Week 4 - Protein Design
This week focuses on how sequence, structure, and energetics can be modeled and manipulated to create or optimize proteins with specified functions.

Part A. Conceptual Questions
Answer any NINE of the following questions from Shuguang Zhang:
- How many molecules of amino acids do you take with a piece of 500 grams of meat? (on average an amino acid is ~100 Daltons)
This paper : “Samicho, Z. , Mutalib, S. R. A. and Abdullah, N. (2013) Amino acid composition of droughtmaster beef at various beef cuts. Agricultural Sciences, 4, 61-64. doi: 10.4236/as.2013.45B012. " We get the information that the different cut of beef meat contains from 10 to 20 grams of amino acis per 100g of meat.
From that we can approximate the number of molecules of amino acids in 500 grams of meat.
Knowing that on average: There is ~15g of amino acid in 100g of meat, amino acid is ~100 Daltons, 1 Dalton is 1,67x10e-24
15 x 5 = 75 (g of amnio acids in 500 g of meat)
75 / 100 x 1,67x10e-24 = 4,49x10e23
So there is approximately 4,5x10e23 amino acid in 500g of meats
- Why do humans eat beef but do not become a cow, eat fish but do not become fish?
We don’t became the animals (or any other aliments) we eat because the proces of digestion break down the protein, cells, fat and other compound into amino acids that are absorbed and then used to create our human cells.
- Why are there only 20 natural amino acids?
There is now 22 and there isn’t an absolute answer from the knowledge we have today. They have been know to appeared a long time ago (The 20 standard amino acids encoded by the Genetic Code were adopted during the RNA World, around 4 billion years ago) and life keep this configuration even if there little expection here and there (Pyrolysine is only found in some extremophiles) and human are know to use 21 since Pyrolysine was discovered in 2002. Some clues I understood were the limited genetic space to encode and decode those amino acids, that this number is likely a large enough panel to have a diversity of properties and possibilities but small enough to be practical to create. And the cost of creating the living pathway to create and use new ones would be really important for the benefice it would provide.
- Can you make other non-natural amino acids? Design some new amino acids.
yes definitly. an amino acid is a compound that contain both amino and carboxylic acid functional groups. There for by adding a C between the CR and COOH I just created a new amino acids that is non-natural because all the natural amino acid are alpha ones so like so: H2N - CR- COOH (R is the amino acid side chain)
- Where did amino acids come from before enzymes that make them, and before life started?
Amino acids likely formed naturally on early Earth before life existed through chemical reactions like: Primitive atmosphere reactions, Gases like methane, ammonia, water vapor, and hydrogen were hit by lightning or UV light, forming amino acids. This was shown in the Miller-Urey experiment. Deep-sea vents: Hot underwater vents provided energy and minerals that helped simple molecules combine into amino acids. From space: Amino acids have been found in meteorites, meaning they can form in space and may have been delivered to Earth. So, amino acids did not need enzymes or life to form they came first through natural chemistry, and life later appeared with a way to use them.
- If you make an α-helix using D-amino acids, what handedness (right or left) would you expect?
In natural proteins, α-helices are formed almost exclusively from L-amino acids. The typical α-helix is a right-handed helix, and this geometry is favored by L-amino acids. If you built a helix from D-amino acids, it would form a left-handed helix instead (mirror image). L and D for amino acids refer to the spatial arrangement (chirality) of the atoms around the α carbon. This shift between right-handed helif to a left-handed one will make the properties of that protein.
- Can you discover additional helices in proteins?
Yes, proteins already have multiple helix types beyond α-helices and in principles new ones can be discovered or designed using physical tools or computational tools like alpha fold to analyse the structure of the protein and find uncomun ones but they’re usually variants rather than entirely new structural families.
- Why are most molecular helices right-handed?
Mort molecular helices are right-handed because the amino acids used by life are L-amino acids, and their geometry naturally packs into right-handed helices with the lowest energy. It’s likely a “choice” made by life at some point, that became the norm.
- Why do β-sheets tend to aggregate?
β-sheets are protein structures where strands lie side by side and are held together by hydrogen bonds, They tend to aggregate because their edges have exposed hydrogen bonds that easily attach to other strands, and their flat shape plus hydrophobic side chains makes them stack and stick together easily.
- What is the driving force for β-sheet aggregation?
The driving force for their aggregation is mainly the formation of more hydrogen bonds between strands and the hydrophobic effect, where nonpolar side chains cluster together to avoid water, making the structure more stable.
10 and 11 skiped
Part B: Protein Analysis and Visualization
- Briefly describe the protein you selected and why you selected it.
