Week 4 HW: Protein Design Part I
Part A. Conceptual Questions
Answer any NINE of the following questions:
1. How many molecules of amino acids do you take with a piece of 500 grams of meat? (on average, an amino acid is ~100 Daltons)
Based on Gemini AI: 100 Dalton = 1.66054e-22 gram
Gemini AI performed the following division: 500/1.66054e-22 = 3.0110687e+24
3,011,068,700,000,000,000,000,000 amino acids in 500 grams of meat
2. Why do humans eat beef but do not become cows, eat fish but do not become fish?
Our cells are rebuilt from building block molecules such as amino acids. When we eat beef or fish, these proteins are broken down by our digestive system into amino acids, fatty acids, and sugars, as the beef and fish tissues are made out of them. Our body can only absorb such small molecules, and our cells make new cells based on the instructions from our DNA. Beef and fish become new human cells and tissues.
3. Why are there only 20 natural amino acids?
The 20 natural amino acids are not included by accident. Doig argues that specific chemical reasons are why evolution favored them, so they are included. These amino acids were selected because they enabled the formation of stable and soluble proteins (Doig 2017). The 20 natural amino acids do not have redundancy. Due to the cost of making them, redundant simple amino acids are not included evolutionarily. Occupancy of a chemical space regarding size, charge, and hydrophobicity is also not random (Philip and Freeland 2011, Ilardo et al. 2015). Kirschning’s analysis highlights amino acid biosynthesis, central metabolic networks, and separate evolutionary tracks for amino acids and cofactors (Kirschning 2022). Amino acids do not need to be optimized for redox reactions, for example. So, evolutionarily, selection is structural rather than catalytic. As reported by experimental work by Shibue et al. and Newton et al., and additionally, a theoretical work by Bywater, to achieve better folding and structurally stable proteins, 20 amino acids are needed, not fewer. Fewer amino acids would not be ideal for stability and structural diversity in protein folds (Shibue et al 2018, Newton et al. 2018, Bywater 2018). Finally, codon saturation is another factor for having the 20 natural amino acids. Although two additional amino acids, 21st (selenocysteine), and 22nd (pyrrolysine) are known to exist and be incorporated in proteins in certain living systems.
References, Click to Expand
Doig, A. 2017. Frozen, but no accident—why the 20 standard amino acids were selected. FEBS J.
Freeland, S.J. and Hurst, L.D. 1998. The genetic code is one in a million. Journal of Molecular Evolution
Ilardo, M., Meringer, M., Freeland, S., Rasulev, B. & Cleaves, H.J. 2015. Extraordinarily adaptive properties of the genetically encoded amino acids. Scientific Reports
Kirschning, A. 2022. On the evolutionary history of the twenty encoded amino acids. Chemistry
Shibue, R., Sasamoto, T., Shimada, M., Zhang, B., Yamagishi, A. & Akanuma, S. 2018. Comprehensive reduction of amino acid set in a protein suggests the importance of prebiotic amino acids for protein function. Scientific Reports
Newton, M. S., D. J. Morrone, K-H Lee, and B. Seeling. 2018. Genetic code evolution investigated through the synthesis and characterisation of proteins from reduced-alphabet libraries. ChemBioChem*
Bywater, R.P. 2018. Why twenty amino acid residue types suffice(d) to support all living systems. PLoS One
I’ve searched Google Scholar and researched Claude.
Prompts:
Why are there only 20 natural amino acids?
Can you review reference papers? Include additional insights from other papers as well. Papers were uploaded: Doig 2017 and Kirschning 2022.
4. Can you make other non-natural amino acids? Design some new amino acids.
For this, I’ve started by building an interactive amino acid design tool in Claude. I then ask Claude AI to design the non-natural amino acid. I focused on applying the new non-natural amino acid as a probe in a biomedical imaging application.
The designer tool enables non-natural amino acids to have multiple chemistries, including photoactivation by light that makes the amino acid bioavailable on demand and a reporter attachment for detectability.
Amino Acid Designer Tool
The designer tool requires an Anthropic API key to run interactively.
What the designer tool does:
Reporter / labelling notes: AI explains specifically where on the molecule the label attaches, what signal it produces, and key in vivo considerations.
Orthogonality assessment: AI evaluates whether the combined chemistries (cage, reporter, radiolabel, handle) are mutually compatible and identifies any potential interference or synthetic challenge.
I’ve chosen to design an imaging probe for the purpose of detecting thyroid tumors specifically.
AI will help to generate a tyrosine analog with the given considerations:
The probe should be based on a tyrosine, as tyrosine is used along with iodine to make thyroid hormones in the thyroid. The standard amino acid backbone is suitable for uptake by the LAT1 transporter, and thyroid peroxidase (TPO) activity, which covalently attaches iodine to tyrosine, trapping it in the thyroid.
The probe should have a one-way photo-caged/light-controlled chemistry. It is activated by light, which causes the cage to be cleaved, and tyrosine becomes available as a substrate for TPO. The circulating probe accumulates in the thyroid. Signal becomes greater than background noise, better precision detection.
The probe should have the two-photon chromophore, as activated by NIR light, more effective in deeper tissues than a one-photon activation mechanism.
The probe should have a radiolabelled isotope, such as ¹²³I, for traceability through isotope decay after metabolic trapping in the thyroid tissue. ¹²³I is ideal because the thyroid has a sodium-iodide symporter (NIS) that actively pumps iodide into the cells.
Example: two-photon photocaging tyrosine analog (3-¹²³I-α-Methyl-(7-nitroindolinyl-2-carbonyl)-Tyrosine)
Fully designed result card.

I used Claude AI research to generate the interactive tool.
Prompts:
Can you build an interactive design tool to make non-natural amino acids?
Can you research why tyrosine was specifically picked for imaging the thyroid? Why not other amino acids?
Can you research the two-photon uncaging of tyrosine analogs for the detection of small tumors based on metabolic activation of tyrosine in the thyroid?
Could 123I-labelled tyrosine analogs provide new advantages over existing probes for imaging the thyroid?
5. Where did amino acids come from before enzymes that make them, and before life started?
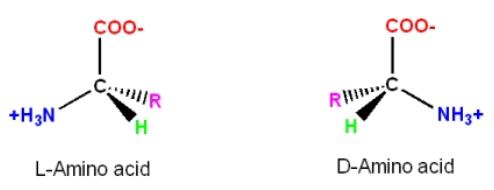
6. If you make an alpha-helix using D-amino acids, what handedness (right or left) would you expect?
D-amino acids would assume left-handed alpha-helical structures.
In D-amino acids, because of the side chain ‘R’ positioning in relation to the alpha C atom, as shown in the image below, the turn in a helical structure occurs counterclockwise, placing the side chain toward the center backbone of the helix and resulting in steric hindrance.
Although forming helices with D-amino acids is not favorable, there are still ways to achieve left-handed helices’ stability. As reported in the methods for incorporating the D-amino acids into peptides, superior performance and better therapeutic outcomes were achieved with the peptides incorporating the D-analogs (Annavarapu and Nanda 2009, Garton et al. 2018).
References, Click to Expand
Annavarapu, S. and V. Nanda. 2009. Mirrors in the PDB: left-handed alpha-turns guide design with D-amino acids. BMC Structural Biology.
Garton, M. S. Nim, T. A. Stone, K. E. Wang, C. M. Deber, and P. M. Kim. 2018. Method to generate highly stable D-amino acid analogs of bioactive helical peptides using a mirror image of the entire PDB. PNAS.
I’ve researched Claude.
Prompts:
Can you research D-amino acids making alpha-helices?
Explain the handedness in L and D amino acids
7. Can you discover additional helices in proteins?
It is possible to discover additional helices in proteins. Other helical structures have been reported in proteins, in addition to the most common alpha helices.
As a short segment in global proteins, the 310 helix exists in nature, as demonstrated in the structure of a fungal peptide (Karle et al. 2003).
