Week 2 HW: DNA, Read, Write and Edit
PART 1
Benchling & In-silico Gel Art
- Make a free account at benchlig.com
- Import the Lambda DNA
Genome sequence of the lambda phage at the NCBI database.
The screenshot below is the linear map of lambda DNA (LAMCG) from Benchling, displaying all enzymes with their cut sites on the DNA.
I have used the accession number to import the DNA.
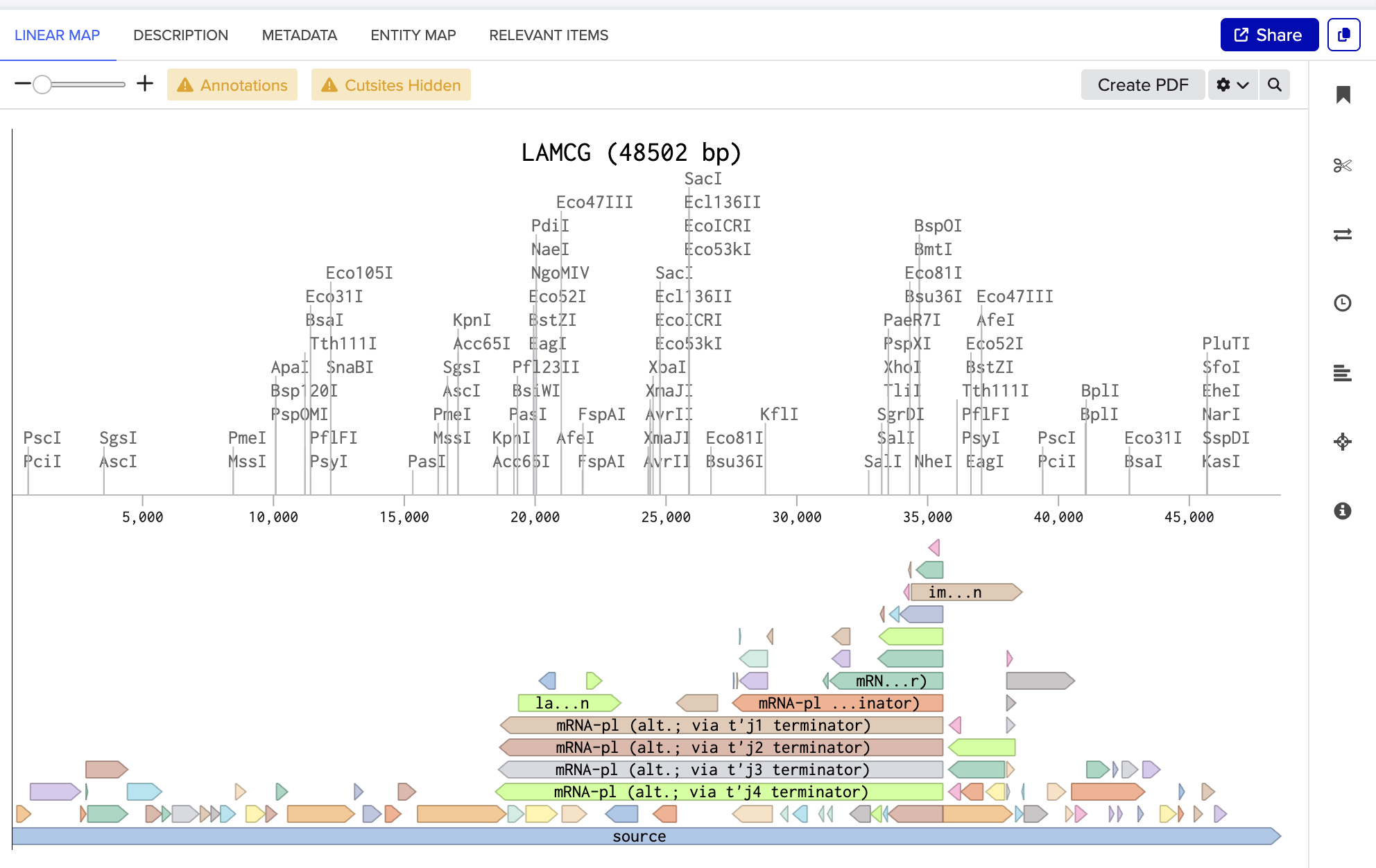
- Simulate Restriction Enzyme Digestion with the following enzymes:
- EcoRI, HindIII, BamHI, KpnI, EcoRV, SacI, SalI
I followed the steps below to simulate restriction enzyme digestion in Benchling:
As shown in figure (B), a new window appears, which allows naming the digest in the text box pointed to by the red arrow. Hit “Save”, and repeat the process for each enzyme digestion.