I’ve chosen Chitodextrinase, it’s an enzyme that permit the degradation of chitin which a polysaccaride that form structure and rigidity of living orgnisme. It’s found mostly in mushrooms and insect, there for chitodextrinase is produced by animal eating insect or plants to protect themselve from fungi. I found it intersting because i worked on mycelium based composite and chitin is on of the main strucuring compond allowing those object to be resistant and light while being biodegradable. So i thought it would be intersting to understand better the process of it’s decomposition by living organisme.
- dentify the amino acid sequence of your protein.
I found 2 differents relevant possibilities :
Uniprot reviewed : https://www.uniprot.org/uniprotkb/P96156/entry - 5/5 score of annotation - 1046 amino acids - Vibrio furnissii, une bactérie
unreviewed : https://www.uniprot.org/uniprotkb/F8UNI5/entry - 1/5 score of annotation - 611 amino acids - modèle 3D de la proteine - Clostridium perfringens, une bacterie
I choosed the 2nd one because of the accurate 3D modèles which is really relevant for this exercice.
Here it is : Chitodextrinase - Clostridium perfringens
MKNSKLKQILITFLTITLTSSYLVTSSNPIETKAKEKFKTTKIKNSSELNRKLVGYFPEWAYSSEAQGYFNVTDLQWDSLTHIQYSFAMVDPSTNKITLSNKHAAIEEDFSEFDLNYNGKKIELDPSLPYKGHFNVLQTMKKNYPDVSLLISVGGWTGTRCFYTMIDTDNRINTFADSCVDFIRKYGFDGVDIDFEYPSSTSQSGNPDDFDLSEPRRTKLNERYNILIKTLREKIDMASKEDGKEYLLTAAVTASPWVLGGISDNTYAKYLDFLSIMSYDYHGGWNEYVEHLAGIYPNKEDIETVTQIMPTLCMDWAYRYYRGVLPAEKILMGIPYYTRGWENVQGGINGLHGSSKTPASGKYNILGDDLNNDGVLEPAGANPLWHVLNLMEQDPNLKVYWDEISKVPYVWQNDKKVFVSFENEKSIDARLEYIQNKNLGGALIWVMNGDYGLNPNYVEGSNKINEGKYTFGDTLTKRLSQGLKKMGVCNKTPDDLNISLEPINVDVKFNGKYDHPNYTYSIDITNYTDKEIKGGWNVSFDLPKSAVFKSSWGGTYSVTDNGDFNTITLTSGAWQNIAPNSTITVQGMIGLCFSGIRNVTFNGMNPIGNDK
- How long is it? What is the most frequent amino acid?
The lenght of the sequence is 611, the most frequent amino acid is Asparagine (N) with 51 apeareance.
- How many protein sequence homologs are there for your protein?
yes there are many homologues from differents many differents bacterias
- Does your protein belong to any protein family?
This protein belongs to the glycoside hydrolase family 18 (GH18) and contains a conserved chitinase catalytic domain along with carbohydrate-binding modules (CBMs), consistent with chitin-degrading enzymes.
- Identify the structure page of your protein in RCBS
- When was the structure solved? Is it a good quality structure? Good quality structure is the one with good resolution. Smaller the better (Resolution: 2.70 Å)
it was solve in 2001 and in 2023 for the specific chitodextrinase from the Clostridium perfringens. the methodology used is X-RAY DIFFRACTION 1.5 Å so yes the quality structure is good. - https://www.rcsb.org/structure/8OTB
- Are there any other molecules in the solved structure apart from protein?
No it’s just the protein. But there is other solved structure with the protein and other molecules like : inhibitor bisdionin C, chitin, chitosan for example.
- Does your protein belong to any structure classification family?