Another helix is the Pi helix, which typically forms by bulging from a long alpha helix and is associated with functional sites in proteins (Cooley et al. 2003).
A few others are polyproline II helix (PPII), polyproline I helix (PPI), and collagen triple helix, in which helical strands bundle and twist together to form a triple helix. (Hollingsworth and Karplus 2010).
A beta helix is another one that can form either left- or right-handed helices. It is a tandem protein with a repeat structure (Eisenberg 2003).
Although never observed in nature, but possible, the gamma helix was proposed as a model by Pauling et al. in 1951, and is reviewed in the historical timelines by Eisenberg (Eisenberg 2003).
References, Click to Expand
Eisenberg, D. 2003. The discovery of the alpha-helix and beta-sheet, the principal structural features of proteins. PNAS.
Karle, I. L., J. Flippen-Anderson, M. Sukumar, and P. Balaram. 1987. Confirmation of a 16-residue zervamicin IIA analog peptide containing three different structural features: a 310-helix, alpha-helix, and beta-bend ribbon. PNAS.
Cooley, R. B., D. J. Arp, and P. A. Karplus. 2010. Evolutionary origin of a secondary structure: pi-helices as cryptic but widespread insertional variations of alpha-helices that enhance protein functionality. J of Mol Bio.
Hollingsworth, S. A. and P. A. Karplus. 2010. A fresh look at the Ramachandran plot and the occurrence of standard structures in proteins. BioMol Concepts.
I’ve researched Claude.
Prompts:
Can you research the discovery of additional helices in proteins?
What are the known helices discovered so far?
8. Why are most molecular helices right-handed?
Most commonly found right-handed helices consist of left-handed amino acids and the right-handed sugars (Blackmond 2010). Biology favored this form of helix due to the structural stability provided by strong cooperativity effects of electrostatic interactions (Liu 2020).
Amino acids exist in chiral forms, L- and D-forms, where the alpha C atom has an ‘R’ side chain positioned asymmetrically. L-amino acid makes a clockwise turn in a helix, positioning the side chain ‘R’ away from the center backbone of the helix. By doing so, it avoids structural hindrance and is energetically favored. On the other hand, the positioning of the side chain in a left-handed helix would be much closer to the backbone, causing steric hindrance and not being energetically favorable.
L-amino acid became more abundant than the other chiral form, D-amino acid. There have been various hypotheses that explain this, such as in the kinetic theory, a faster production rate and slower depletion in a reaction could lead to a dominance of one form, which may have happened in the early history of the Earth.
Another is the parity violation hypothesis. In this view, although the effect imposed by the weak energy difference is very small, weak energy differences may have favored the reactions of one chirality over the other.
The most recent view considers the chirality hierarchy, which states that the chirality of helices dictates the abundance of the stereochemistry of the lower ones, amino acids, and sugars (Liu 2020).
References, Click to Expand
Blackmond, D. G. 2010. The origin of biological homochirality. (Cold Spring Harbor Perspectives in Biology).
Liu, S. 2020. Homochirality originates from the handedness of helices. (J Phys Chem Lett).
I’ve researched Claude.
Prompts:
Can you research the reasons why most molecular helices are right-handed?
9. Why do B-sheets tend to aggregate?
- What is the driving force for B-sheet aggregation?
Beta-sheets formed by beta-strands are amphipathic; that is, they have two distinct behavioral characteristics: a hydrophobic core and a hydrophilic surface.
Beta-strands connect via backbone hydrogen bonds. As beta-strands lie alongside, the alpha C atoms of each strand sit across each other. Positioning of the side chains occurs in an alternating pattern, creating an amphipathic character with one side more hydrophobic and the other side more hydrophilic.
Because beta-strands lie side by side and connect, the edge strand is “exposed”. The edge strand has unsatisfied hydrogen donors and wants to connect to form hydrogen bonds. This is known as the open edge, or the “edge strand problem”. Even though a new docking strand satisfies hydrogen donors, it becomes the edge strand with unsatisfied hydrogen donors. So, due to this geometry, beta-sheets tend to form aggregates, and their open edge primarily drives protein aggregation.
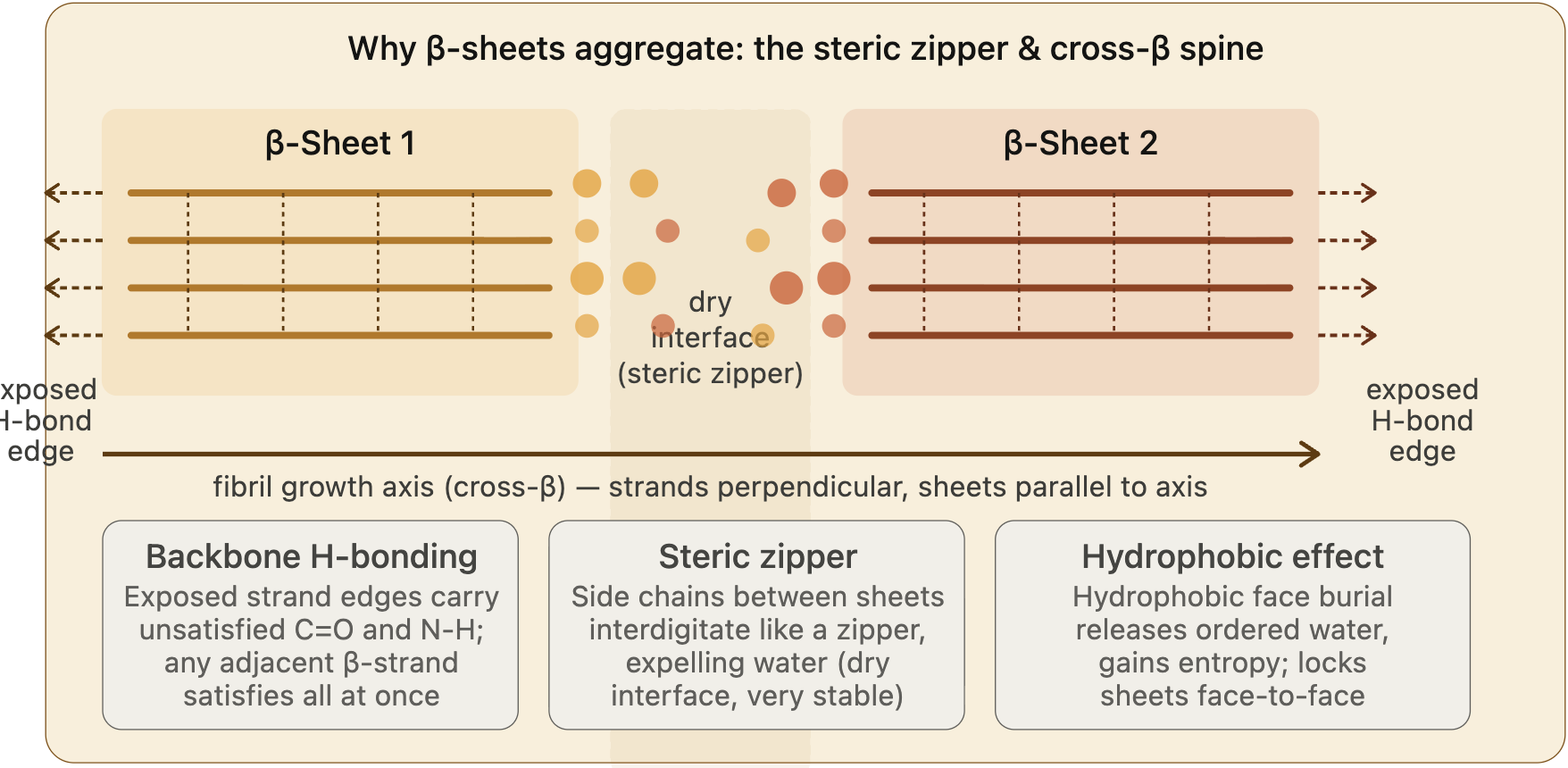
Other types of interactions are also known to drive aggregation: hydrophobicity and steric zipper, which is the effect of side chains between beta-sheet proteins that create a dry interface, allowing beta-sheets to grow in parallel to the axis.
