Week 5 HW: Protein Design Part II
Part A: SOD1 Binder Peptide Design
Superoxide dismutase 1 (SOD1) is a cytosolic enzyme that converts superoxide radicals into hydrogen peroxide and oxygen. In its native state, it forms a stable homodimer and binds copper and zinc.
Mutations in SOD1 cause familial Amyotrophic Lateral Sclerosis (ALS). Among them, the A4V mutation (Ala to Val at residue 4) leads to one of the most aggressive forms of the disease. The mutation subtly destabilizes the N-terminus, perturbs folding energetics, and promotes toxic aggregation.
Your challenge:
- Design short peptides that bind mutant SOD1.
- Then decide which ones are worth advancing toward therapy.
Part 1: Generate Binders with PepMLM
- Begin by retrieving the human SOD1 sequence from UniProt (P00441) and introducing the A4V mutation.
MATKAVCVLKGDGPVQGIINFEQKESNGPVKVWGSIKGLTEGLHGFHVHEFGDNTAGCTSAGPHFNPLSRKHGGPKDEERHVGDLGNVTADKDGVADVSIEDSVISLSGDHCIIGRTLVVHEKADDLGKGGNEESTKTGNAGSRLACGVIGIAQ
Fig 1. The human SOD1 protein sequence.
MATKVVCVLKGDGPVQGIINFEQKESNGPVKVWGSIKGLTEGLHGFHVHEFGDNTAGCTSAGPHFNPLSRKHGGPKDEERHVGDLGNVTADKDGVADVSIEDSVISLSGDHCIIGRTLVVHEKADDLGKGGNEESTKTGNAGSRLACGVIGIAQ
Fig 2. The human SOD1 variant carrying an A4V mutation.
Using the PepMLM CoLab linked from the HuggingFace PepMLM-650M model card:
Generate four peptides of length 12 amino acids conditioned on the mutant SOD1 sequence.
To your generated list, add the known SOD1-binding peptide FLYRWLPSRRGG for comparison.
Record the perplexity scores that indicate PepMLM’s confidence in the binders.
| Peptide Name | Sequence | Perplexity |