| (A) Select enzyme | (B) Save digest |
|---|---|
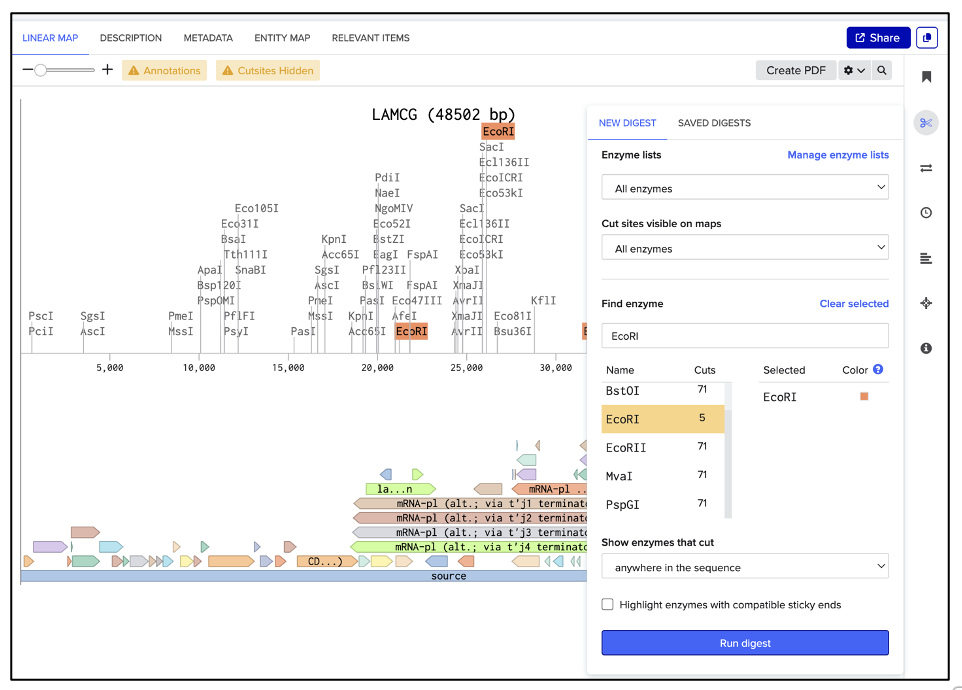
| 
|
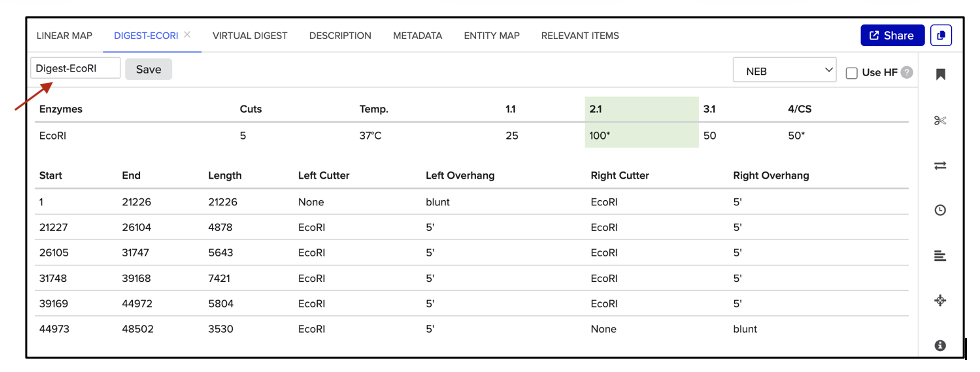
Go to VIRTUAL DIGEST, then select a DNA ladder and previously saved enzyme digestions to display as shown in figure (C).
| (C) Simulated enzyme digestion of lambda DNA |
|---|

|
- Create a design/pattern artwork.
Perform a virtual digest in combinations of single, double, and triple enzyme digestions of lambda DNA as shown in figure (D).
| (D) Simulated enzyme digestion of lambda DNA in a pattern |
|---|
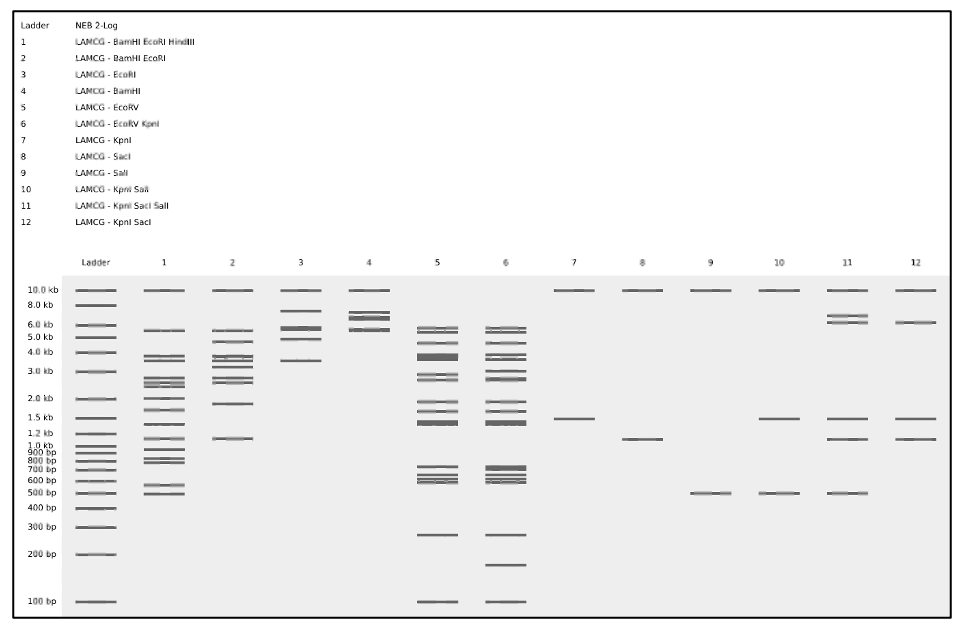
|
PART 2
GelArt - Restriction Digests and Gel Electrophoresis
This was run virtually; see figures, (C) and (D) above.