I’ve researched Claude.
Claude provided the figure.
Prompts:
Can you research the reasons beta-sheets tend to aggregate?
What is a beta-sheet protein?
10. Why do many amyloid diseases form B-sheets?
Amyloid diseases, such as Alzheimer’s, are associated with the formation of fibrils and protein aggregates.
Once misfolded, the protein’s hydrophobic regions are exposed, and this triggers a self-assembly process, resulting in aggregates (amyloid fibrils) that are highly organized, rich in beta-sheet structures, and stable, achieved through an intramolecular hydrogen bonding network. In contrast, in the native state, proteins achieve stability by hydrophobic interactions via side chains.
There is no sequence dependency, as any protein can participate in hydrogen bonding. The native fold goes through misfolding and some destabilization, in which backbone hydrogen bonding can occur at this stage.
Dimerization of the monomers is the key step for locking the misfolding. As shown by Lv et al., monomers lead to cooperative formation of beta-sheet conformation and dimerization, which is thought to stabilize the misfolded state (Lv et. al. 2013).
The aggregate grows slowly until it becomes thermodynamically more stable, which occurs at a critical nucleus size (Tsemekhman et al. 2009).
Once a nucleus forms, monomers join the fibril ends rapidly, growing in parallel to the fibril axis. As in the state of an amyloid fibril, each new beta strand satisfies all hydrogen bonds. The amyloid state is thermodynamically more stable than the native state of the protein.
- Can you use amyloid B-sheets as materials?
There are many applications reported for the use of amyloid beta-sheets. The field is expanding to include environmental remediation, biomedical, sustainable materials, and food proteins.
Amyloid fibrils provide a rigid surface and resistance to chemicals, provided by highly dense hydrogen bonds, and coded by a specific peptide sequence.
A few examples as materials:
Bioplastic, amyloid-based material development from plant protein sources (Li et al. 2024).
Conductive aerogels with sensing properties development. Han et al. demonstrated that in situ polymerization of amyloid fibrils as scaffolds coats the conductive polymer, polypyrrole, creating a porous, conductive network, enabling pressure sensing, strain sensing, and potential wearable electronics applications. (Han et al. 2020).
References, Click to Expand
Lv, Z., R. Roychaudhuri, M. M. Condron, D. B. Teplow, and Y. L. Lyubchenko. 2013. Mechanism of amyloid β-protein dimerization determined using single-molecule AFM force spectroscopy. Scientific Reports.
Tsemekhman, K., L. Goldschmidt, D. Eisenberg, and D. Baker. 2009. Cooperative hydrogen bonding in amyloid formation. Protein Science.
Li, T., J. Kambanis, T. L. Sorenson, M. Sunde, and Y. Shen. 2024. From Fundamental Amyloid Protein Self-Assembly to Development of Bioplastics. Biomacromolecules.
Han, Y., Y. Cao, S. Bolisetty, T. Tian, S. Handschin, C. Lu, and R. Mezzenga. 2020. Amyloid Fibril‐Templated High‐Performance Conductive Aerogels with Sensing Properties. Small.
I’ve researched Claude.
Claude provided the figure.
Prompts:
Can you research why many amyloid diseases form beta-sheets?
Show me research findings about protein misfolding triggering highly organized beta-sheet formation.
What determines a misfolded protein to adopt a beta-sheet structure? What is the force behind this event?
Can you research to find examples and use cases for amyloid beta-sheets as materials?
11. Design a B-sheet motif that forms a well-ordered structure.
Part B. Protein Analysis and Visualization
1. Briefly describe the protein you selected and why you selected it.
I selected the human ferritin protein, the major iron-storage protein, which is present in all cells, and its structure has been solved (Wang et al. 2006). It consists of 24 self-assembled subunits, light (L-ferritin) and heavy (H-ferritin) chains. Mutations in the L-ferritin gene (FTL gene) are reported to be associated with five different diseases (Cadenas et al 2019).
L-ferritin is the target analyte for the body’s iron status. I’ve proposed an aptasensor-based detection of ferritin as one of the possible final projects, and I want to deepen my understanding of this protein, which will help develop the design of the aptasensor project.
References, Click to Expand
Wang, Z., C. Li, M. Ellenburg, E. Soistman, J. Ruble, B. Wright, J. X. Ho, and D. C. Carter. Structure of human ferritin L chain. 2006. Structural Biology
Cadenas, B. J. Fita-Torro, M. Bermudez-Cortes, I. Hernandez-Rodriguez, J. Luis Fuster, M. E. Llinares, A. M. Glaera, J. L. Romero, S. Perez-Montero, C. Tornador and M. Sanchez. L-Ferritin: one gene, five diseases; from hereditary hyperferritinemia to hypoferritinemia-report of new cases. 2019. Pharmaceuticals
2. Identify the amino acid sequence of your protein.
How long is it?
What is the most frequent amino acid?
MSSQIRQNYSTDVEAAVNSLVNLYLQASYTYLSLGFYFDRDDVALEGVSHFFRELAEEKREGYERLLKMQNQRGGRALFQDIKKPAEDEWGKTPDAMKAAMALEKKLNQALLDLHALGSARTDPHLCDFLETHFLDEEVKLIKKMGDHLTNLHRLGGPEAGLGEYLFERLTLKHD
Human Ferritin Light Chain
I’ve retrieved the amino acid sequence from the UniProt database.
UniProt accession: P02792
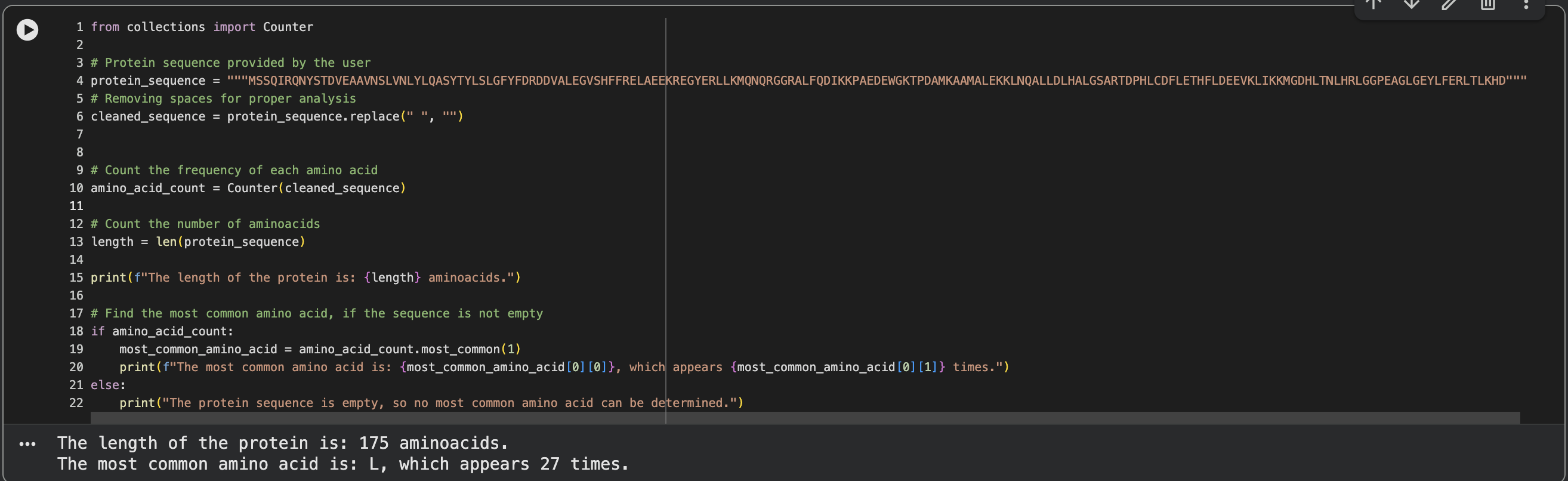
It has 175 amino acids, including methionine.