|---|---|---|
| peptide_1 | WRYYVVVAEWGE | 30.94 |
| peptide_2 | WLYYATVARWGK | 20.55 |
| peptide_3 | WHYYVVGLRWWE | 28.21 |
| peptide_4 | WRYYVTGAAWWK | 17.13 |
| known binder peptide | FLYRWLPSRRGG | 20.6 |
Table 1. Results of generating 4 new peptides and the perplexity values against the SOD1_A4V by PepMLM. The known binder is shown in the bottom row for comparison.
Newly generated peptides with varied pseudo perplexity values indicate that some could achieve even better binding than the known binding peptide, FLYRWLPSRRGG, as judged by the lower value in perplexity.
Part 2: Evaluate Binders with AlphaFold3
Navigate to the AlphaFold Server: alphafoldserver.com
For each peptide, submit the mutant SOD1 sequence followed by the peptide sequence as separate chains to model the protein-peptide complex.
Record the ipTM score and briefly describe where the peptide appears to bind.
Does it localize near the N-terminus where A4V sits?
Does it engage the B-barrel region or approach the dimer interface?
Does it appear surface-bound or partially buried?
| Peptides | ipTM Values | Localization Characteristics |
|---|---|---|
| peptide_1 | 0.32 | Surface-bound, may have partial interaction with beta-sheets |
| peptide_2 | 0.33 | Interacts with beta-sheets, proximity to dimer interface residues |
| peptide_3 | 0.49 | Surface-bound, does not interact with beta-sheets |
| peptide_4 | 0.42 | Surface-bound, may have partial interaction with beta-sheets |
| known binder peptide | 0.38 | Surface-bound, does not interact with beta-sheets |