it seems not to belong to any structure classification family because i don’t have any return from SCOP, on RCSB PDB i have those information : Global Symmetry : Asymmetric - C1 Global Stoichiometry: Monomer - A1
- Open the structure of your protein in any 3D molecule visualization software:
1st Visualisation
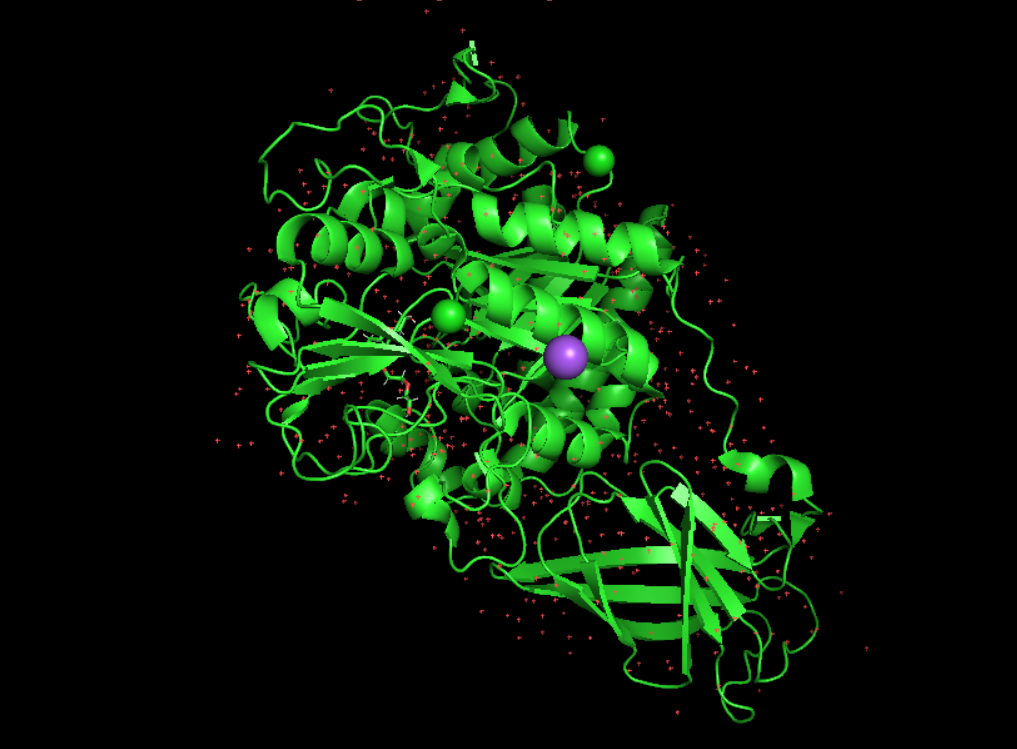
Visualisation as cartoon, riboon and ball and stick