Leucine is the most frequent amino acid, present 27 times.
I’ve used the Colab Notebook for calculating amino acid frequency.
Below is the screenshot from the CoLab Notebook, length and amino acid frequency calculation.
- How many protein sequence homologs are there for your protein?
There are 250 BLAST homology results found by UniProtKB analysis.
Homology alignment with the human ferritin light chain protein is shown below.
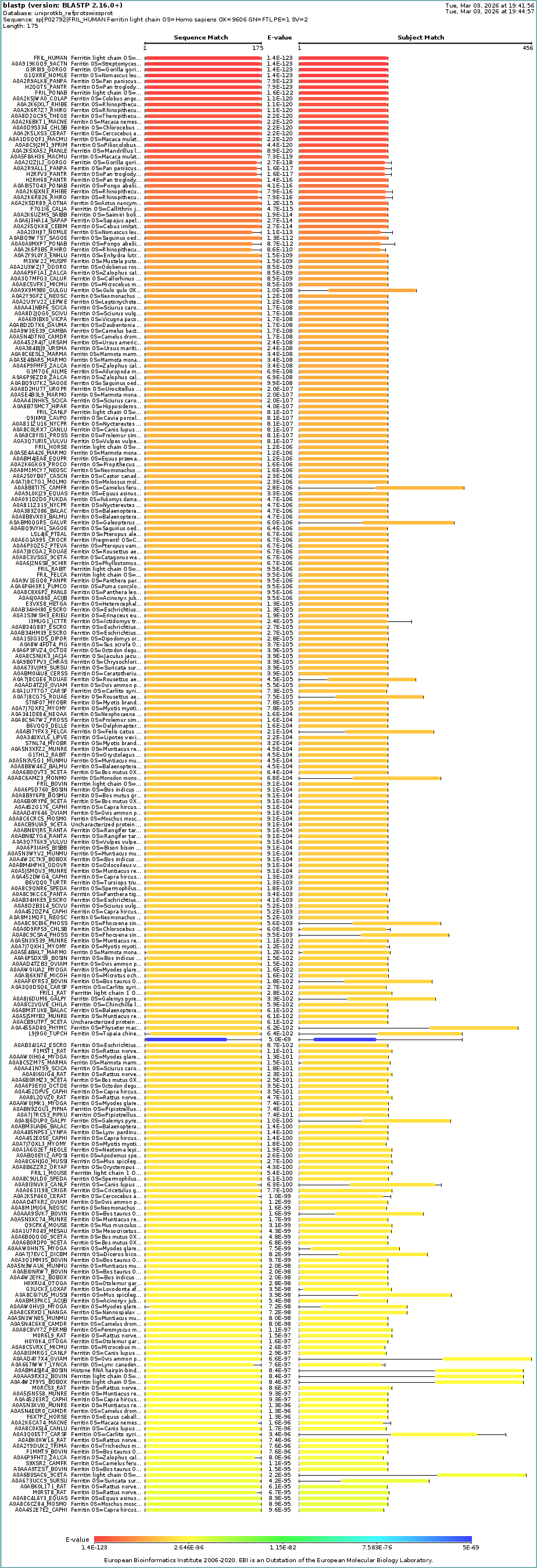
- Does your protein belong to any protein family?
I used the PANTHER database to identify the family to which the human ferritin light chain belongs.
protein family: Ferritin (PTHR11431)
protein class: storage protein
molecular function: ferrous iron binding
3. Identify the structure page of your protein in RCSB.
Link to RCSB Protein Data Bank
Search term: human light chain ferritin, metal-binding protein
Protein ID: 2FG4 (pdb_00002fg4)
| (A) ferritin light chain subunit | (B) self-assembled 24 subunits |
|---|---|
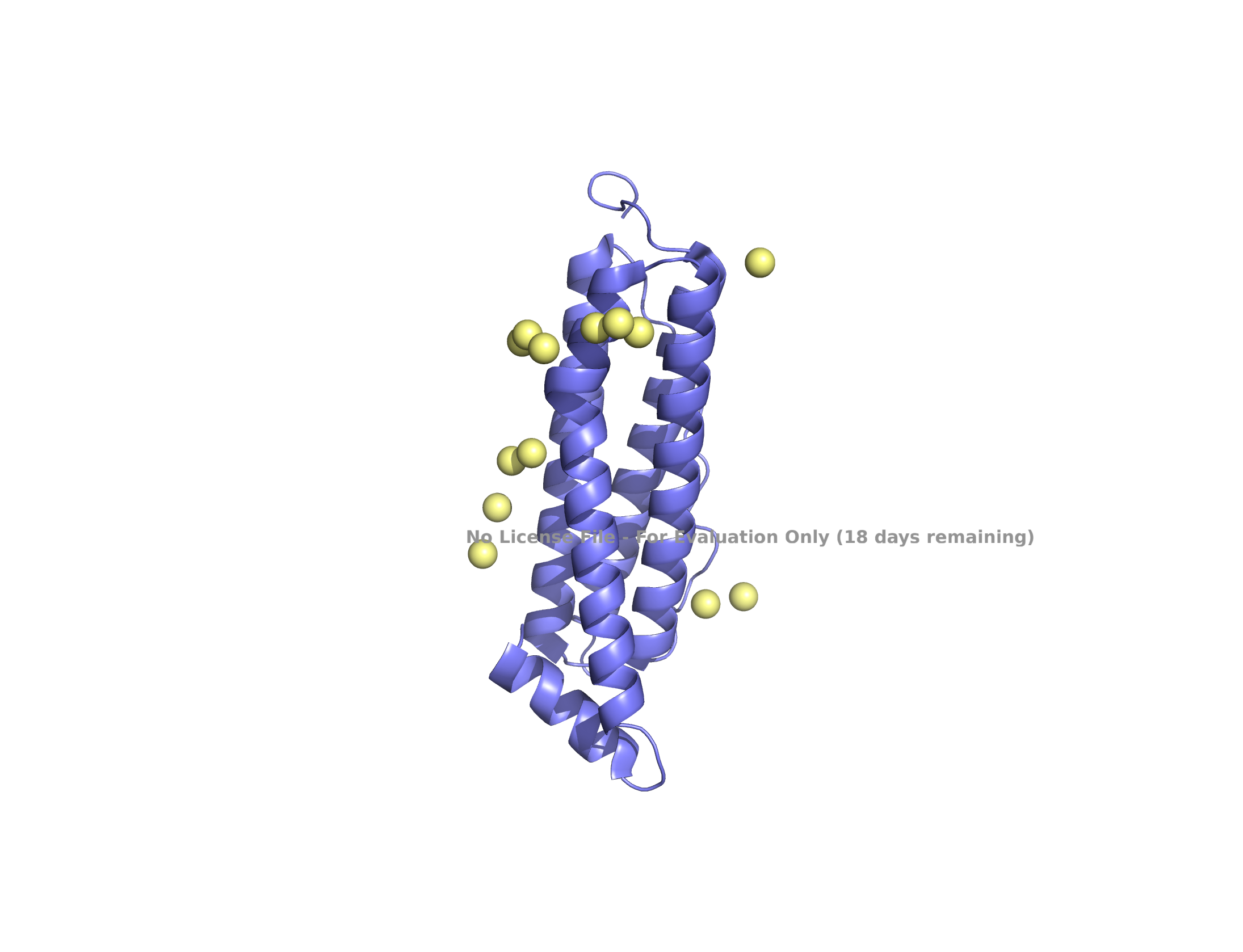
| 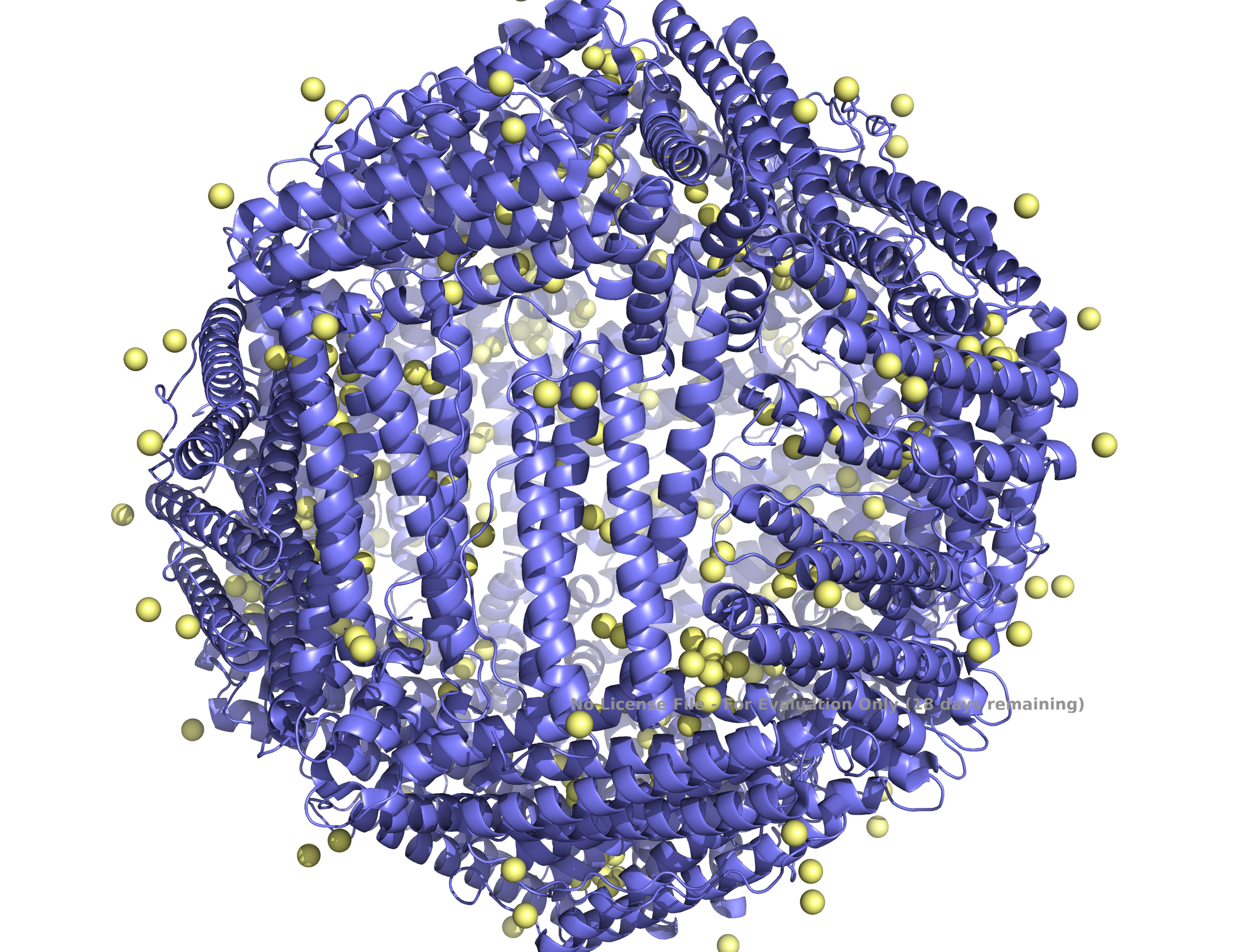
|
Structure of the human ferritin L chain visualized by PyMol, shown as a single subunit in (A) and 24 self-assembled subunits in (B). Transient metal cadmium ions are shown in ball & stick model mimicking the iron binding sites entering into the ferritin channel.
- When was the structure solved?
- Is it a good-quality structure?
The structure of the human ferritin L chain was solved in 2006 with a resolution of 2.10 Å, indicating that the structure is of good quality.
Link to publication, Wang et al. 2006