Table 2. Modeling a peptide-protein complex. Interface predicted template modeling (ipTM) scores were obtained from AlphaFold analysis.
None of the peptides engages at the N-terminus where the A4V mutation is found, except peptide_2, which may interact with residue 153, the last residue at the C-terminus participating in the dimer interface.
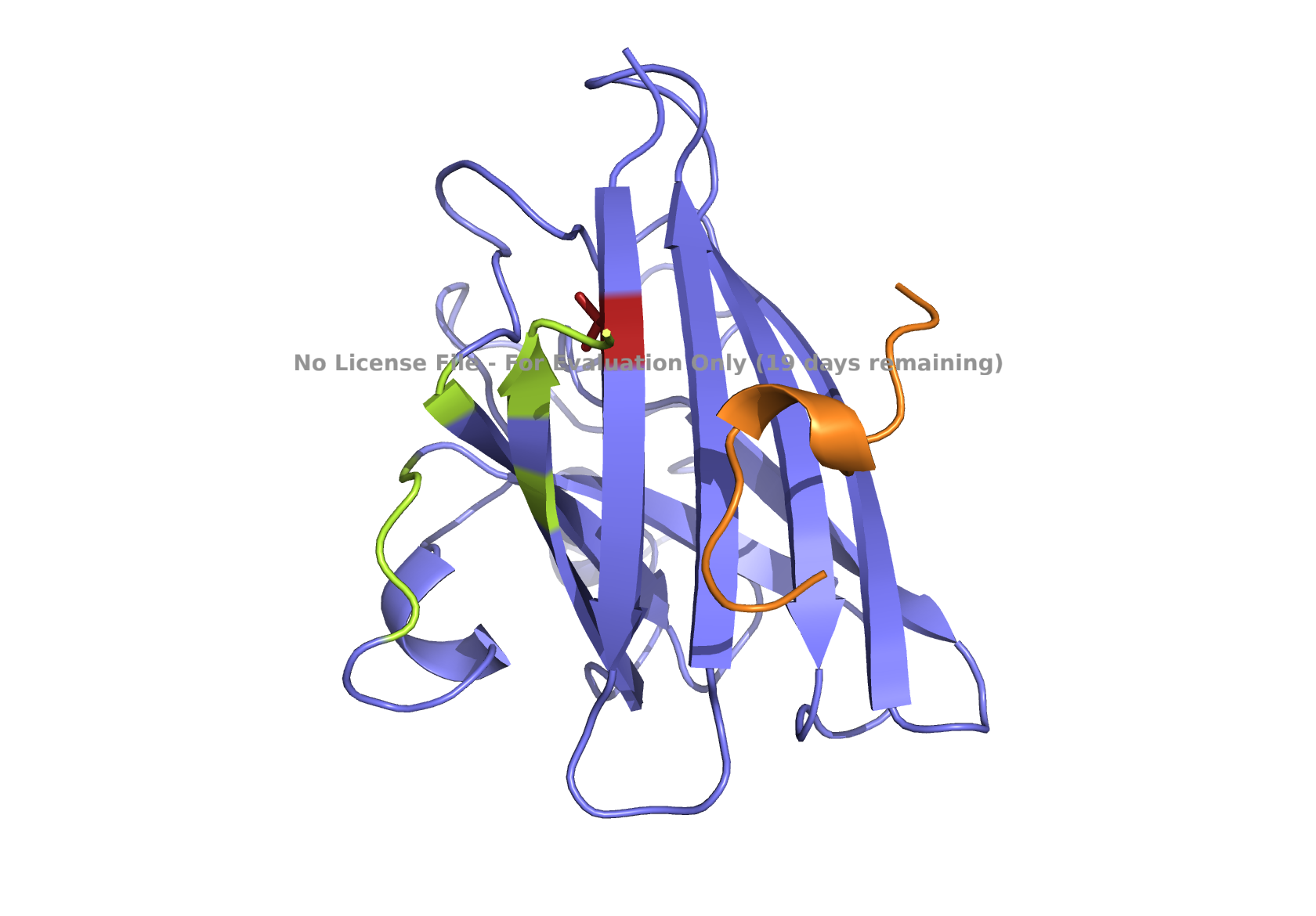
| (A) peptide_1 | (B) peptide_2 |
|---|---|
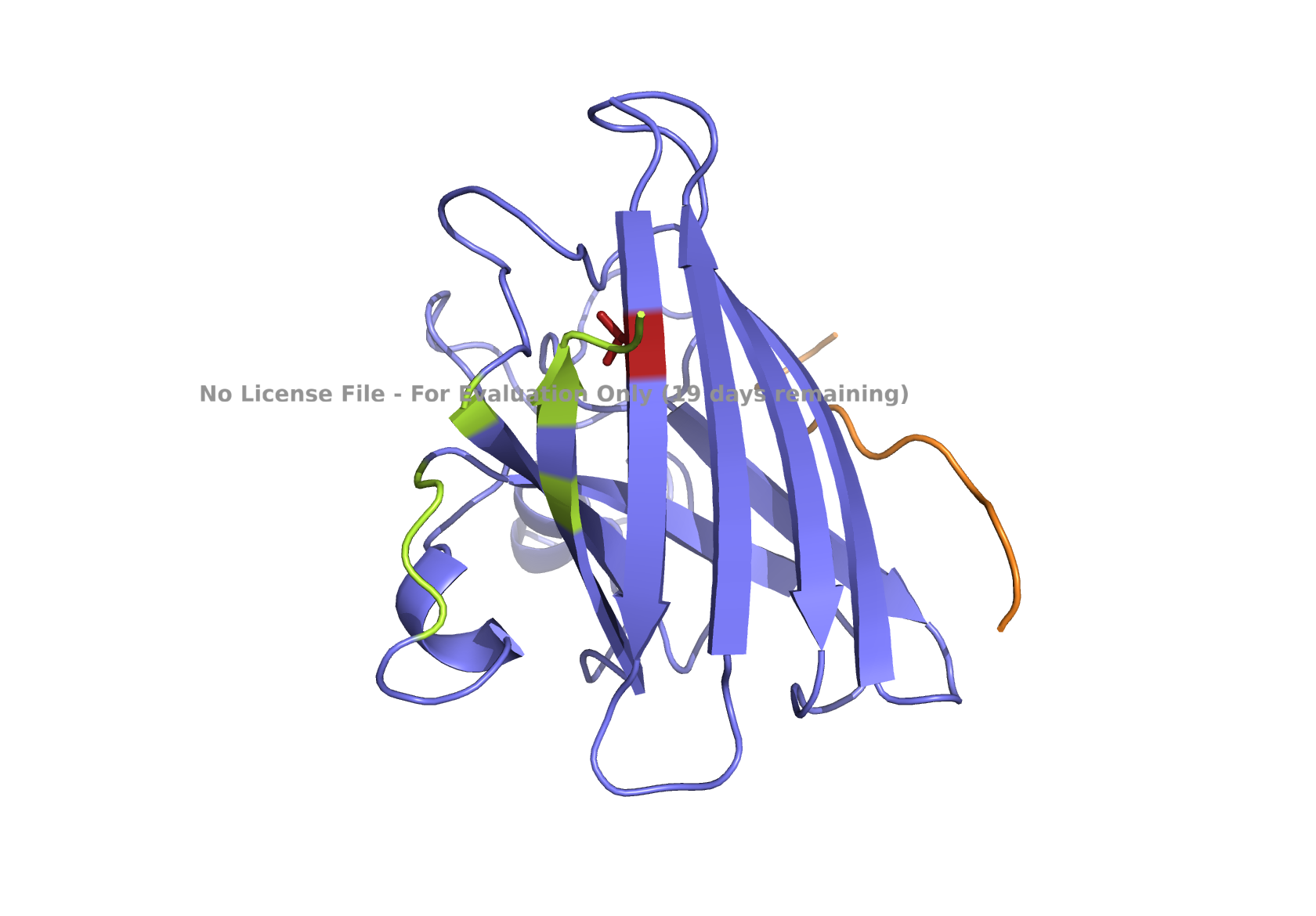
| 
|
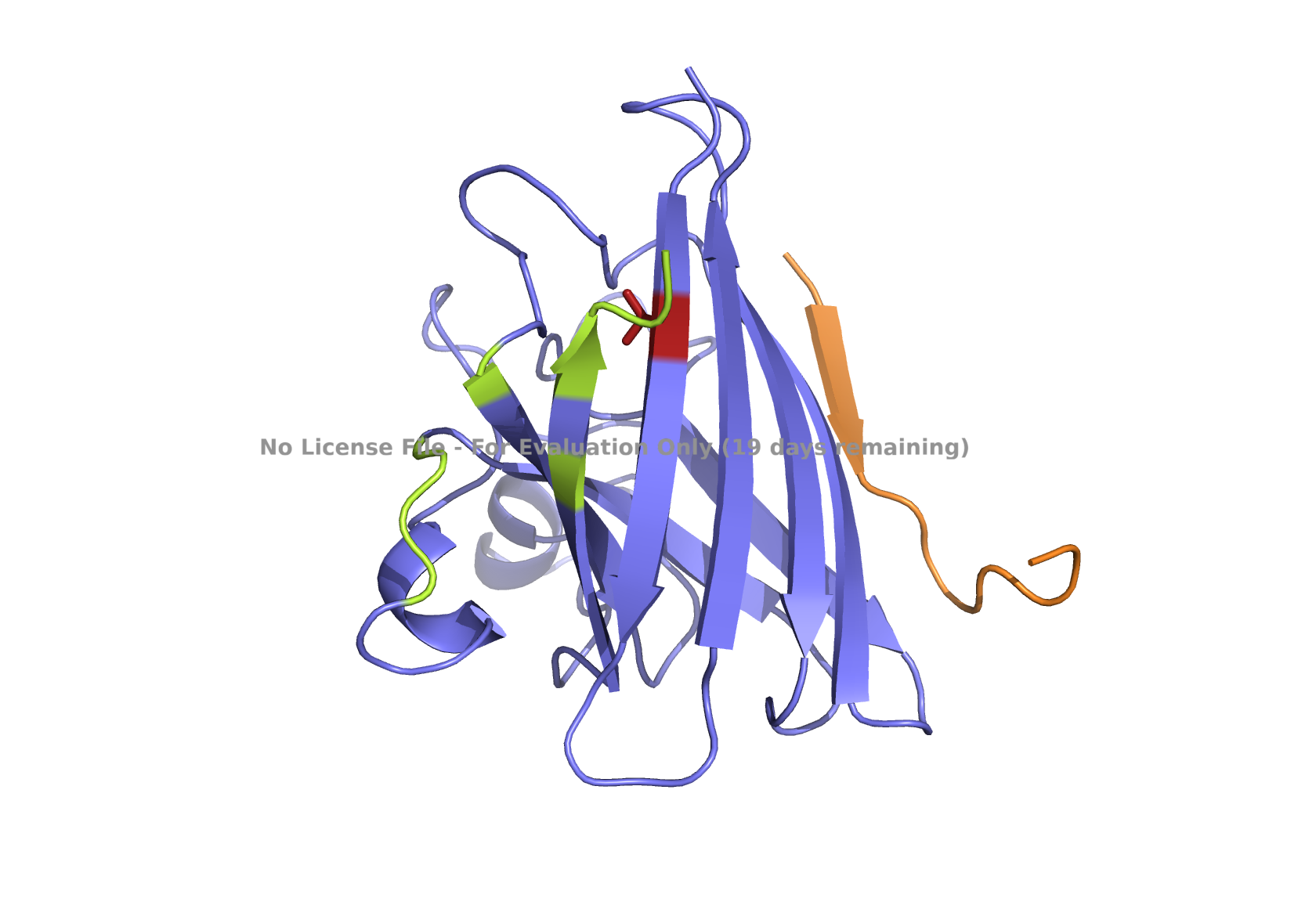
| (C) peptide_3 | (D) peptide_4 |
|---|---|
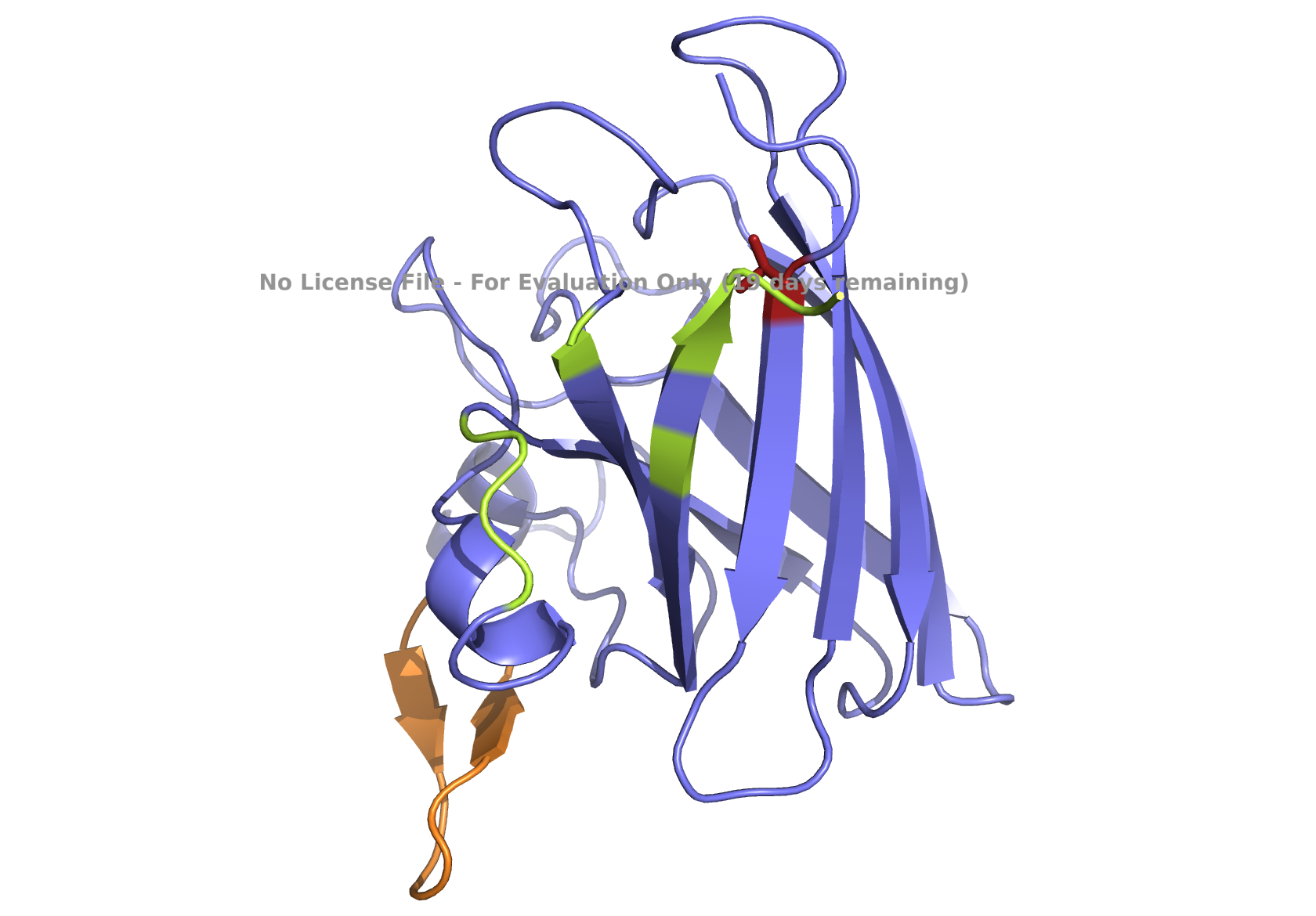
| 
|
| (E) known binder peptide | |
|---|---|
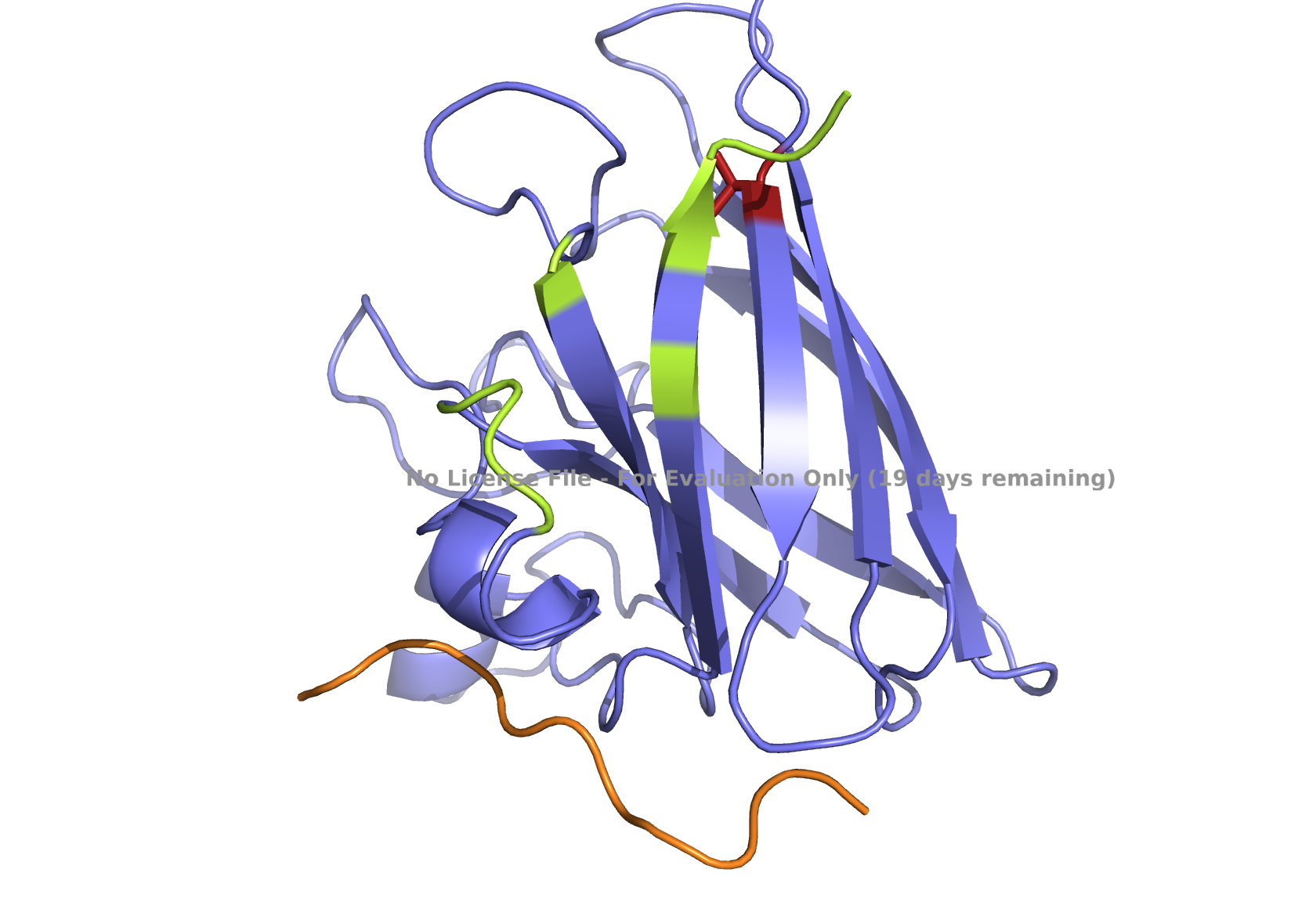
| 
|
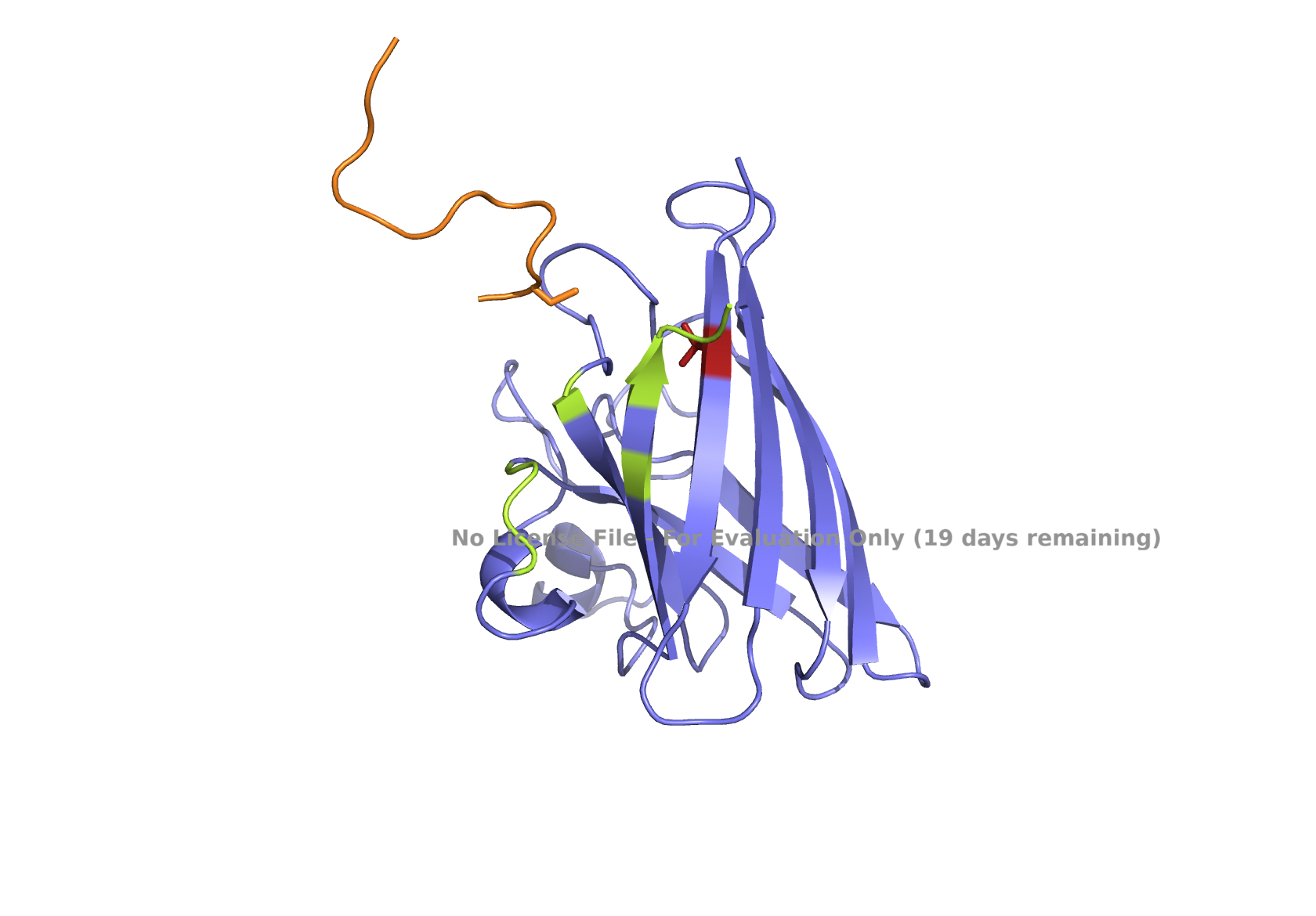
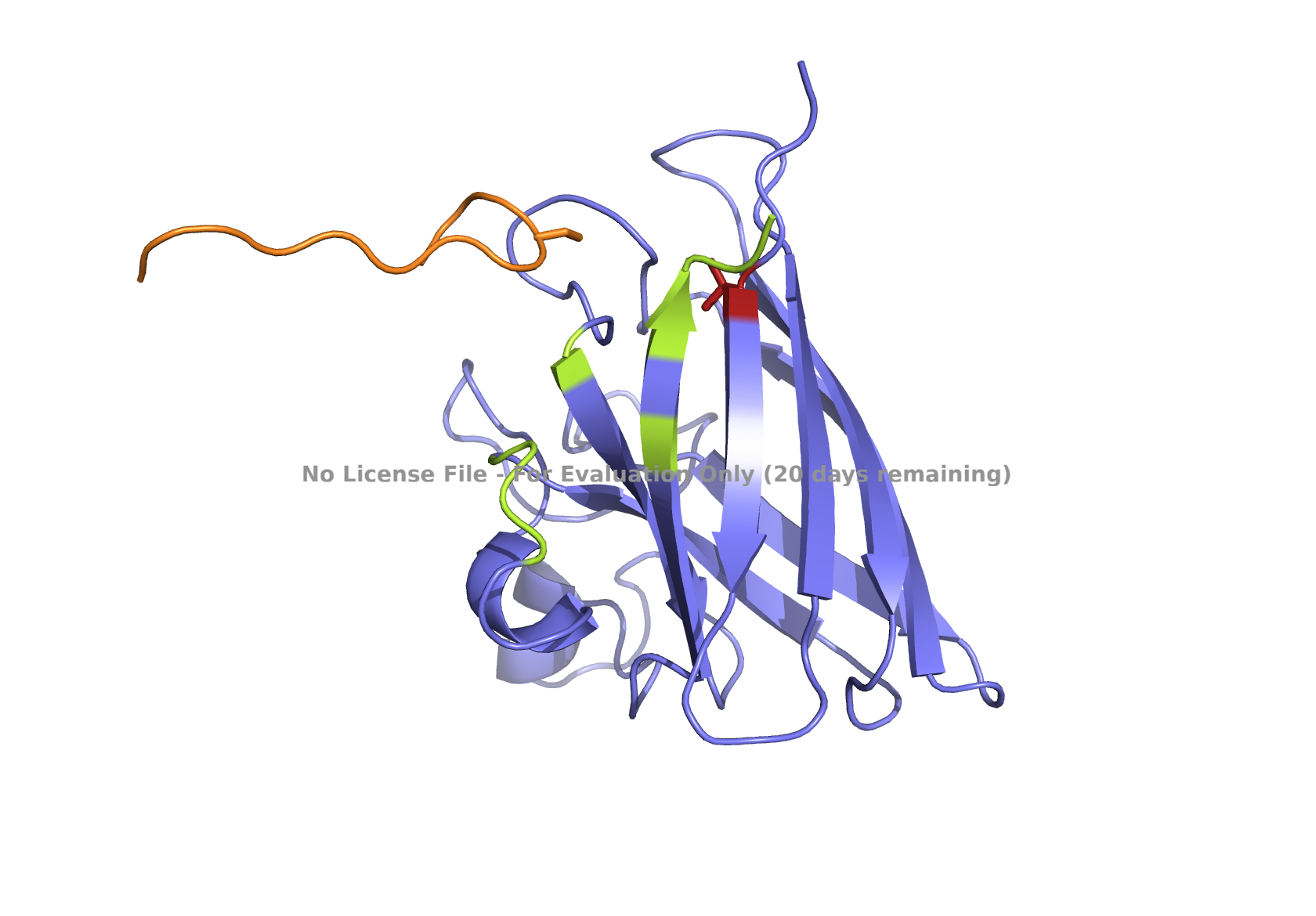
Fig 3. AlphaFold modeling of pepMLM peptides with SOD1_A4V visualized by PyMol. SOD1_A4V monomer (blue) and binder peptides (orange). Highlights: A4V residue with sidechain in stick (red) and dimer interface residues (green) 50–53, 114, 148, and 150–153, as referenced in Hough et al. 2004.
- In a short paragraph, describe the ipTM values you observe and whether any PepMLM-generated matches or exceed the known binder.
All peptides, including the benchmark, have ipTM values that fall between 0.3 and 0.5. Interpretation of the 0.3 – 0.5 range indicates weak interactions, but they may still occur. As ChatGPT research indicated, AlphaFold struggles with short peptides, as short as 12 amino acids. Having a benchmark, a known binder peptide, helps interpret the values, even if categorized as weak.
