetic Circuits Part I: Assembly Technologies
This week we learn core molecular biology tools and techniques for processing and assembling DNA, including PCR and Gibson Assembly.
Assignment: DNA Assembly
What are some components in the Phusion High-Fidelity PCR Master Mix and what is their purpose?
Phusion High-Fidelity PCR Master Mix usually contains the DNA polymerase, dNTPs, buffer, and MgCl2. Its purpose is to copy DNA accurately and efficiently during PCR.
What are some factors that determine primer annealing temperature during PCR?
Primer annealing temperature depends on the primer melting temperature, primer length, GC content, salt concentration, and additives like DMSO. Primers with higher Tm usually need a higher annealing temperature.
There are two methods from this class that create linear fragments of DNA: PCR, and restriction enzyme digests. Compare and contrast these two methods, both in terms of protocol as well as when one may be preferable to use over the other.
PCR and restriction digest both make linear DNA fragments, but PCR amplifies a chosen region using primers, while restriction enzymes cut DNA at specific recognition sites. PCR is best when you want a precise fragment without depending on restriction sites, and restriction digestion is best when the cut sites already exist in the DNA.
How can you ensure that the DNA sequences that you have digested and PCR-ed will be appropriate for Gibson cloning?
To make DNA suitable for Gibson cloning, the insert and vector need matching overlapping ends, usually about 20–30 bp of homology. The PCR product should also be clean and in the correct orientation and size
How does the plasmid DNA enter the E. coli cells during transformation?
During transformation, plasmid DNA enters E. coli cells through the membrane after the cells are made competent, usually by heat shock or electroporation. The treatment helps the DNA cross into the cell.
Describe another assembly method in detail (such as Golden Gate Assembly)
Explain the other method in 5 - 7 sentences plus diagrams (either handmade or online).
Model this assembly method with Benchling or Asimov Kernel!
Golden Gate Assembly uses type IIS restriction enzymes that cut outside their recognition sites, creating custom overhangs. You design primers so each DNA fragment has recognition sites at the ends and matching overhangs between fragments.In one reaction, the enzymes cut all fragments and DNA ligase joins the compatible overhangs in the correct order.The final product has no restriction sites left, so you can repeat the process for hierarchical assembly. This method is great for assembling 5+ fragments quickly and precisely without needing specific restriction sites.
Assignment: Asimov Kernel
Create a Repository for your work
Create a blank Notebook entry to document the homework and save it to that Repository
Explore the devices in the Bacterial Demos Repo to understand how the parts work together by running the Simulator on various examples, following the instructions for the simulator found in the “Info” panel (click the “i” icon on the right to open the Info panel)
Create a blank Construct and save it to your Repository
Recreate the Repressilator in that empty Construct by using parts from the Characterized Bacterial Parts repository
Search the parts using the Search function in the right menu
Drag and drop the parts into the Construct
Confirm it works as expected by running the Simulator (“play” button) and compare your results with the Repressilator Construct found in the Bacterial Demos repository
Document all of this work in your Notebook entry - you can copy the glyph image and the simulator graphs, and paste them into your Notebook
Build three of your own Constructs using the parts in the Characterized Bacterials Parts Repo
Explain in the Notebook Entry how you think each of the Constructs should function
Run the simulator and share your results in the Notebook Entry
If the results don’t match your expectations, speculate on why and see if you can adjust the simulator settings to get the expected outcome
I’m late and I was finishing the other HM before doing this one so far, i’m doing it soon.
Week 7 — Genetic Circuits Part II: Neuromorphic Circuits
This week covers neuromorphic genetic circuits, showing how engineered gene networks can implement neural-network “perceptron”-like computation and learning.
Assignment Part 1: Intracellular Artificial Neural Networks (IANNs)
What advantages do IANNs have over traditional genetic circuits, whose input/output behaviors are Boolean functions?
IANNs (Intracellular Artificial Neural Networks) can handle analog, continuous signals instead of just on/off Boolean logic.They learn complex patterns through training, while traditional circuits need hand-designed logic gates. IANNs approximate nonlinear functions better than simple threshold-based genetic gates. They can generalize from noisy inputs, unlike rigid Boolean functions.
Describe a useful application for an IANN; include a detailed description of input/output behavior, as well as any limitations an IANN might face to achieve your goal.
I could use an IANN to detect different glucose concentrations and output varying GFP fluorescence levels. Inputs would be glucose sensors (X1, X2) feeding into hidden nodes, with GFP as the output. Low glucose → low GFP, medium → medium GFP, high → high GFP for a smooth analog response.
Limitation: slow response times due to transcription/translation delays, and sensitivity to cellular noise.
Assignment Part 2: Fungal Materials
What are some examples of existing fungal materials and what are they used for? What are their advantages and disadvantages over traditional counterparts?
Mycelium pannels and tilling for home interior, fungal leather for fashion (replaces animal leather), mycelium packaging (replaces styrofoam). Mogu and Ecovative are actual companies selling those products.
Advantages: biodegradable, grown from waste, carbon-negative; Disadvantages: different production pipeline, consumers rejection/fear/not used to it, problematics of producting out of living organisms.
What might you want to genetically engineer fungi to do and why? What are the advantages of doing synthetic biology in fungi as opposed to bacteria?
I would engineer fungi to capture airborne pollutants even after the mycelium composite dries and dies. Many pollutants like VOCs (volatile organic compounds) and heavy metals remain in the air long-term, so I’d add genes for metallothioneins (metal-binding proteins) and enzymes like laccases that degrade pollutants. The dead mycelium would act like a passive filter that keeps working after growth stops.
I’d also make fungi more resistant to mechanical stress, water, and flame for stronger construction materials. For stress resistance, add genes for chitin synthase or hydrophobins to create a tougher outer cell wall. For water resistance, engineer hydrophobin overproduction to make a waterproof surface layer. For flame resistance, add phosphate accumulation genes could enhance the natural fire retardant capacities of the overgrowth outer layer.
Advantages of synthetic biology in fungi vs bacteria:
Better protein secretion: Fungi naturally secrete large amounts of enzymes and structural proteins into the environment
Eukaryotic modifications: Fungi properly fold and modify complex proteins like hydrophobins that need glycosylation
Scalable material production: Fungal mycelium grows on cheap waste substrates and forms bulk materials directly
Assignment Part 3: First DNA Twist Order
Review the Individual Final Project documentation guidelines.
Submit this Google Form with your draft Aim 1, final project summary, HTGAA industry council selections, and shared folder for DNA designs. DUE MARCH 20 FOR MIT/HARVARD/WELLESLEY STUDENTS
Review Part 3: DNA Design Challenge of the week 2 homework. Design at least 1 insert sequence and place it into the Benchling/Kernel/Other folder you shared in the Google Form above. Document the backbone vector it will be synthesized in on your website.
Same as for the 6th, all the AI and comptute part took me a lot of time and therefore i’m quite late but I’ll catch up quick