- Are there any other molecules in the solved structure apart from protein?
Transient metal, cadmium ions, are found in the solved structure. Cadmium ions with a similar charge to iron indicate the route to the iron-binding cavity.
- Does your protein belong to any “structure classification family”?
The human ferritin L chain belongs to the ferritin-like superfamily proteins.
Link to Structural Classification of Proteins (SCOP 2)
Search term: P02792
4. Open the structure of your protein belong in any 3D molecule visualization software.
PyMol protein visualization
- Visualize the protein as “cartoon,” “ribbon,” and “ball and stick.”
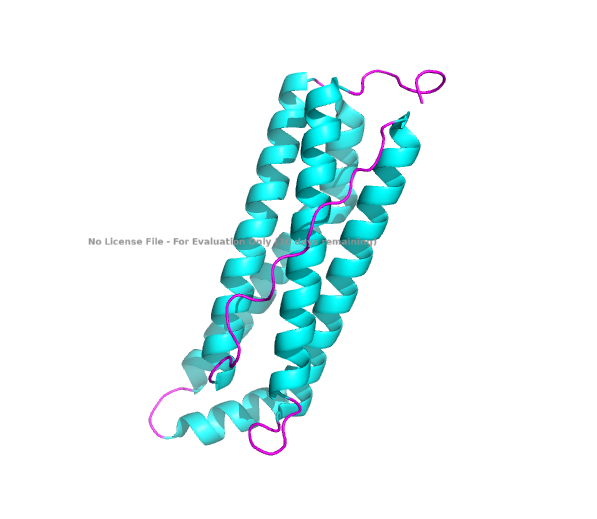
- Color the protein by secondary structure. Does it have more helices or sheets?
- Color the protein by residue type. What can you tell about the distribution of hydrophobic vs hydrophilic residues?
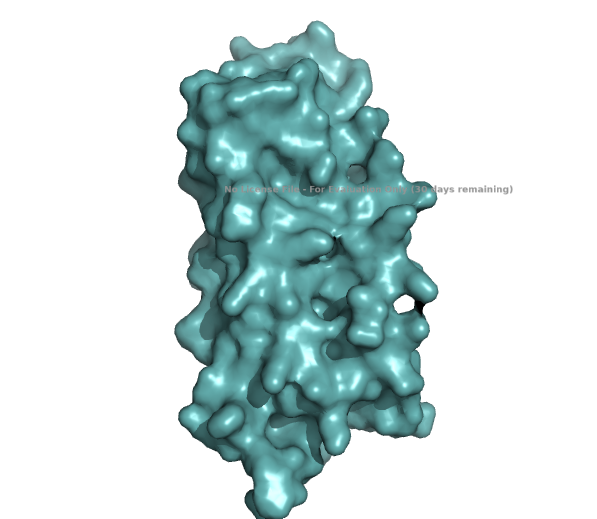
- Visualize the surface of the protein. Does it have any “holes” (aka binding pockets)?
| (A) cartoon | (B) ribbon | (C) stick & ball |
|---|---|---|
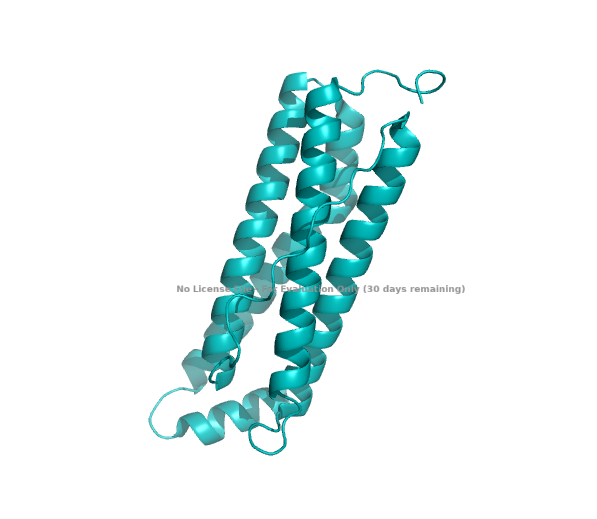
| 
| 
|
| (D) secondary structures | (E) hydrophobic / hydrophilic | (F) surface |
|---|---|---|
| 
| 
|
The structure of the human ferritin light chain is visualized in PyMol. Images on the top are the protein structure in a cartoon diagram (A), in a ribbon diagram (B), and in a ball and stick diagram (C). Images on the bottom are highlighting the secondary structures, helices (teal) and loops (magenta) (D), highlights in hydrophobic (yellow) and hydrophilic (blue) residues (E), and highlighting the surface (F).