PART 3
DNA Design Challenge
3.1. Choose your protein
- Which protein have you chosen?
I’ve chosen Histidine Ammonia-Lyase (HAL) enzyme. HAL converts L-histidine to trans-urocanic acid (trans-UCA) and ammonia.
- Why?
Found in human skin, the enzyme, HAL, catalyzes trans-urocanic acid (trans-UCA) formation, which has skin moisturizing properties and is implicated in skin disease management. Topical application of a cis-urocanic acid (UV-induced isomeric form) in combination with orally administered histidine has been shown to be effective in the management of atopic dermatitis (AD), the most common form of eczema (Peltonen et al 2014).
Trans-UCA is naturally liberated from histidine-rich filaggrin monomers of a major epidermis protein in skin. As part of the skin’s natural moisturizing factor, trans-UCA provides important functions in maintaining the skin’s hydration, pH balance, epidermal barrier integrity, and skin’s microbial community balance (Debinska 2021, Kim and Lim 2021).
AD is caused by dysfunction in the epithelial barrier and the overactivation of the immune system. Pathology of the disease is viewed as beginning with a dysfunction in the epithelial barrier. There is no cure, but management includes the application of daily moisturizers and corticosteroids to improve the skin barrier function. In severe cases, biologics-based therapies such as monoclonal antibodies targeting cytokine signaling pathways are available (Debinska 2021).
As a synthetic biology application, I have designed a microbial expression system to produce trans-UCA from yeast for clinical use as an active ingredient in a lotion. HAL homologs are found in microbes, such as bacteria and certain groups of fungi. But common yeast, Saccharomyces cerevisiae, does not have a HAL homolog. I chose the yeast as the host organism to produce trans-UCA because yeast has several advantages as an expression system: the GRAS status and lack of endotoxin production. The Pseudomonas putida hutH gene, which encodes for HAL, is the microbial source for trans-UCA production (Hernandez and Phillips 1993).
References, Click to Expand
Peltonen, J. M., L. Pylkkanen, C. T. Jansen, I. Volanen, T. Lehtinen, J. K. Laihia, and L. Leino. 2014. Three randomized phase 1/IIa trials if 5 % cis-urocanic acid emulsion cream in healthy adult subjects and in patients with atopic dermatitis. Acta Derm Venereol.
Debinska, A. 2021. New treatments for atopic dermatitis targeting skin barrier repair via the regulation of FLG expression. J. of Clinical Medicine.
Kim, Y. and KM Lim. 2021. Skin barrier dysfunction and filaggrin. Arch. Pharm. Res.
Peltonen, J. M., L. Pylkkanen, C. T. Jansen, I. Volanen, T. Lehtinen, J. K. Laihia, and L. Leino. 2014. Three randomized phase 1/IIa trials if 5 % cis-urocanic acid emulsion cream in healthy adult subjects and in patients with atopic dermatitis. Acta Derm Venereol.
Hernandez D. and A. T. Phillips. 1993. Purification and characterization of Pseudomonas putida histidine ammonia-lyase expressed in Escherichia coli. Protein Expression and Purification.
I’ve used Google, ChatGPT, and Claude searches.
Prompts:
1. Can you research proteins that can be developed as therapies for skin diseases such as eczema?
2. Can you research the development of the production of cis-urocanic acid in a biotechnologically relevant host system? What genes are needed to produce cis-urocanic acid?
3. Can you compare the stability of trans-UCA and cis-UCA? Can you research what the best ammonia removal strategies are in a cell-free production system that primarily forms trans-UCA?
4. Can you research what the possible formulation components should be for trans-UCA as the active pharmaceutical ingredient applied topically for atopic dermatitis therapy? What buffer system, cryoprotectant, surfactant, and excipients should be used for achieving maximum effectiveness in treatment and shelf-life stability?
- Obtain the protein sequence from UniProt:
I’ve retrieved the amino acid sequence of the histidine ammonia lyase from Pseudomonas putida from the UniProt database.
UniProt accession: P21310
MTELTLKPGTLTLAQLRAIHAAPVRLQLDASAAPAIDASVACVEQIIAEDRTAYGINTGFGLLASTRIASHDLENLQRSLVLSHAAGIGAPLDDDLVRLIMVLKINSLSRGFSGIRRKVIDALIALVNAEVYPHIPLKGSVGASGDLAPLAHMSLVLLGEGKARYKGQWLSATEALAVAGLEPLTLAAKEGLALLNGTQASTAYALRGLFYAEDLYAAAIACGGLSVEAVLGSRSPFDARIHEARGQRGQIDTAACFRDLLGDSSEVSLSHKNCDKVQDPYSLRCQPQVMGACLTQLRQAAEVLGIEANAVSDNPLVFAAEGDVISGGNFHAEPVAMAADNLALAIAEIGSLSERRISLMMDKHMSQLPPFLVENGGVNSGFMIAQVTAAALASENKALSHPHSVDSLPTSANQEDHVSMAPAAGKRLWEMAENTRGVLAIEWLGACQGLDLRKGLKTSAKLEKARQALRSEVAHYDRDRFFAPDIEKAVELLAKGSLTGLLPAGVLPSL
3.2. Reverse Translate: Protein (amino acid) sequence to DNA (nucleotide) sequence
- The original sequence of the histidine ammonia lyase from the Pseudomonas putida genome
The original DNA sequence of the Pseudomonas putida histidine ammonia lyase can be retrieved from the NCBI database.