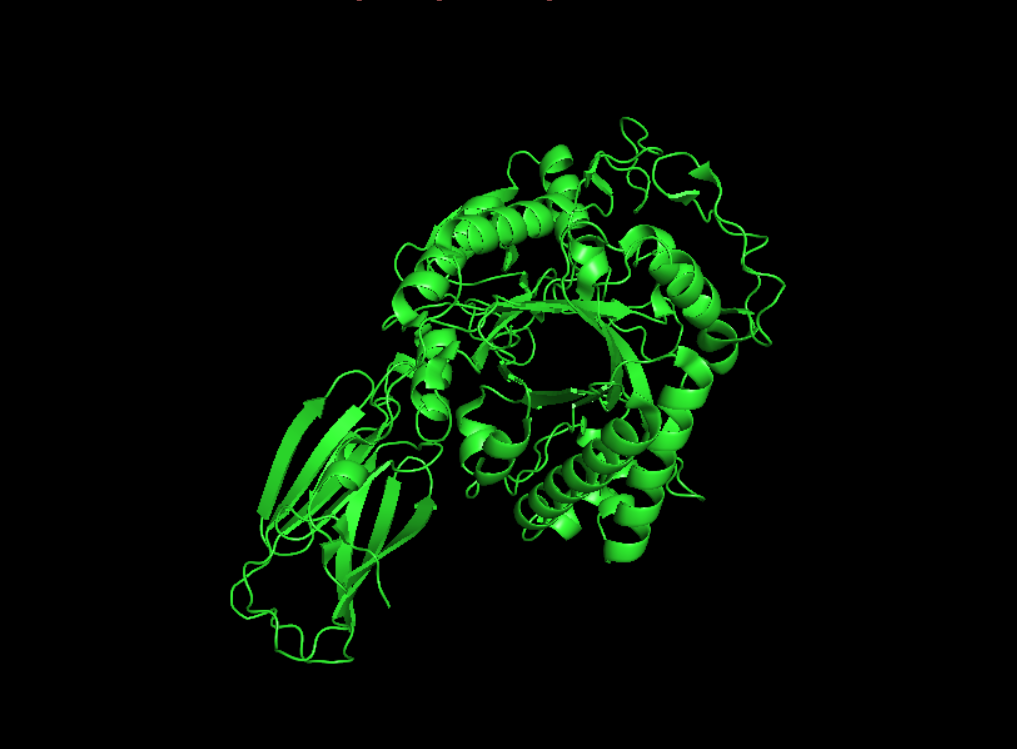
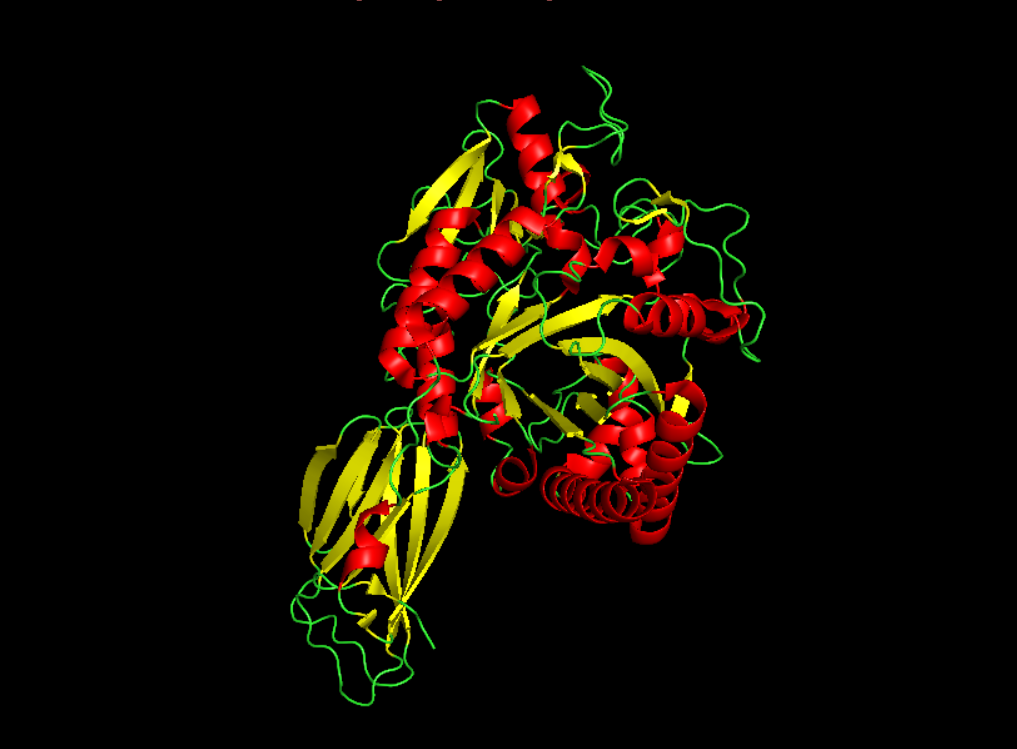
- Color the protein by secondary structure. Does it have more helices or sheets?
It seems to have more few more helices than sheets.
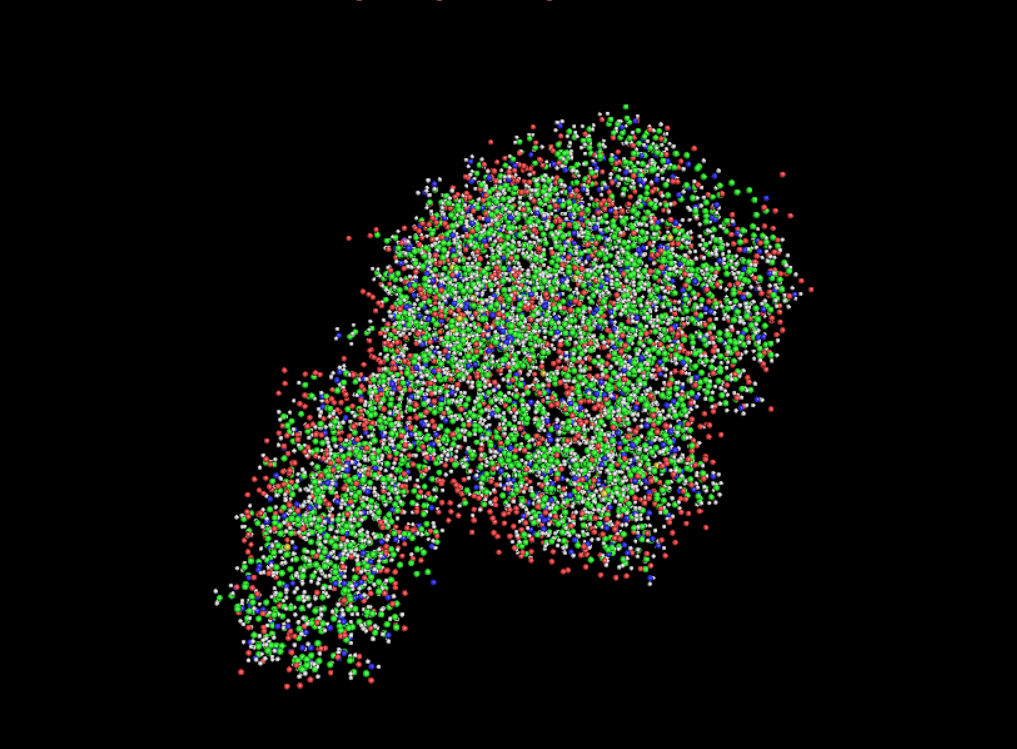
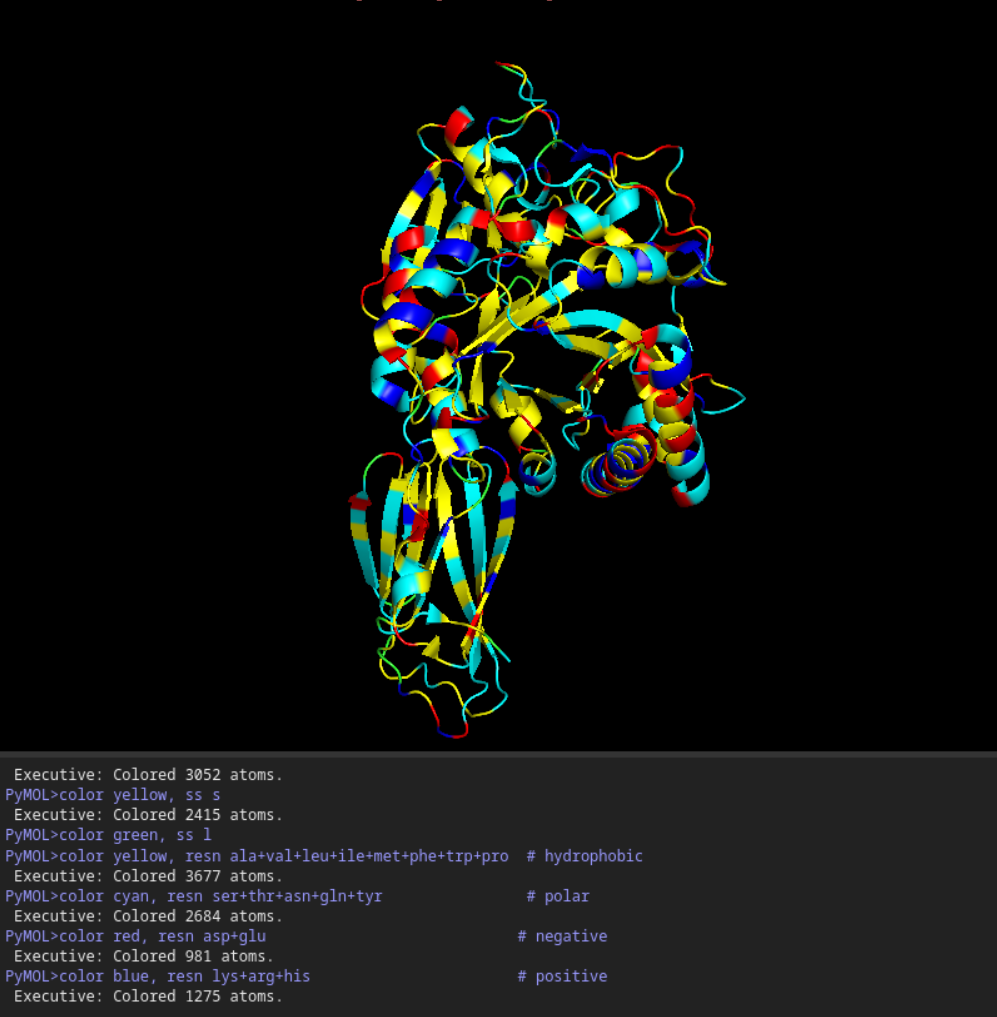
- Color the protein by residue type. What can you tell about