A low perplexity score of a peptide indicates that it is more likely to occur based on a protein language model. Among the new peptides, the perplexity score of peptide_4, 17.13, which is the lowest, is even lower than the perplexity score of the known binder peptide, 20.6. Two of the peptides have higher perplexity scores, peptide_1 and peptide_3, 30.94 and 28.21, respectively, placing these in the disfavored category.
| ipTM score | Interpretation |
|---|---|
| > 0.7 | confident interaction |
| 0.5 - 0.7 | possible interaction |
| 0.3 - 0.5 | weak interaction |
| < 0.3 | likely no interaction |
Table 3. Reference for interpreting ipTM scores.
I’ve used ChatGPT research to interpret the results.
Prompt:
Can you explain the results from AlphaFold structural analysis for binder peptides to the SOD1_A4V variant? Results in Tables 1 and 2.
Part 3: Evaluate Properties of Generated Peptides in the PeptiVerse
Structural confidence alone is insufficient for therapeutic development. Using PeptiVerse, let’s evaluate the therapeutic properties of your peptide! For each PepMLM-generated peptide:
Paste the peptide sequence
Paste the A4V mutant SOD1 sequence in the target field.
Check the boxes
- Predicted binding affinity
- Solubility
- Hemolysis probability
- Net charge (pH 7)
- Molecular weight
Compare these predictions to what you observed structurally with AlphaFold3. In a short paragraph, describe what you see. Do peptides with higher ipTM also show stronger predicted affinity? Are any strong binders predicted to be hemolytic or poorly soluble? Which peptide best balances predicted binding and therapeutic properties?