Accession: M35140.1 GI: 151273
2,249 bp
The coding sequence of the hutH gene starts at position 316 and ends at position 1848, with the initiation codon GTG for methionine.
GTGACCGAACTGACCCTGAAACCGGGCACCCTGACCCTGGCGCAGCTGCGCGCGATTCATGCGGCGCCGGTGCGCCTGCAGCTGGATGCGAGCGCGGCGCCGGCGATTGATGCGAGCGTGGCGTGCGTGGAACAGATTATTGCGGAAGATCGCACCGCGTATGGCATTAACACCGGCTTTGGCCTGCTGGCGAGCACCCGCATTGCGAGCCATGATCTGGAAAACCTGCAGCGCAGCCTGGTGCTGAGCCATGCGGCGGGCATTGGCGCGCCGCTGGATGATGATCTGGTGCGCCTGATTATGGTGCTGAAAATTAACAGCCTGAGCCGCGGCTTTAGCGGCATTCGCCGCAAAGTGATTGATGCGCTGATTGCGCTGGTGAACGCGGAAGTGTATCCGCATATTCCGCTGAAAGGCAGCGTGGGCGCGAGCGGCGATCTGGCGCCGCTGGCGCATATGAGCCTGGTGCTGCTGGGCGAAGGCAAAGCGCGCTATAAAGGCCAGTGGCTGAGCGCGACCGAAGCGCTGGCGGTGGCGGGCCTGGAACCGCTGACCCTGGCGGCGAAAGAAGGCCTGGCGCTGCTGAACGGCACCCAGGCGAGCACCGCGTATGCGCTGCGCGGCCTGTTTTATGCGGAAGATCTGTATGCGGCGGCGATTGCGTGCGGCGGCCTGAGCGTGGAAGCGGTGCTGGGCAGCCGCAGCCCGTTTGATGCGCGCATTCATGAAGCGCGCGGCCAGCGCGGCCAGATTGATACCGCGGCGTGCTTTCGCGATCTGCTGGGCGATAGCAGCGAAGTGAGCCTGAGCCATAAAAACTGCGATAAAGTGCAGGATCCGTATAGCCTGCGCTGCCAGCCGCAGGTGATGGGCGCGTGCCTGACCCAGCTGCGCCAGGCGGCGGAAGTGCTGGGCATTGAAGCGAACGCGGTGAGCGATAACCCGCTGGTGTTTGCGGCGGAAGGCGATGTGATTAGCGGCGGCAACTTTCATGCGGAACCGGTGGCGATGGCGGCGGATAACCTGGCGCTGGCGATTGCGGAAATTGGCAGCCTGAGCGAACGCCGCATTAGCCTGATGATGGATAAACATATGAGCCAGCTGCCGCCGTTTCTGGTGGAAAACGGCGGCGTGAACAGCGGCTTTATGATTGCGCAGGTGACCGCGGCGGCGCTGGCGAGCGAAAACAAAGCGCTGAGCCATCCGCATAGCGTGGATAGCCTGCCGACCAGCGCGAACCAGGAAGATCATGTGAGCATGGCGCCGGCGGCGGGCAAACGCCTGTGGGAAATGGCGGAAAACACCCGCGGCGTGCTGGCGATTGAATGGCTGGGCGCGTGCCAGGGCCTGGATCTGCGCAAAGGCCTGAAAACCAGCGCGAAACTGGAAAAAGCGCGCCAGGCGCTGCGCAGCGAAGTGGCGCATTATGATCGCGATCGCTTTTTTGCGCCGGATATTGAAAAAGCGGTGGAACTGCTGGCGAAAGGCAGCCTGACCGGCCTGCTGCCGGCGGGCGTGCTGCCGAGCCTGTAA
- Using Reverse Translation Tools:
I’ve used a reverse translation tool, BCCM Gene Corner Sequence Manipulation Suite, to translate a likely DNA sequence of the P. putida histidine ammonia lyase.
The reverse translation tool generated the same DNA coding sequence for the protein.