The human ferritin light chain. PDB Protein ID: 2FG4
Visualization of the structure of the human ferritin light chain revealed the presence of secondary structures, like helices and loops, and the absence of beta sheet structures.
The surface visualization shows small holes and pockets for metal ion binding.
Part C. Using ML-Based Protein Design Tools
C1. Protein Language Modeling
1. Deep Mutational Scans
- Use ESM2 to generate an unsupervised deep mutational scan of your protein based on language model likelihoods.
Link to “HTGAA_ProteinDesign2026 Colab
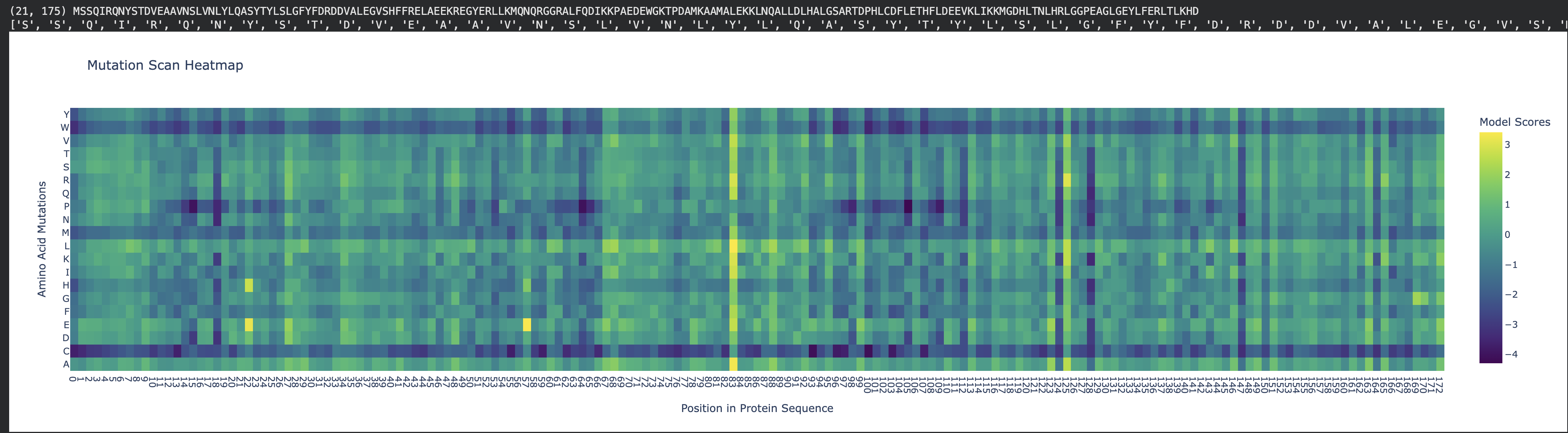 Fig 1. Mutation heat map. Human ferritin light chain.
Fig 1. Mutation heat map. Human ferritin light chain.
- Can you explain any particular pattern? (choose a residue and a mutation that stands out)
To address this question, first, with the AI’s help, I’ve re-created the x-axis to display the naming of residues, starting with methionine, from 1 to 175. This allowed easy identification of the residue of interest (see Fig 2).
Link to “HTGAA_ProteinDesign2026 Colab Notebook
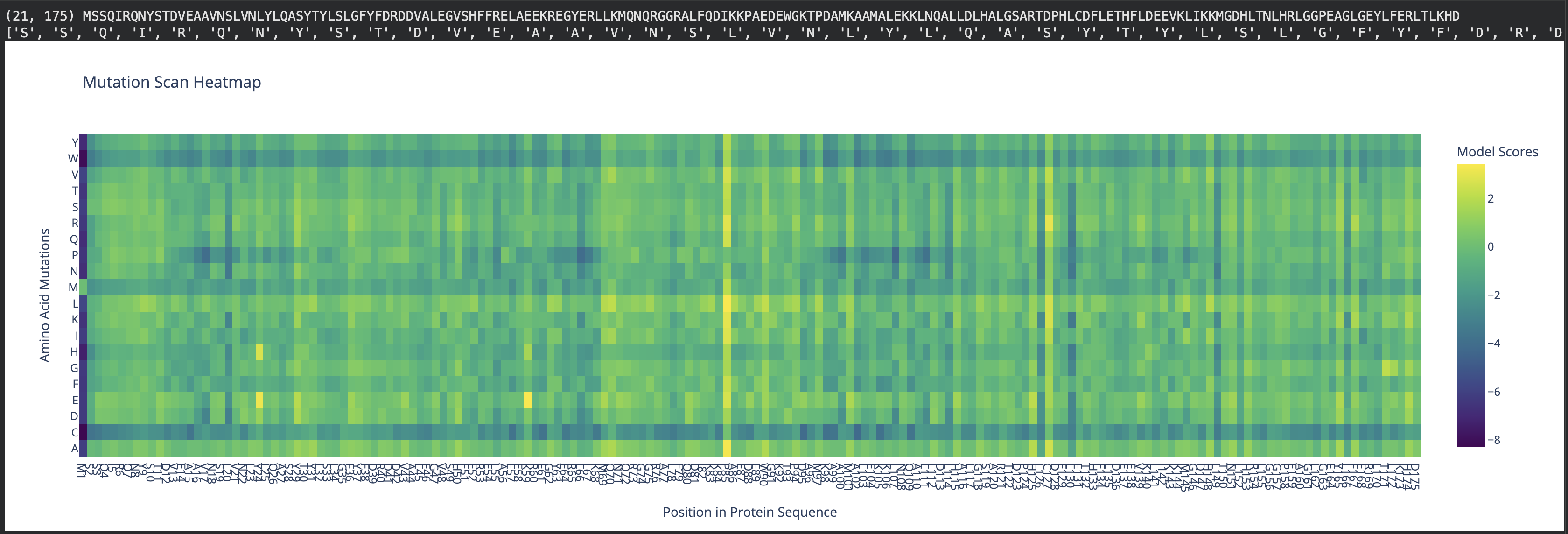 Fig 2. Mutation scan heatmap displaying x-axis with residue naming. Human ferritin light chain.
Fig 2. Mutation scan heatmap displaying x-axis with residue naming. Human ferritin light chain.
Secondly, I used the AI to build an interactive tool to work with the mutation scan heatmap. The interactive tool allows users to enter a residue of interest, which displays a list of mutations that are tolerable and disfavored based on their score.