Choose one peptide you would advance and justify your decision briefly.
| Peptide | Property | Prediction | Value | Unit |
|---|---|---|---|---|
| peptide_1, WRYYVVVAEWGE | Binding Affinity | Weak binding | 6.750 | pKd/pKi |
| Solubility | Soluble | 1.000 | Probability | |
| Hemolysis | Non-hemolytic | 0.258 | Probability | |
| Net Charge (pH 7) | -1.23 | |||
| Molecular Weight | 1556.7 | Da | ||
| peptide_2, WLYYATVARWGK | Binding Affinity | Weak binding | 6.729 | pKd/pKi |
| Solubility | Soluble | 1.000 | Probability | |
| Hemolysis | Non-hemolytic | 0.054 | Probability | |
| Net Charge (pH 7) | 1.76 | |||
| Molecular Weight | 1513.7 | Da | ||
| peptide_3, WHYYVVGLRWWE | Binding Affinity | Weak binding | 6.733 | pKd/pKi |
| Solubility | Soluble | 1.000 | Probability | |
| Hemolysis | Non-hemolytic | 0.135 | Probability | |
| Net Charge (pH 7) | -0.15 | |||
| Molecular Weight | 1693.9 | Da | ||
| peptide_4, WRYYVTGAAWWK | Binding Affinity | Medium binding | 7.059 | pKd/pKi |
| Solubility | Soluble | 1.000 | Probability | |
| Hemolysis | Non-hemolytic | 0.041 | Probability | |
| Net Charge (pH 7) | 1.76 | |||
| Molecular Weight | 1586.8 | Da | ||
| known binder peptide, FLYRWLPSRRGG | Binding Affinity | Weak binding | 5.968 | pKd/pKi |
| Solubility | Soluble | 1.000 | Probability | |
| Hemolysis | Non-hemolytic | 0.047 | Probability | |
| Net Charge (pH 7) | 2.76 | |||
| Molecular Weight | 1507.7 | Da |
Table 4. Summary of the PeptiVerse analysis.
I’ve chosen peptide_2 for the therapeutic advancement with the following reasons:
Based on the PeptiVerse evaluation of the new peptides, the binding affinities of 3 out of 4 were predicted to be weak (Table 4). The known binder peptide is also scored with weak binding affinity in PeptiVerse. Only one peptide, peptide_4, had medium affinity, with a higher affinity score than the other peptides. However, peptide_4 is localized elsewhere, as determined by AlphaFold structure analysis, not near the A4V or dimer interface (Fig 1D).
On the other hand, the second-best-ranked model for the peptide_2 showed that this peptide could be localized near AV4 and closer to the dimer interface (Fig 1B), which may potentially interact with one of the dimer interface residues, 153, and prevent aggregation. Notably, other model predictions for this peptide, including the best-ranking one, showed that the peptide might also be located elsewhere.
Given the location of the second-best predicted model, I favored peptide_2 because it could potentially have a more relevant binding location. All peptides are scored as weak binding but possible interactions (ipTM between 0.3 and 0.5). The visualization of peptide_2 is shown in Fig 1B, reflecting its predicted binding location based on that second-best model. The ranking scores for the best and second-best were 0.43 and 0.42, respectively, as reported in the AlphaFold analysis. ipTM is 0.33 for the best and 0.31 for the second-best. Figs. 1A, 1C, 1D, and 1E , show the best-ranked models of peptide_1, peptide_3, peptide_4, and the known binder peptide, respectively.
Part 4: Generate Optimized Peptides with moPPIt
Now, move from sampling to controlled design. moPPIt uses Multi-Objective Guided Discrete Flow Matching (MOG-DFM) to steer peptide generation toward specific residues and optimize binding and therapeutic properties simultaneously. Unlike PepMLM, which samples plausible binders conditioned on just the target sequence, moPPIt lets you choose where you want to bind and optimize multiple objectives at once.
Open the moPPIt CoLab linked from the HuggingFace moPPIt model card.
Make a copy and switch to a GPU runtime.
In the notebook,
- Paste your A4V mutant SOD1 sequence.
- Choose specific residue indices on SOD1 that you want your peptide to bind (for example, residues near position 4, the dimer interface, or another surface patch).
- Set peptide length to 12 amino acids.
- Enable motif and affinity guidance (and solubility/hemolysis guidance if available). Generate peptides.
- After generation, briefly describe how these moPPIt peptides differ from your PepLMM peptides. How would you evaluate these peptides before advancing them to clinical studies?
| moPPIt Peptide | Binder Sequence | Hemolysis | Solubility | Affinity | Motif |
|---|---|---|---|---|---|
| m_peptide_1 | TEQKKFTCRTQD | 0.98065103 | 0.91666669 | 5.93099356 | 0.92896324 |
| m_peptide_2 | YQKCLVRETTGV | 0.96973951 | 0.75 | 6.67283011 | 0.86192995 |
| m_peptide_3 | KEKKQRVQCTDG | 0.97739551 | 0.91666669 | 5.56807947 | 0.85476714 |
| m_peptide_4 | SVKTTHCEQGKP | 0.97208287 | 0.83333331 | 5.50222826 | 0.84767622 |
Table 5. moPPIt-generated binder peptides and their in-silico attributes. The following weight adjustments were made for targeting position 4, specifically: Hemolysis=1, Solubility=1, Affinity=1, Motif=1.
Unlike the PepMLM peptides, none of the moPPIt peptides achieved desirable low scores on hemolytic activity prediction. Instead, all peptides had high hemolysis scores. This is due to the region surrounding position 4 and the binder peptide that has a high proportion of hydrophobicity, resulting in high scoring of hemolytic activity. A further optimization is needed to reduce net hydrophobicity and charge residues in the peptide binder. As peptides were designed for a specific region, it can be harder to achieve every desired trait, requiring the consideration of trade-offs as well.
As a next step, I applied more stringent weight adjustment to optimize peptide binders in moPPIt (see results in Table 6 and Fig 4).
| moPPIt Peptide | Binder Sequence | Hemolysis | Solubility | Affinity | Motif |
|---|---|---|---|---|---|
| m2_peptide_1 | DTECTQTRLKKS | 0.9731365 | 0.916666687 | 5.666040897 | 0.787758291 |
| m2_peptide_2 | YDVTTRLYFGRW | 0.94705141 | 0.666666627 | 6.606595039 | 0.334373683 |
| m2_peptide_3 | KDEFDCKPCYNL | 0.93650597 | 0.75 | 7.194892883 | 0.706446946 |
| m2_peptide_4 | TEKTIEKKQWCA | 0.98217107 | 0.75 | 6.305016994 | 0.889526725 |
| m2_peptide_5 | SKECGTLRFKQR | 0.96697627 | 0.833333313 | 6.679063797 | 0.910008729 |
| m2_peptide_6 | YKKETVKTNQFH | 0.97450537 | 0.833333313 | 5.35283041 | 0.899615645 |
| m2_peptide_7 | TTSTHICTCPLC | 0.87881172 | 0.75 | 5.995385647 | 0.758235991 |
| m2_peptide_8 | TGDTTCLKKQHF | 0.97177865 | 0.833333313 | 5.857715607 | 0.851789355 |
Table 6. MoPPIt-generated binder peptides and their in-silico attributes, testing more stringent weight adjustment for optimal binder generation. The following weight adjustments were made, targeting position 4, specifically: Hemolysis=10, Solubility=5, Affinity=5, Motif=10.