atgaccgaactgaccctgaaaccgggcaccctgaccctggcgcagctgcgcgcgattcatgcggcgccggtgcgcctgcagctggatgcgagcgcggcgccggcgattgatgcgagcgtggcgtgcgtggaacagattattgcggaagatcgcaccgcgtatggcattaacaccggctttggcctgctggcgagcacccgcattgcgagccatgatctggaaaacctgcagcgcagcctggtgctgagccatgcggcgggcattggcgcgccgctggatgatgatctggtgcgcctgattatggtgctgaaaattaacagcctgagccgcggctttagcggcattcgccgcaaagtgattgatgcgctgattgcgctggtgaacgcggaagtgtatccgcatattccgctgaaaggcagcgtgggcgcgagcggcgatctggcgccgctggcgcatatgagcctggtgctgctgggcgaaggcaaagcgcgctataaaggccagtggctgagcgcgaccgaagcgctggcggtggcgggcctggaaccgctgaccctggcggcgaaagaaggcctggcgctgctgaacggcacccaggcgagcaccgcgtatgcgctgcgcggcctgttttatgcggaagatctgtatgcggcggcgattgcgtgcggcggcctgagcgtggaagcggtgctgggcagccgcagcccgtttgatgcgcgcattcatgaagcgcgcggccagcgcggccagattgataccgcggcgtgctttcgcgatctgctgggcgatagcagcgaagtgagcctgagccataaaaactgcgataaagtgcaggatccgtatagcctgcgctgccagccgcaggtgatgggcgcgtgcctgacccagctgcgccaggcggcggaagtgctgggcattgaagcgaacgcggtgagcgataacccgctggtgtttgcggcggaaggcgatgtgattagcggcggcaactttcatgcggaaccggtggcgatggcggcggataacctggcgctggcgattgcggaaattggcagcctgagcgaacgccgcattagcctgatgatggataaacatatgagccagctgccgccgtttctggtggaaaacggcggcgtgaacagcggctttatgattgcgcaggtgaccgcggcggcgctggcgagcgaaaacaaagcgctgagccatccgcatagcgtggatagcctgccgaccagcgcgaaccaggaagatcatgtgagcatggcgccggcggcgggcaaacgcctgtgggaaatggcggaaaacacccgcggcgtgctggcgattgaatggctgggcgcgtgccagggcctggatctgcgcaaaggcctgaaaaccagcgcgaaactggaaaaagcgcgccaggcgctgcgcagcgaagtggcgcattatgatcgcgatcgcttttttgcgccggatattgaaaaagcggtggaactgctggcgaaaggcagcctgaccggcctgctgccggcgggcgtgctgccgagcctgtaa
Here, I’ve translated the input DNA sequence into an amino acid sequence by Expasy Translate, validating the DNA sequence by in-silico reverse translation.
3.3. Codon optimization
Codon optimize your sequence.
Describe why you need to optimize codon usage.
Which organism have you chosen to optimize the codon sequence for, and why?
The efficiency of protein production is strongly influenced by the host organism’s codon usage. Preferences for a codon recognition sequence and the rate of synthesis for amino acid carriers, transfer RNA, are expected to differ in each lineage of living systems. To take care of the codon usage barrier, one could take this into account in the design, synthesizing the DNA code and avoiding codon usage limitations.
I am using a yeast species as the host organism for expressing the hutH gene for histidine ammonia lyase production. I decided on the yeast expression system due to its well-developed protein expression systems, which are robust and have a “generally safe” status. The yeast-derived protein products would be easily accepted for clinical use since they have the GRAS status. The histidine ammonia lyase is the catalyst for making trans-uroconic acid for skin therapeutic use.
The hutH gene will be sourced from the bacterial species Pseudomonas putida, where the gene is well-characterized. Since the hutH coding sequence is bacteria-sourced, it should be optimized for codon usage in the yeast, the production strain, not bacteria.
I’ve used the IDT Codon Optimization Tool to generate the codon-optimized DNA sequence for yeast.
GTGACCGAACTAACATTAAAACCAGGCACTCTAACCCTGGCACAACTTAGAGCTATTCATGCCGCCCCAGTGAGATTGCAACTAGATGCTAGCGCTGCACCTGCTATTGATGCTTCTGTGGCTTGTGTCGAACAGATTATTGCCGAGGACCGTACGGCTTATGGTATTAATACAGGGTTTGGTTTATTAGCGTCTACAAGAATTGCATCACATGATTTGGAAAATCTACAAAGGTCTTTGGTTTTAAGTCATGCAGCGGGTATTGGTGCTCCACTAGATGACGACTTAGTAAGGTTAATTATGGTGTTAAAGATTAACAGTTTGTCTAGGGGCTTTTCAGGAATTAGGCGTAAGGTCATTGATGCTCTGATTGCGCTGGTGAATGCAGAAGTGTACCCACACATACCATTGAAAGGTAGTGTTGGTGCTAGTGGTGACTTGGCACCTTTAGCCACGATGTCTTTGGTACTGCTGGGAGAAGGCAAAGCAAGGTACAAAGGTCAATGGCTGTCAGCAACAGAGGCTTTGGCCGTTGCTGGTCTAGAACCTTTGACCCTAGCTGCCAAGGAAGGTTTAGCGTTGCTAAATGGCACGCAAGCAAGTACTGCCTATGCTTTAAGAGGGCTTTTCTACGCCGAAGACTTGTACGCAGCAGCCATTGCTTGCGGTGGATTGTCAGTGGAAGCCGTGTTGGGCTCCAGAAGTCCATTCGATGCAAGAATTCACGAAGCAAGAGGTCAGCGTGGCCAAATTGACACAGCTGCGTGTTTTAGAGATTTACTGGGGGATAGCAGCGAGGTGTCTTTAAGCCATAAGAACTGCGATAAAGTCCAAGATCCATACAGCTTGAGATGCCAACCTCAGGTGATGGGTGCATGTCTGACCCAATTGAGACAAGCTGCCGAAGTCTTAGGTATTGAAGCGAACGCTGTGTCTGATAACCCCTTGGTATTCGCAGCTGAGGGAGATGTCATATCTGGCGGCAATTTTCATGCAGAACCAGTTGCCATGGCGGCTGATAACTTAGCGCTTGCTATCGCTGAGATAGGGTCTCTAAGCGAAAGAAGAATCAGCTTGATGATGGACAAACATATGTCCCAACTGCCACCCTTCCTAGTTGAAAATGGCGGAGTTAACAGCGGGTTTATGATCGCACAGGTTACTGCAGCCGCATTGGCTTCCGAGAACAAGGCTCTGTCACACCCGCATTCTGTCGATAGTTTGCCAACATCAGCAAATCAAGAGGACCACGTTTCTATGGCTCCAGCTGCTGGTAAGAGGCTATGGGAGATGGCAGAAAATACTAGAGGGGTGCCTGCAATTGAATGGTTGGGGGCGTGCCAAGGGTTGGATTTGAGAAAAGGTCTAAAAACCAGCGCAAAATTAGAAAAGGCGAGACAGGCTCTTCGTAGTGAAGTCGCCCATTATGATAGAGACAGATTTTTCGCACCAGATATTGAAAAAGCCGTTGAACTTTTGGCCAAAGGTTCACTTACTGGTCTTTTGCCAGCCGGAGTGTTACCATCCTTGTAA
Download PDF to view full alignment