the distribution of hydrophobic vs hydrophilic residues?
Once again it is quite balanced but it seems to have more hydrophilic residues.
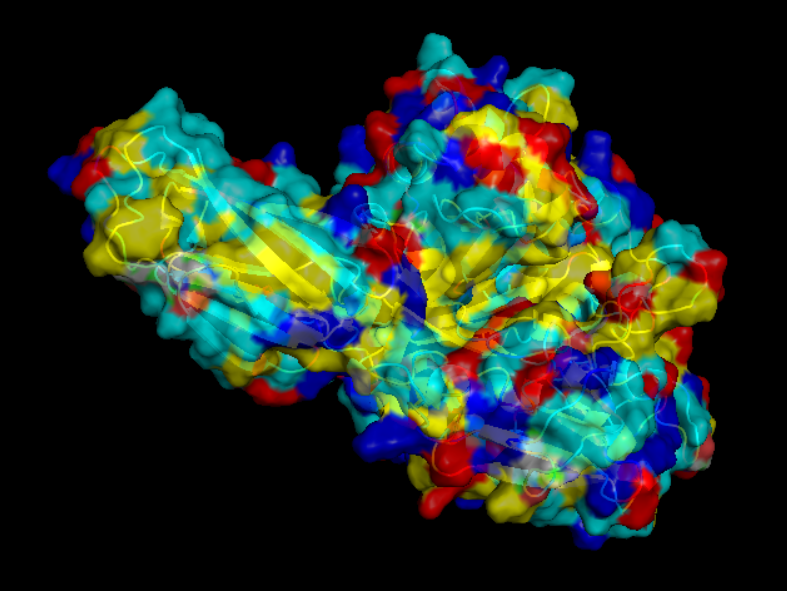
- Visualize the surface of the protein. Does it have any “holes” (aka binding pockets)?
yes it look like it does have binding pockets, I don’t know if it is many. I can notice that some are quite deep.