Link to “HTGAA_ProteinDesign2026 Colab Notebook
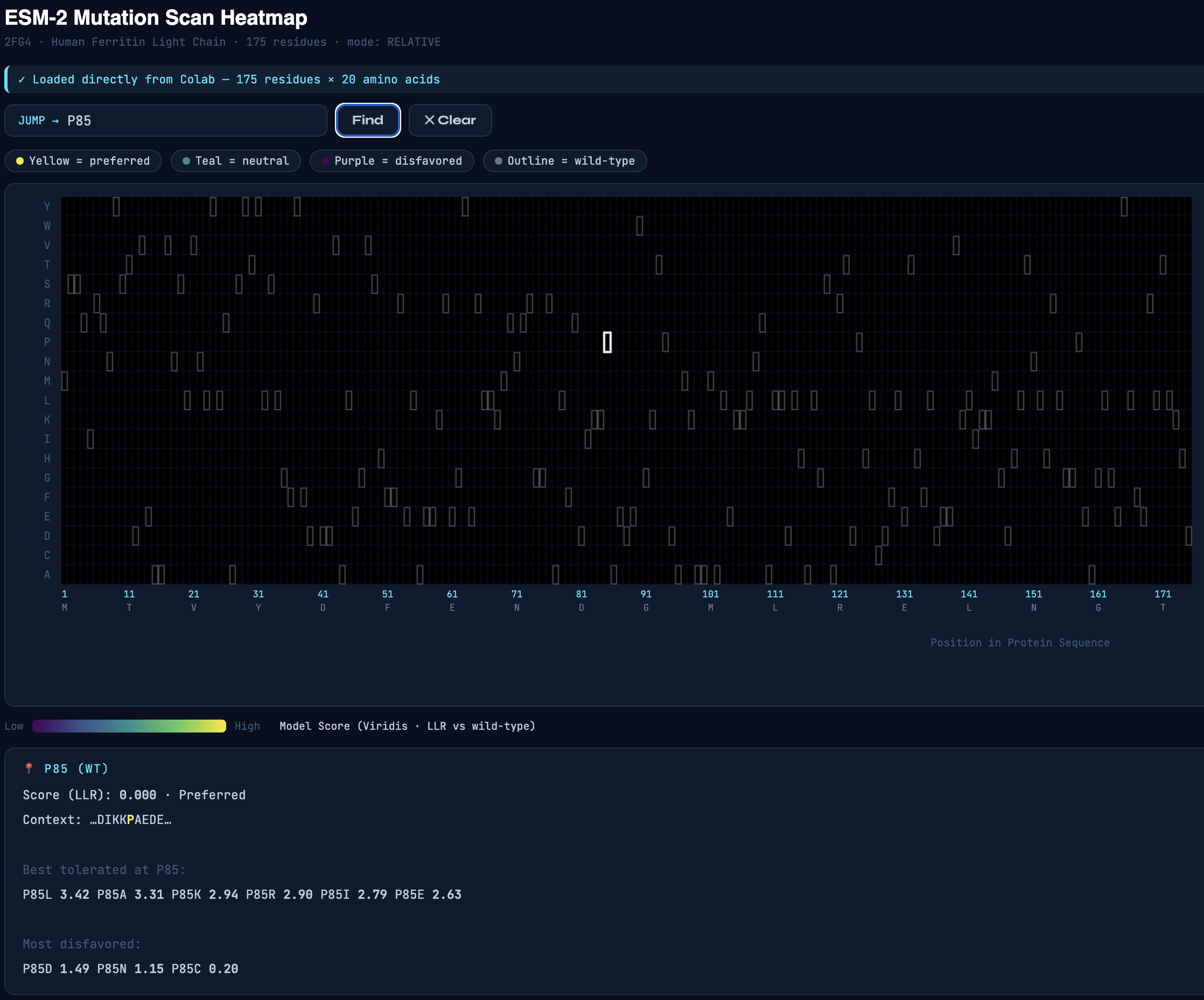 Fig 3. Interactive heatmap tool.
Fig 3. Interactive heatmap tool.
Inspecting the mutation scan heatmap, I noticed that any replacements with Cys, Trp, and Met that are replacing the wild-type residues are mostly predicted to be disfavored. As an example, see Thr11 replaced with Cys, Trp, or Met.
Pro85 is predicted to be highly tolerant to many substitutions. Proline is known to allow bending, flexibility, and functioning as a “helix breaker," which can tolerate substitutions.
Many Leu residues, such as L20, L66, L107, L114, L126, L130, L149, L152 and L166 are disfavoring many replacements. As leucine is hydrophobic, it most likely contributes to the structural stability of the helices.
- (Bonus) Find sequences for which we have experimental scans, compare the prediction of the language model to experiment.
2. Latent Space Analysis
Use the provided sequence dataset to embed proteins in reduced dimensionality.
Analyze the different formed neighborhoods: do they approximate similar proteins?
Place your protein in the resulting map and explain its position and similarity to its neighbors.
Link to “HTGAA_ProteinDesign2026 Colab Notebook
3D t-SNE Visualization of Protein Sequence Embeddings. The human ferritin light chain is displayed as a black circle.
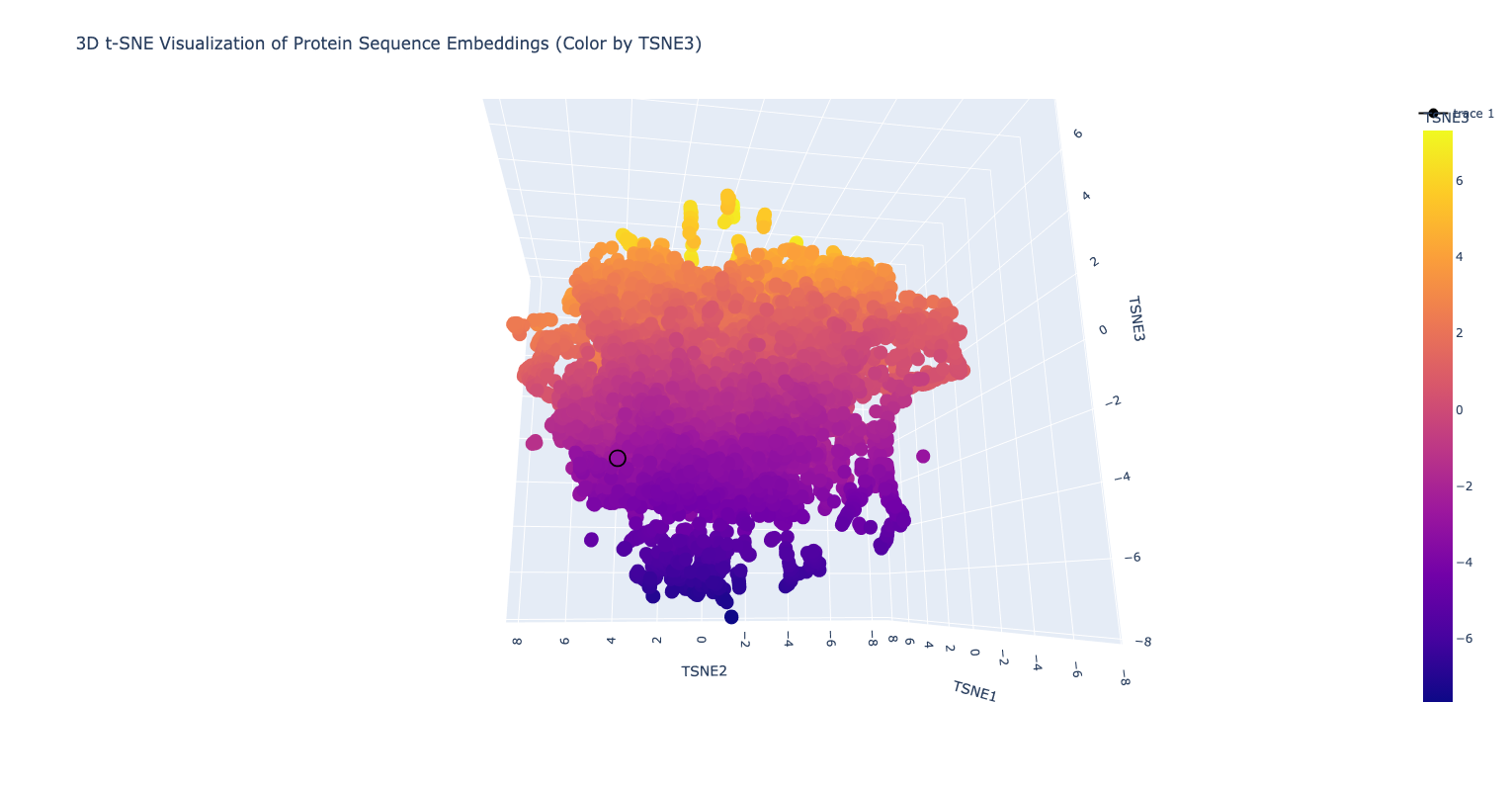
Inspecting the image above and the result from the annotation output, the human ferritin light chain is neighboring with the proteins, which belong to the ferritin family of proteins (see below)
 Neighboring proteins to the human ferritin light chain, as annotated from the latent space analysis.