3.4. You have a sequence! Now what?
Codon-optimized gene expression will allow a robust production of the protein in the host. It will be expressed from a well-characterized promoter in the yeast expression plasmid. Depending on the copy size of the expression plasmid, we expect more proteins to be produced from a self-replicating plasmid in cells, increasing the production yield. The expression system targets the enzyme to be exported outside of cells, creating more efficient downstream processing.
3.5. [Optional] How does it work in nature/biological systems?
Describe how a single gene codes for multiple proteins at the transcriptional level.
Try aligning the DNA sequence, the transcribed RNA, and also the resulting translated Protein!!! See example below.
[Example shows the biomolecular flow in central dogma from DNA to RNA to Protein] Special note that all “T” were transcribed into “U” and that the 3-nt codon represents 1-AA.
PART 4
Prepare a Twist DNA Synthesis Order
4.1. Create a Twist account and a Benchling account
4.2. Build Your DNA Insert Sequence
I will use the yeast expression vector from Twist Bio.
AOX1 promoter, methanol-inducible
Alpha-factor secretion signal sequence, which contains its translation initiation signal, Kozak-like sequence, and the initiation codon (ATG)
AOX1 terminator
The codon-optimized hutH gene insert:
The initiation codon (ATG) is removed due to the existing initiation codon in the alpha factor secretion sequence in pTwist_PIC9.
Unique restriction enzyme sites added: SnaBI (TACGTA) at the N-terminus for in-frame cloning with the alpha-factor secretion signal sequence and NotI (GCGGCCGC) at the C-terminus.
7x his-tag is added at the C-terminus site: CATCACCATCACCATCATCAC
Stop codon is added: TAA
Link to the linear map of the final expression plasmid at Benchling: pTwist_PIC9_hutH_histag
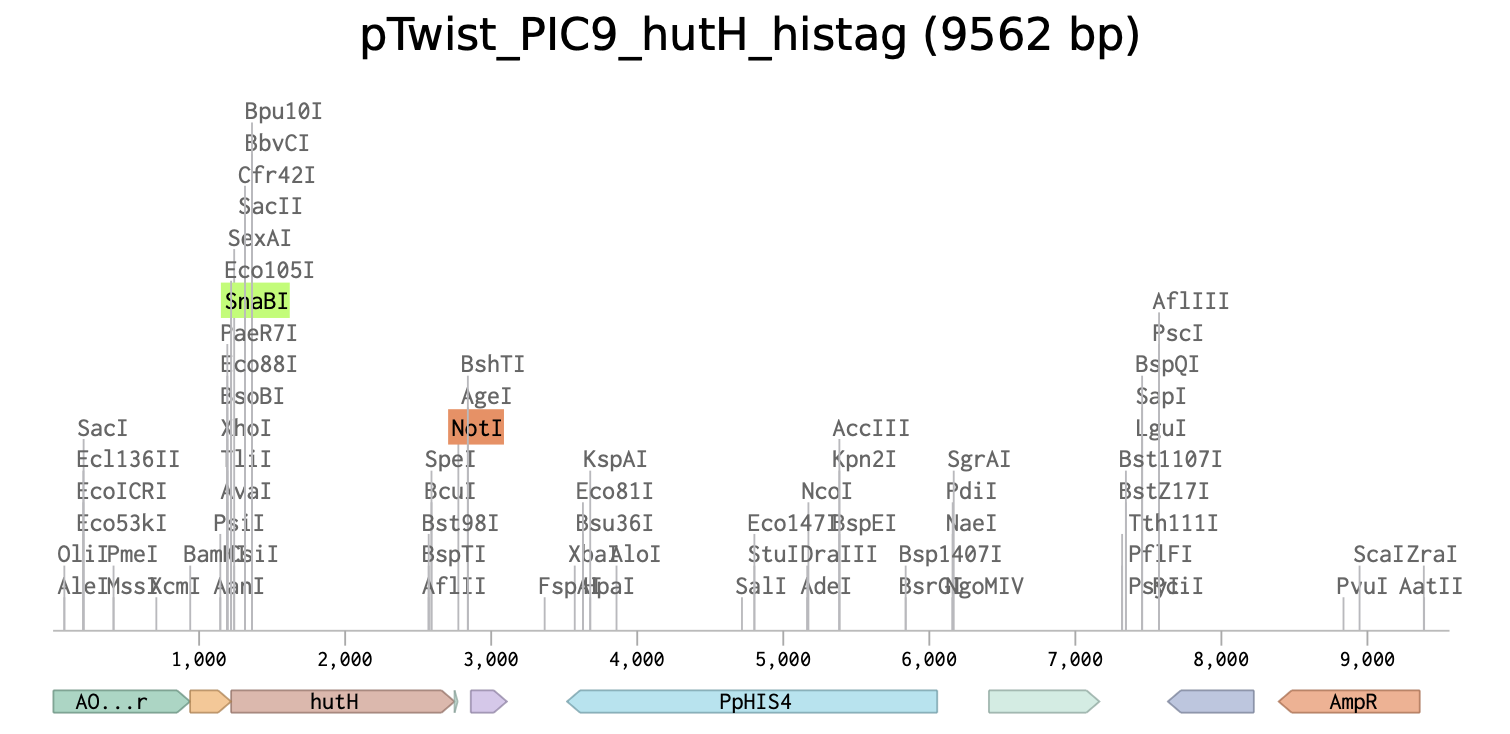
PART 5
DNA/Read/Write/Edit
5.1 DNA Read
(i) What DNA would you want to sequence (e.g., read) and why? This could be DNA related to human health (e.g., genes related to disease research), environmental monitoring (e.g., sewage wastewater, biodiversity analysis), and beyond (e.g., DNA data storage, biobanks).
I’d like to read the DNA from the human skin microbiome and compare it with healthy and diseased conditions. Findings from comparative studies would reveal insight into dysbiosis and could potentially lead to rationally designed probiotics as therapeutics, which may help reverse disease conditions.
(ii) What technology or technologies would you use to perform sequencing on your DNA, and why?
Also, answer the following questions:
- Is your method first-, second-, or third-generation or other? How so?
- What is your input? How do you prepare your input (e.g., fragmentation, adapter ligation, PCR)? List the essential steps.
- What are the essential steps of your chosen sequencing technology? How does it decode the bases of your DNA sample (base calling)?
- What is the output of your chosen sequencing technology?