Part C. Using ML-Based Protein Design Tools
To continue, I choosed to keep working with the Chitodextrinase.
- Deep Mutational Scans
- Use ESM2 to generate an unsupervised deep mutational scan of your protein based on language model likelihoods.
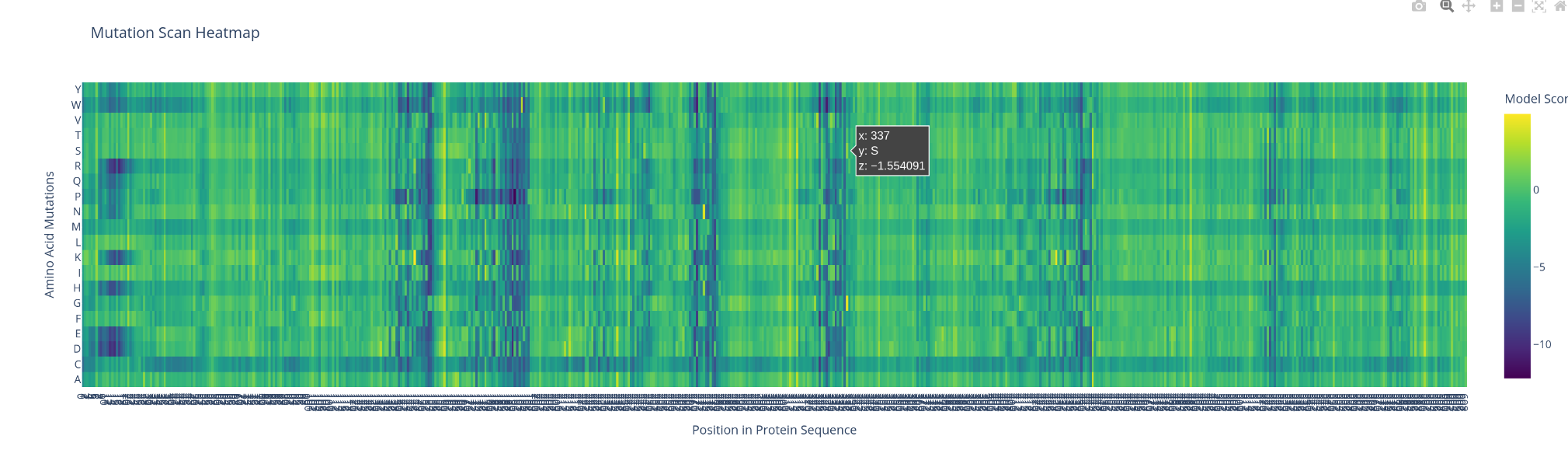
- Can you explain any particular pattern? (choose a residue and a mutation that stands out)