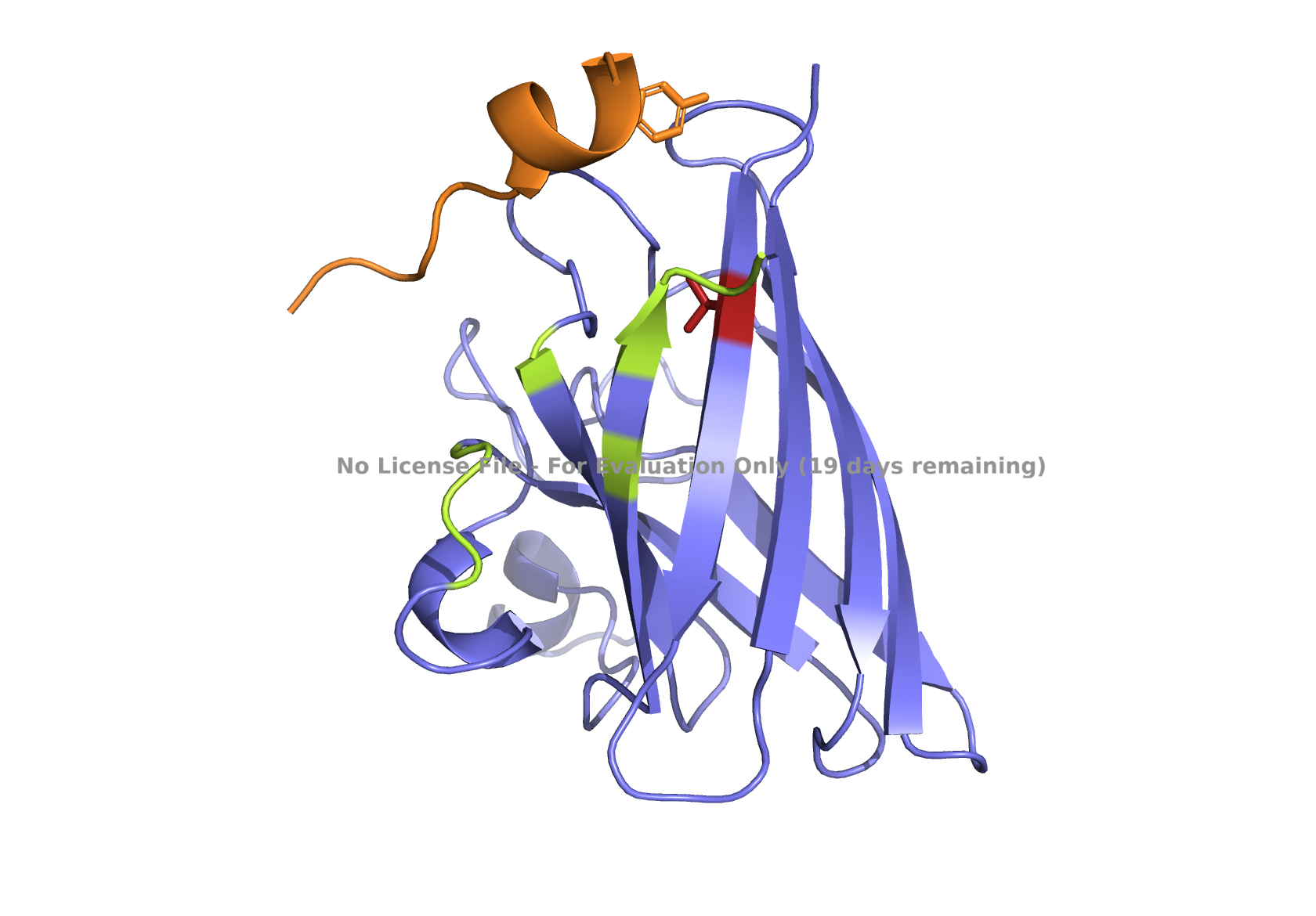
| (A) m2_peptide_1 | (B) m2_peptide_2 |
|---|---|
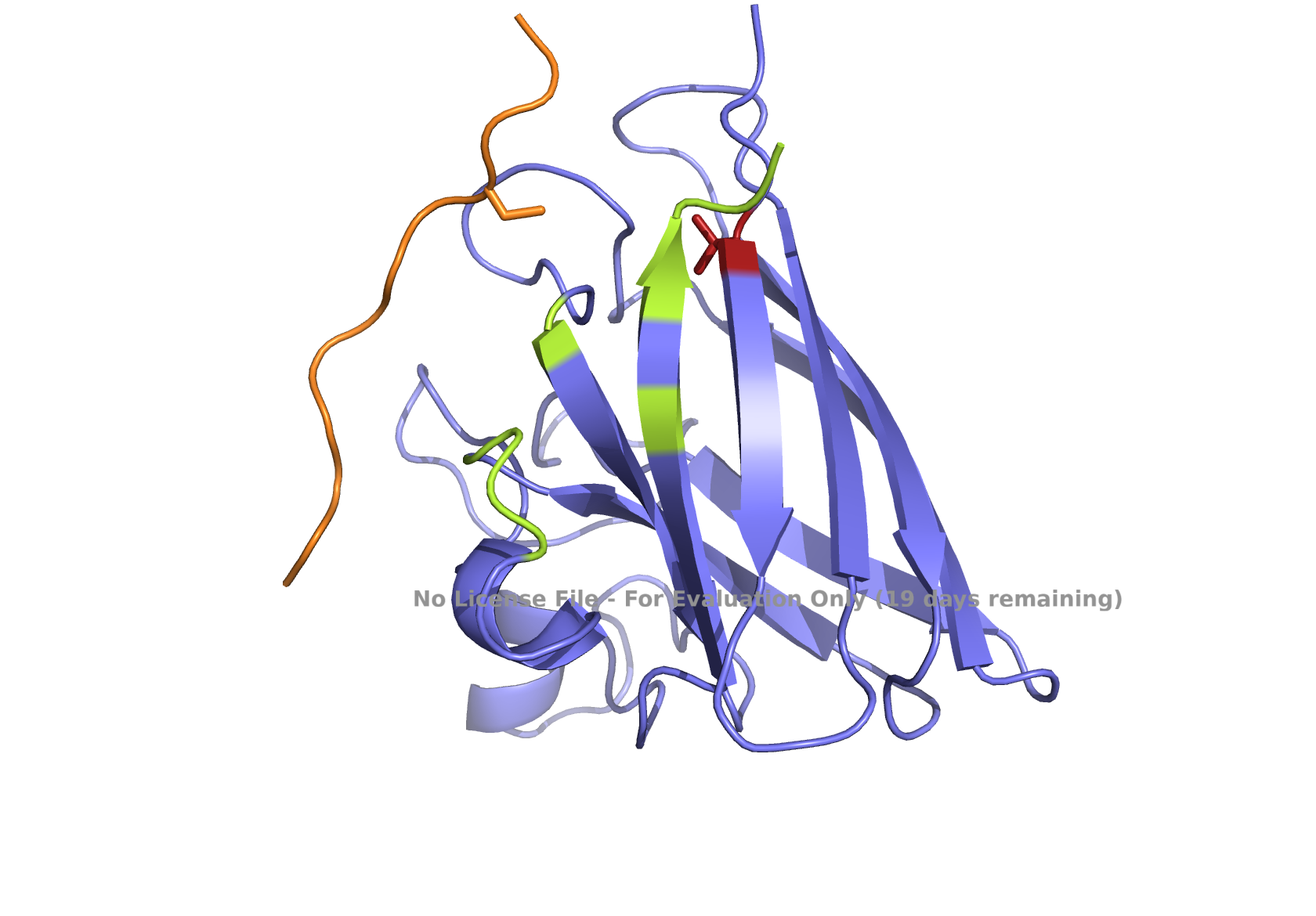
| 
|
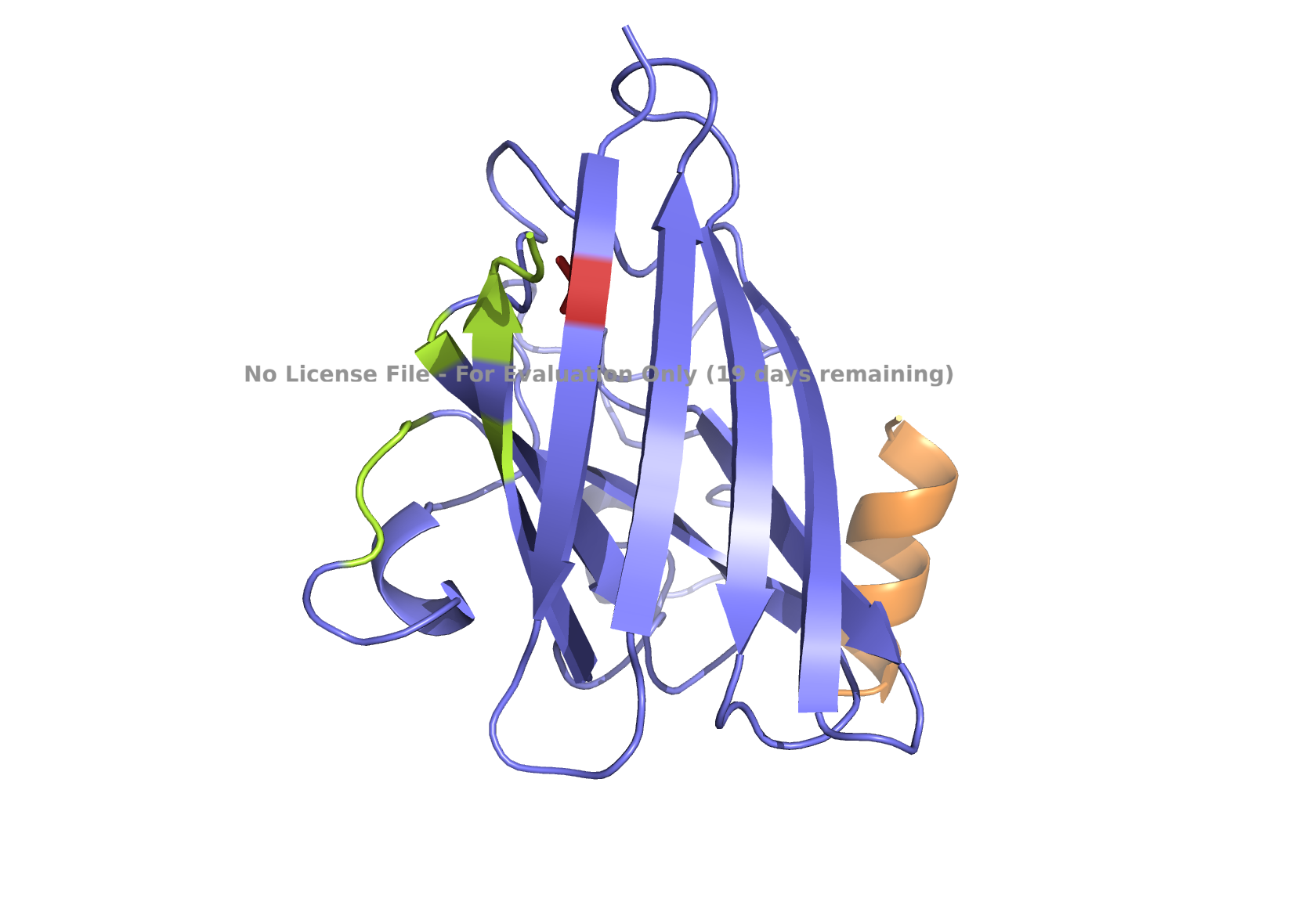
| (C) m2_peptide_3 | (D) m2_peptide_4 |
|---|---|
| 
|
| (E) m2_peptide_5 | (F) m2_peptide_6 |
|---|---|
| 
|
| (G) m2_peptide_7 | (H) m2_peptide_8 |
|---|---|
| 
|
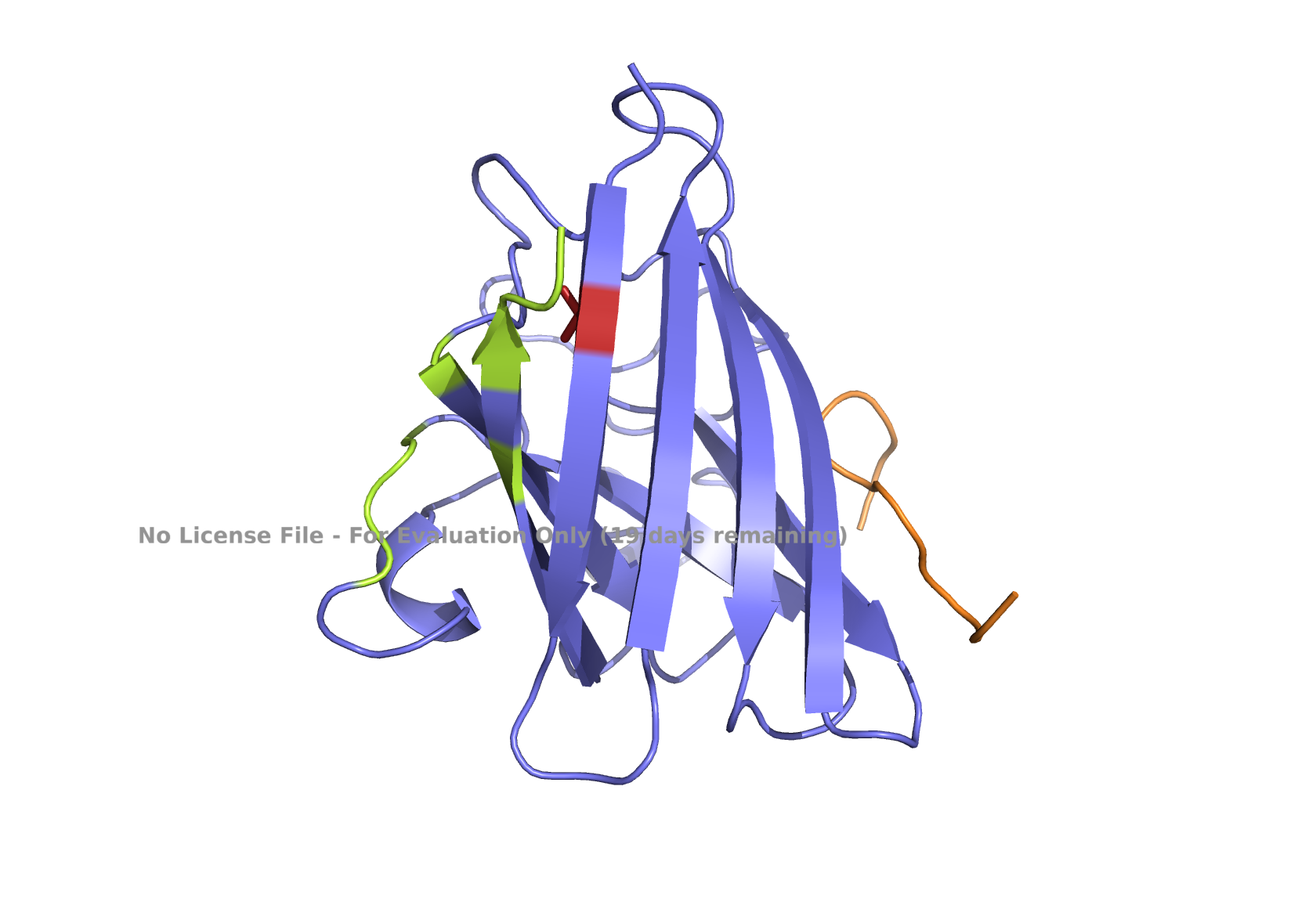
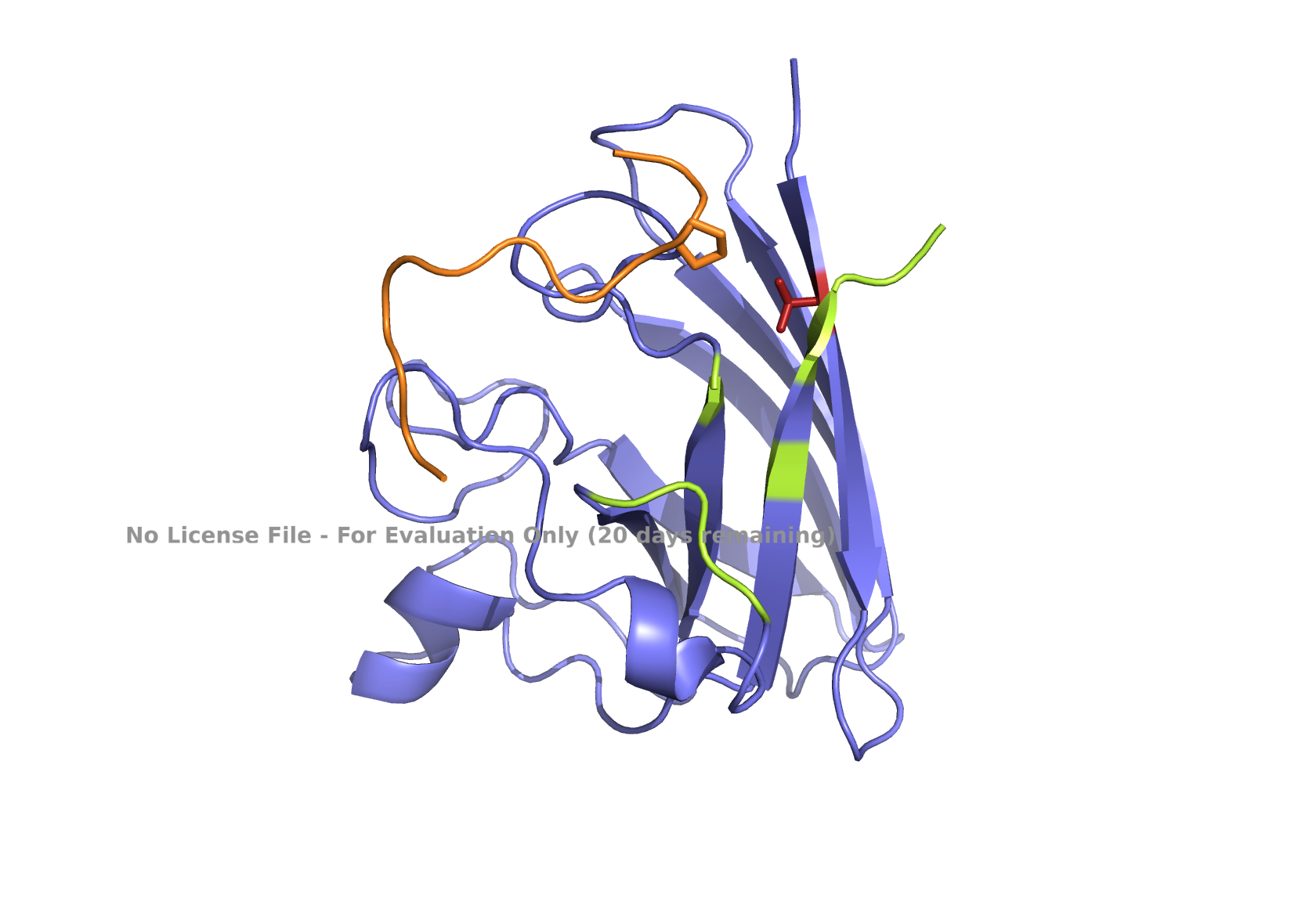
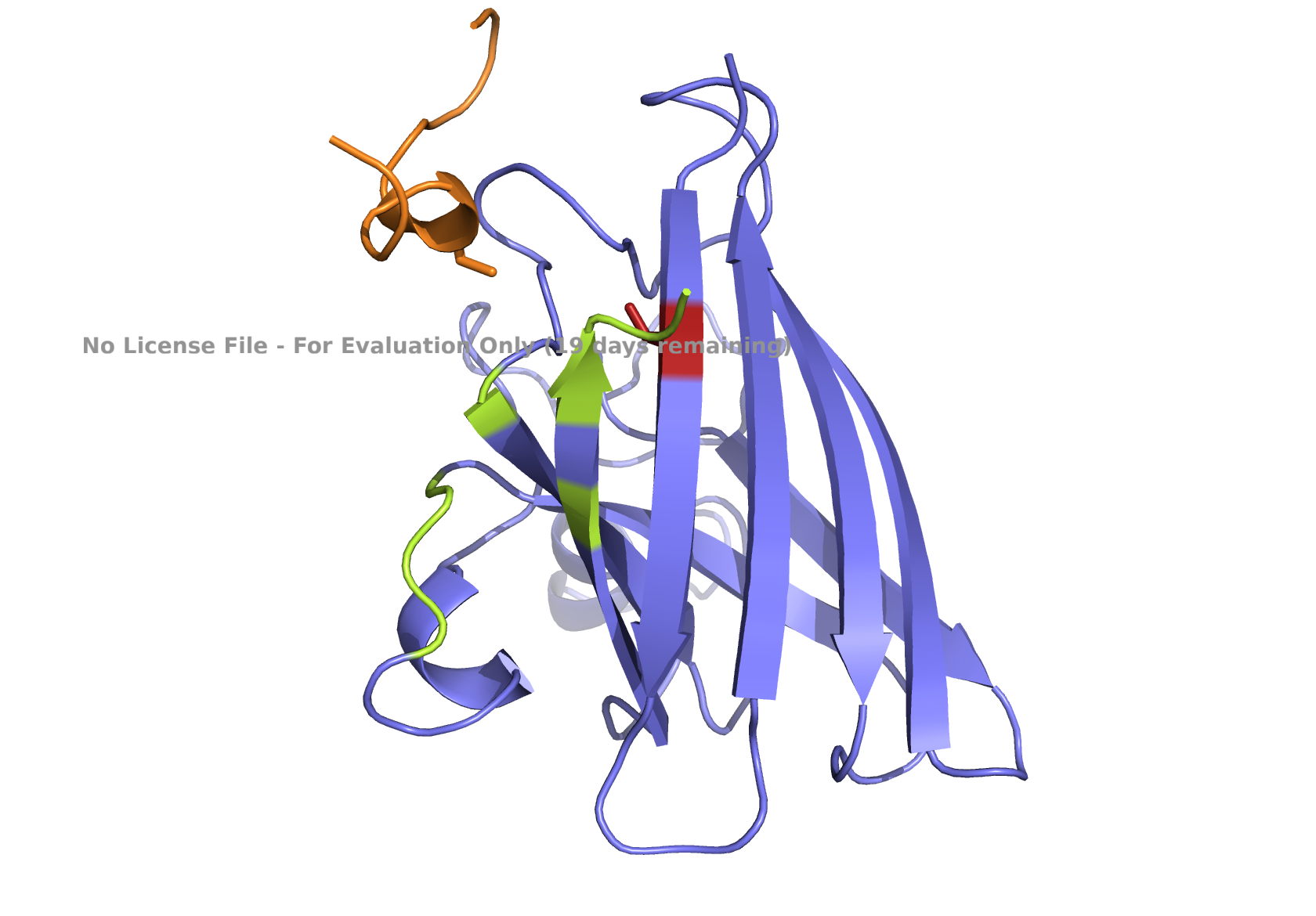
Fig 4. AlphaFold modeling of moPPIt peptides with SOD1_A4V visualized by PyMol. SOD1_A4V monomer (blue) and binder peptides (orange). Highlights: A4V residue with sidechain in stick (red), dimer interface residues (green) 50-53, 114, 148, and 150-153, and sidechains of residues highlighted in peptide binders possibly interacting with A4V in stick (orange): m2_peptide_1, residue 4C; m2_peptide_3, residue 10V; m2_peptide_4, residue 11C; m2_peptide_5, residue 4C; m2_peptide_7, residue 10P, and m2_peptide_8, residue 7C. Based on the best-scoring models of m2_peptide_2 and m2_peptide_6, these binders did not interact with residue A4V.
A new set of moPPIt-generated binder peptides targeting position 4 has still fallen short in achieving low hemolysis scores under the conditions under which the moPPIt generator was run. This indicates further optimization is still needed.