• Method: I would use the second-generation sequencing method (Illumina shotgun metagenomic sequencing) because it is a more economical and applicable method in metagenomic sequencing, constructing metagenome-assembled genomes, and whole metagenome analysis, capturing the entire sequence of the skin microbiome and analysis. (Yang et al. 2021, Chen et al. 2023)
• Input DNA and preparation. The input is genomic DNA of the microbiome from skin. The skin microbiome has low microbial biomass. Working with low-abundance samples can be challenging, leading to poor-quality downstream genomic DNA and data analysis. Additionally, human DNA is often a contaminant that needs to be removed before sequencing. (Bjerre et al. 2019.)
After isolating genomic DNA from the skin samples and removing the human DNA, I would fragment the genomic DNA for the sequencing library construction. For this, I would use an enzymatic method such as Tn5 transposase to randomly cleave DNA. Because the Illumina sequencing can read between 75 and 300 bp, fragmented DNA should be selected appropriately by selectively capturing the DNA fragments on beads.
Next, DNA ends should be repaired to have blunt ends, and a single adenine is added to the 3’ ends, creating A-tailing, which helps to establish directionality.
In the following steps, I would add Illumina adapters, where the adapters are ligated to the DNA at both ends. Illumina adaptors will serve as an anchor for the DNA to be immobilized on the flow cell surface. Next is the PCR amplification step to increase the copies of the adaptor-ligated DNA to ensure that the library has sufficient copies of the DNA fragments. Finally, PCR-amplified DNA is size-selected, and quality is assessed by a fragment analyzer.
• Sequencing technology. It is a synthesis-based technology, sequencing by synthesis (SBS). The flow cell is pre-loaded with oligos that have complementarity to the Illumina adapters. Once the library is loaded on the flow cell, DNA hybridizes with both ends to the complementary oligos, forming a curved arch.
The next step is bridge PCR to make about 1000 clones of each molecule, as necessary for signal amplification.
The following step is the SBS, where DNA polymerase incorporates 3’-blocked fluorescently labeled nucleotides. Blocker is reversible, allowing incorporation of nucleotides in each cycle, with only a single base per cycle.
Simultaneously, in each cycle of incorporation, the camera captures images as generated by the excitation and emission of fluorescent dye. Image analysis software processes and calls the base.
Once the DNA polymerase completes the incorporation of nucleotides, the synthesized DNA strand is washed away, and the entire process is repeated from the other end, so it is paired-end, superior to single-read sequencing because both ends of sequencing are performed in separate rounds, providing better data accuracy on the same fragment.
• Output. On a MiSeq, the output is 250-300 bp, with paired-end reads, 600 reads per DNA fragment.
References, Click to Expand
Chen, Y., R. Knight, and R. Gallo. 2023. Evolving approaches to profiling the microbiome in skin disease. Frontiers in Immunology.