what catch my sigh first is the apparition of line and cluster of vertical line, the bigger ones are negative impact ones. There is also a horitonzal negative line for multiple amino acids like C W R and H. I could spot few bright spot were the switch might be positive.
- Latent Space Analysis
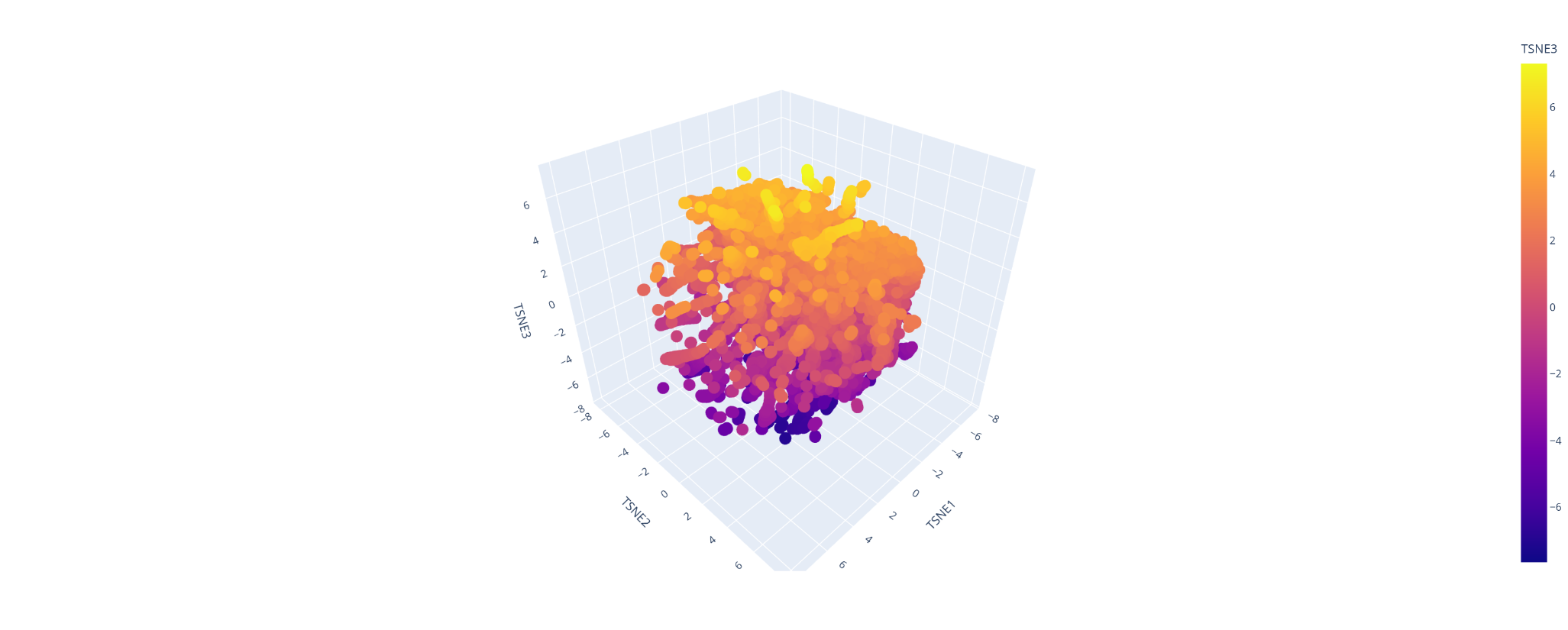
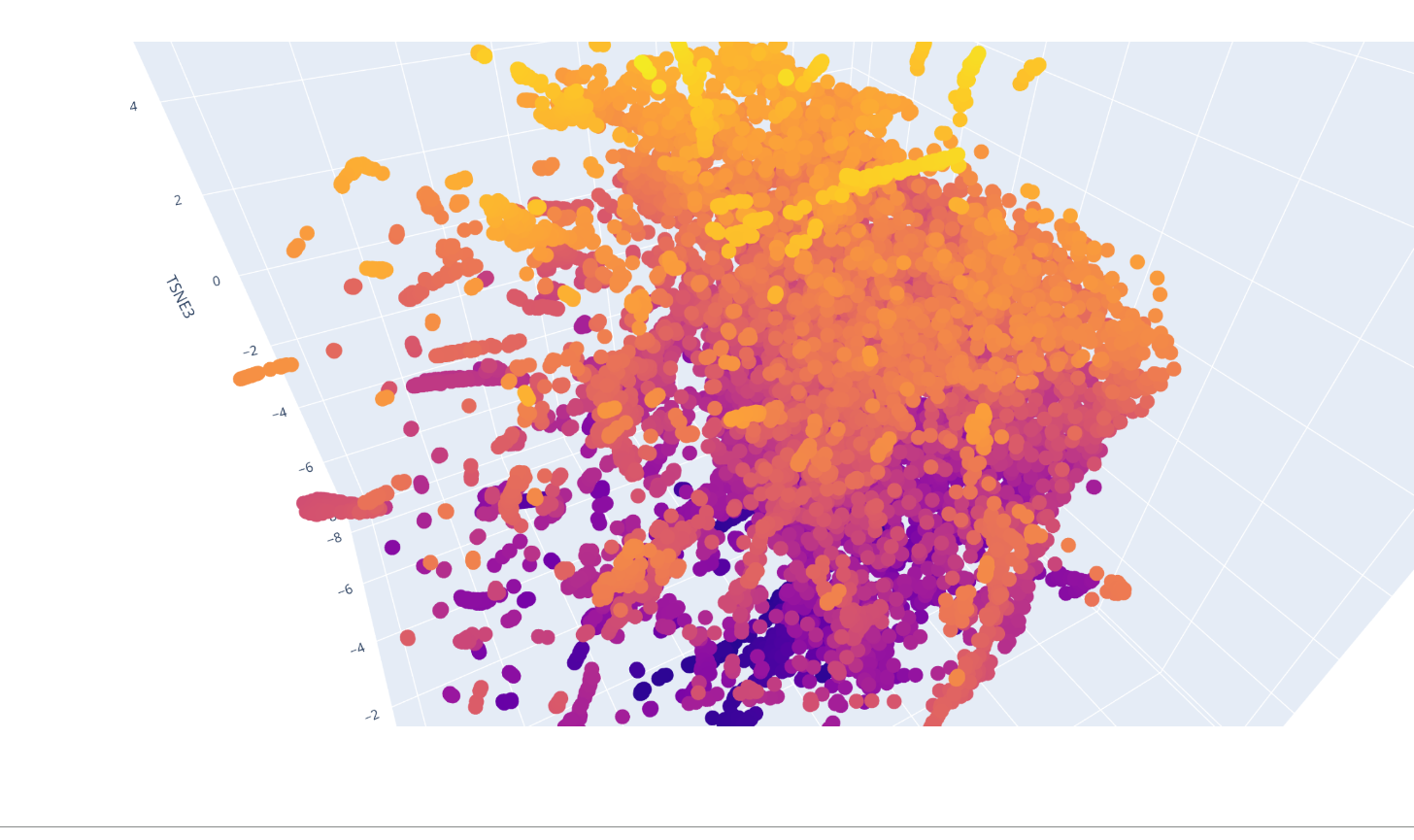
I was able to make the calculus of the space but not to explore it and understand which were the protein forming clusters.