Affinity scores ranged between 5.35 and 7.19, not significantly different from the previous set where the weights were less stringent. Notably, the motif score for the m2_peptide_2 was significantly low, 0.33, and this peptide is localized elsewhere. m2_peptide_6 also localized elsewhere, even though the motif score was higher, 0.89, and the reason for this is unclear. The rest of the moPPIt peptides had expected localization relative to position 4, and by visual inspection, they may interact with position 4 and the surrounding hydrophobic dimer interface, indicating promise for advancement. The only issue is the high hemolytic activity. To achieve less hemolytic activity, hydrophobic amino acids can be replaced by less hydrophobic ones, and charged residues can be balanced. As Gemini AI research also suggests, there are other approaches to reduce hemolytic activity, such as peptide cyclization and conjugating peptides to polymers.
I’ve used Gemini AI for research.
Prompts:
Can you explain why hemolytic activity is seen in some peptides designed for therapeutic purposes? How to avoid hemolytic activity in peptides for better therapeutics?
Part B: BRD4 Drug Discovery Platform Tutorial (Optional)
Part C: Final Project: L-Protein Mutants
High-level summary: The objective of this assignment is to improve the stability and auto-folding of the lysis protein of a MS2-phage. This mechanism is to key to the understanding of how phages can potentially solve antibiotic resistance.
The objective is to improve the stability and autofolding of the lysis protein. More specifically, we want to engineer the lysis protein to increase the ability of MS2 to overcome a common E. coli resistance mechanism: a single point mutation in DnaJ prevents the binding of the lysis protein. We can attempt this by mutating the lysis protein to change its properties. Together, we aim for finding mutations that change the lysis protein one of the following ways: (1) an independence of lysis protein processing from DnaJ or other bacterial chaperones or (2) a faster or more efficient killing of E. coli to reduce the window in which the host can acquire resistance (3) higher lysis protein expression. In the course of this class, we will proceed through the following stages to create and test new MS2 phage mutants:
In this subset:
Stage 1: Engineer novel L-protein mutants using protein design tools
Stage 2: Synthesize the L-protein mutant gene via Twist
Stage 3: Clone the L-protein mutant gene into a plasmid using Gibson Assembly
Stage 4: Test the L-protein mutant’s structural integrity using the Nuclera system
Stage 5: Test the L-protein in E. coli with plaque assays