Yang, C., D. Chowdhury, Z. Zhang, W. K. Cheung, A. Lu, Z. Bian, and L. Zhang. 2021. A review of computational tools for generating metagenome-assembled genomes from metagenomic sequencing data. Computational and Structural Biotechnology Journal.
Bjerre, R. D., L. Warchavchick Hugerth, F. Boulund, M. Seifert, J. D. Johansen, and L. Engstrand. 2019. Effects of sampling strategy and DNA extraction on human skin microbiome investigations. Nature.
I’ve used Claude research.
Prompts:
Can you research sequencing methods used in the skin microbiome research? I want to know about the functionality of the microbiome in disease states. Did they perform de novo genome assemblies to learn about functional pathways (loss of activity or hyperactivity) in disease conditions?
Can you research the second-generation sequencing method? Explain the preparation steps and how the method does base calling.
5.2 DNA Write
(i) What DNA would you want to synthesize (e.g., write) and why? These could be individual genes, clusters of genes or genetic circuits, whole genomes, and beyond. As described in class thus far, applications could range from therapeutics and drug discovery (e.g., mRNA vaccines and therapies) to novel biomaterials (e.g., structural proteins) to sensors (e.g., genetic circuits for sensing and responding to inflammation, environmental stimuli, etc.) to art (DNA origami).
I want to synthesize an enzyme for therapeutic development purposes. One example is histidine ammonia lyase (HAL), which converts histidine into trans-urocanic acid (trans-Uro) and ammonia. Trans-Uro has an important function in maintaining skin barrier function. The product can be used as a topical application to manage skin disease such as atopic dermatitis. (Debinska 2021, Peltonen et al. 2014)
Trans-Uro can be produced from a cell-free expression system. To express and purify the catalyst HAL, I’d like to create a yeast expression system expressing the gene hutH encoding HAL. A gene block synthesis can provide the hutH gene to be cloned into an existing yeast expression vector.
(ii) What technology or technologies would you use to perform this DNA synthesis, and why?
Also, answer the following questions:
- What are the essential steps of your chosen sequencing methods?
- What are the limitations of your sequencing method (if any) in terms of speed, accuracy, scalability?
• Technologies: The hutH gene for the HAL coding sequence alone is 1530 bp (Hernandez and Phillips 1993). The DNA of this size, as a gene block, cannot be synthesized. It needs to be synthesized as short, overlapping oligos and later assembled.
Synthesis technologies: the most widely used are the 1st-generation or the 2nd-generation technologies based on phosphoramidite chemistry, and they achieve 200 nt. The emerging new technology, the 3rd generation, is based on the terminal deoxynucleotidyl transferase (TdT) enzyme, which achieves 300 nt.
Assembly technologies: OE-PCR (overlap extension PCR) or PCA (polymerase cycling assembly) are applied to most fragments under 1000 bp. Gibson assembly is applied to fragments larger than 1500 bp.
• Essential steps in enzymatic synthesis. Although the phosphoramidite chemistry is widely used and available, the 3rd-generation enzyme synthesis technology is more environmentally friendly, as it has no toxic organic solvent use. I would consider the enzymatic method.
The TdT enzyme adds 3’-blocked nucleotides to the 3’-OH of the growing chain. A wash step removes unincorporated nucleotides. Then, a free 3’-OH is generated by cleaving the 3’-blocking group. The cycle is considerably shorter, 10-40 seconds, in comparison to the phosphoramidite method, which is 4-10 minutes.
• Limitations. TdT enzyme adds nucleotides randomly. So, the synthesis must be controlled by temporarily blocking the 3’-OH. Modifications are not as easy as the phosphoramidite method. As it emerges, its availability is currently limited.
• Essential steps in OE-PCR and Gibson assembly. First, a set of overlapping oligos is designed for both strands. Primers are synthesized by the phosphoramidite method, pooled, and assembled in a thermocycling extension reaction. The full-length fragment is then amplified by PCR with two flanking outer primers.
• Limitations. The error rate is 1 in 500 - 1000 bp assembled products, which require sequencing for verification.
For a gene block larger than 1500 bp, the Gibson assembly is used by creating multiple sub-fragments of 300-800 bp size by OE-PCR. The sub-fragments would have 20-40 bp overlapping ends. Final assembly is performed in a single isothermal reaction.
I’ve used Claude research.
Prompts:
Can you research what technologies are used for synthesizing SNA (DNA write)? To make a gene block, explain what technologies can be used. To make a construct, including the vector, explain what technologies can be used.
References, Click to Expand
Debinska, A. 2021. New treatments for atopic dermatitis targeting skin barrier repair via the regulation of FLG expression. Journal of Clinical Medicine.
Peltonen, J. M., L. Pylkkanen, C. T. Jansen, I. Volanen, T. Lehtinen, J. K. Laihia, and L. Leino. 2014. Three randomised phase I/IIa trials of 5% cis-urocanic acid emulsion cream in healthy adult subjects and in patients with atopic dermatitis. Clinical Report Acta Derm Venereol.
Hernandez D. and A. T. Phillips. 1993. Purification and characterization of Pseudomonas putida histidine ammonia-lyase expressed in Escherichia coli. Protein Expression and Purification.
5.3 DNA Edit
(i) What DNA would you want to edit and why?
(ii) What technology or technologies would you use to perform these DNA edits, and why?