- Folding a protein
- Fold your protein with ESMFold. Do the predicted coordinates match your original structure?
Yes, it does match quite closely to the original structure.
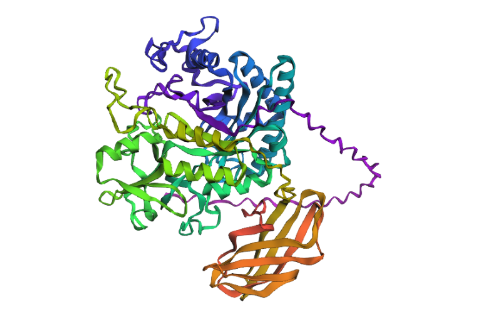
- Try changing the sequence, first try some mutations, then large segments. Is your protein structure resilient to mutations?
First I just tried with some mutations: switching 2 D to E and 2 P to A, it gave me thise result :
Which seems to have barely change the estimated structure
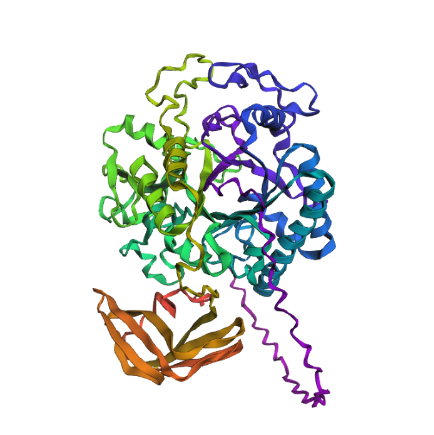
Then I changed an relativaly important chunk of amino acid to a random one, from : QIMPTLCMDWAYRYYRGVLPAEKILMGIAYYT to AADRYGVPYMIITWCYQPCFSKSFCEDRGAG in the middle of the protein’s amino acid code. It gave me this result :
Which seems also quite similare to what we would get before execpt few helix turning flat.
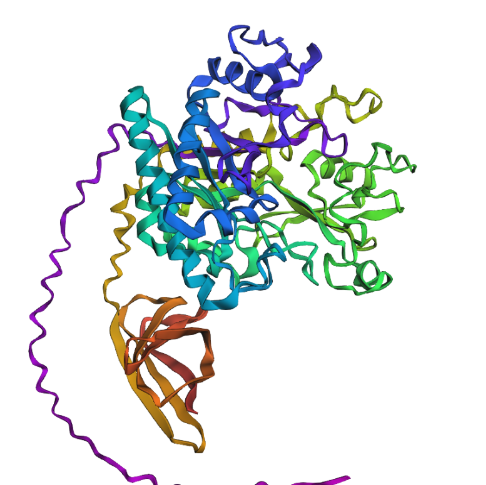
From my thin experience I have the impression that this protein structure would be quite resilient.
- Inverse-Folding a protein:
Didn’t worked out.
Part D. Group Brainstorm on Bacteriophage Engineering
i was late and didn’t organise properly so I’ve done it alone.
- Goal: Increase stability of the MS2 L protein, as this is the most computationally tractable and directly linked to reliable folding and function.
- Approach:
- Use BLAST + Clustal Omega to identify conserved vs. mutable residues → avoid destabilizing core regions.
- Predict structure with ESMFold, then analyze functional/interaction sites with PeSTo.
- Apply ProteinMPNN / ESM-IF to redesign sequences that better fit the backbone and improve stability.
- Use ESM2/ESM-3 for in silico mutagenesis and ranking of mutations.
- Re-validate top variants with ESMFold / AlphaFold to ensure proper folding.
- Why this works: Stability is well-modeled by current AI tools, and inverse folding methods are specifically designed to optimize sequences without altering overall structure.
- Limits: Predictions may be inaccurate, stability ≠ function, and models don’t capture full cellular context (e.g., chaperones, membrane effects), so experimental validation is essential.