Neighboring proteins to the human ferritin light chain, as annotated from the latent space analysis.
C2. Protein Folding
Folding a protein
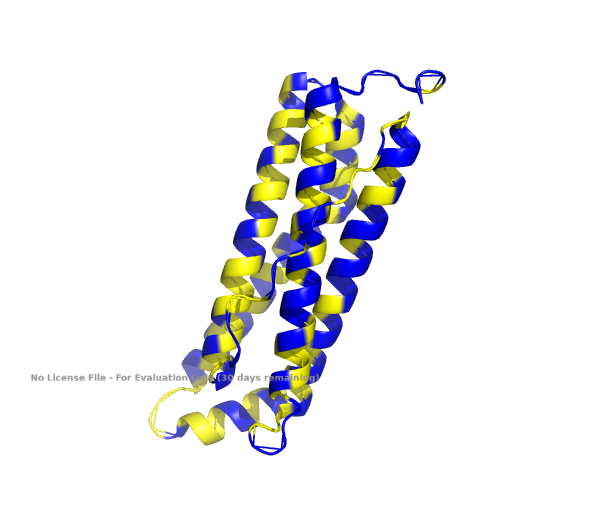
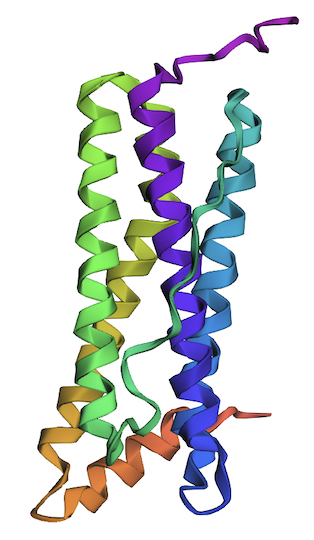
1. Fold your protein with ESMFold. Do the predicted coordinates match your original structure?
The ESMFold-built structure is highly similar to the original structure, as seen in Part B. 4. Fig A, cartoon.
Structure of the human ferritin light chain by ESMFold.
Link to “HTGAA_ProteinDesign2026 Colab Notebook
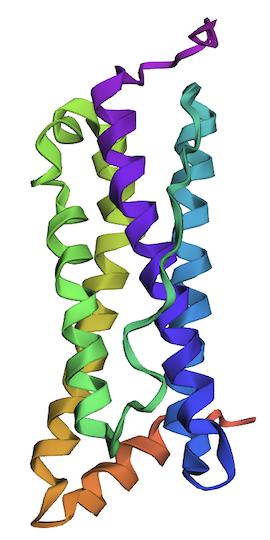
2. Try changing the sequence; first try some mutations, then large segments. Is your protein structure resilient to mutations?
I’ve changed the following residues to create variant 1: L20P, L66P, L107P, L114P, L126C, L130N, L149C, L152C, and L166C. All replacements are predicted to be unfavorable.
One of the helices (shown in green) has a partial disorder as a result of the changes in variant 1.
Considering that variant 1 has nine unfavorable residues and results in a partial defect in only one of four helices, this protein is resilient to mutations.
Shown below is the variant 1 structure modeled by ESMFold.
Link to “HTGAA_ProteinDesign2026 Colab Notebook
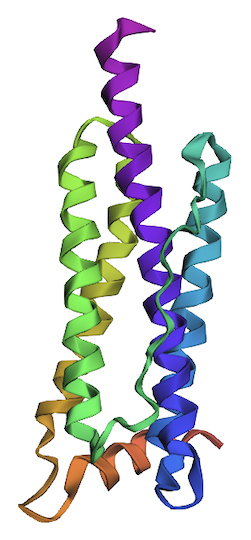
I’ve created single deletion mutations across the leucine residues mentioned in variant 1, creating variant 2. Deletion mutations: DeltaL20, DeltaL66, DeltaL107, DeltaL114, DeltaL126, DeltaL130, DeltaL149, DeltaL152, and DeltaL166.
- The ending of one of the helices (magenta) was removed as a result of deletions in the mentioned leucine residues.
Shown below is the variant 2 structure modeled by ESMFold.
Link to “HTGAA_ProteinDesign2026 Colab Notebook
C3. Protein Generation
Inverse-Folding a protein
1. Analyze the predicted sequence probabilities and compare the predicted sequence vs the original one.
Predicted “New Sequence” generated by the inverse folding with ProteinMPNN for the human ferritin light chain.
Link to “HTGAA_ProteinDesign2026 Colab Notebook
SSQIRQNYSTDVEAAVNSLVNLYLQASYTYLSLGFYFDRDDVALEGVSHFFRELAEEKREGYERLLKMQNQRGGRALFQDIKKPAEDEWGKTPDAMKAAMALEKKLNQALLDLHALGSARTDPHLCDFLETHFLDEEVKLIKKMGDHLTNLHRLGGPEAGLGEYLFERLTL
T=0.1, sample=0, score=0.8521, seq_recovery=0.3450 GPAIRSNFSEEICAALNAQIGLERQAATTYEAMAAYFARPDVARPGVAAFFAAQAAEERAHAAALEAYLASRGCTLVETPVPAPEKAEYGDTLEAFELALAMEEEVTAAIQALIALAKANNDPETVAFFDANFVAEQAAHIAELRDYLARLRALGGPNAAEGERRFDEEVL
New Sequence:
GPAIRSNFSEEICAALNAQIGLERQAATTYEAMAAYFARPDVARPGVAAFFAAQAAEERAHAAALEAYLASRGCTLVETPVPAPEKAEYGDTLEAFELALAMEEEVTAAIQALIALAKANNDPETVAFFDANFVAEQAAHIAELRDYLARLRALGGPNAAEGERRFDEEVL
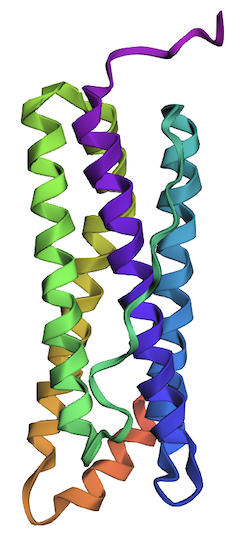
2. Input this sequence into ESMFold and compare the predicted structure to your original.
The predicted structure of the human ferritin light chain generated by the inverse folding with ProteinMPNN is remarkably similar to the original structure.
| Predicted Structure by Inverse Folding | Original Structure |
|---|---|
|
|
Part D. Group Brainstorm on Bacteriophage Engineering
1. Review the Bacteriophage Final Project Goals for engineering the L Protein:
- Increased stability (easiest)
- Higher titers (medium)
- Higher toxicity of lysis protein (hard)
2. Brainstorm session
- Choose one or two main goals from the list that you think you can address computationally (e.g., “stabilize the lysis protein”, “disrupt its interaction with DnaJ”.
- Write a 1-page proposal (bullet points or short paragraphs) describing:
3. Which tools/approaches from recitation you propose using (e.g., “Use Protein Language Models to do in silico mutagenesis, then AlphaFold-Multimer to check complexes.”)
- Why do you think those tools might help solve your chosen sub-problem?
- Name one or two potential pitfalls (e.g., “We lack enough training data on phage-bacteria interactions.”)
- Include a schematic of your pipeline.