Week 2 HW: DNA read, write and edit
Part 1: Benchling & In-silico Gel Art
The process of simulation of Lambda genome Restriction Enzyme Digestion in Benchling:
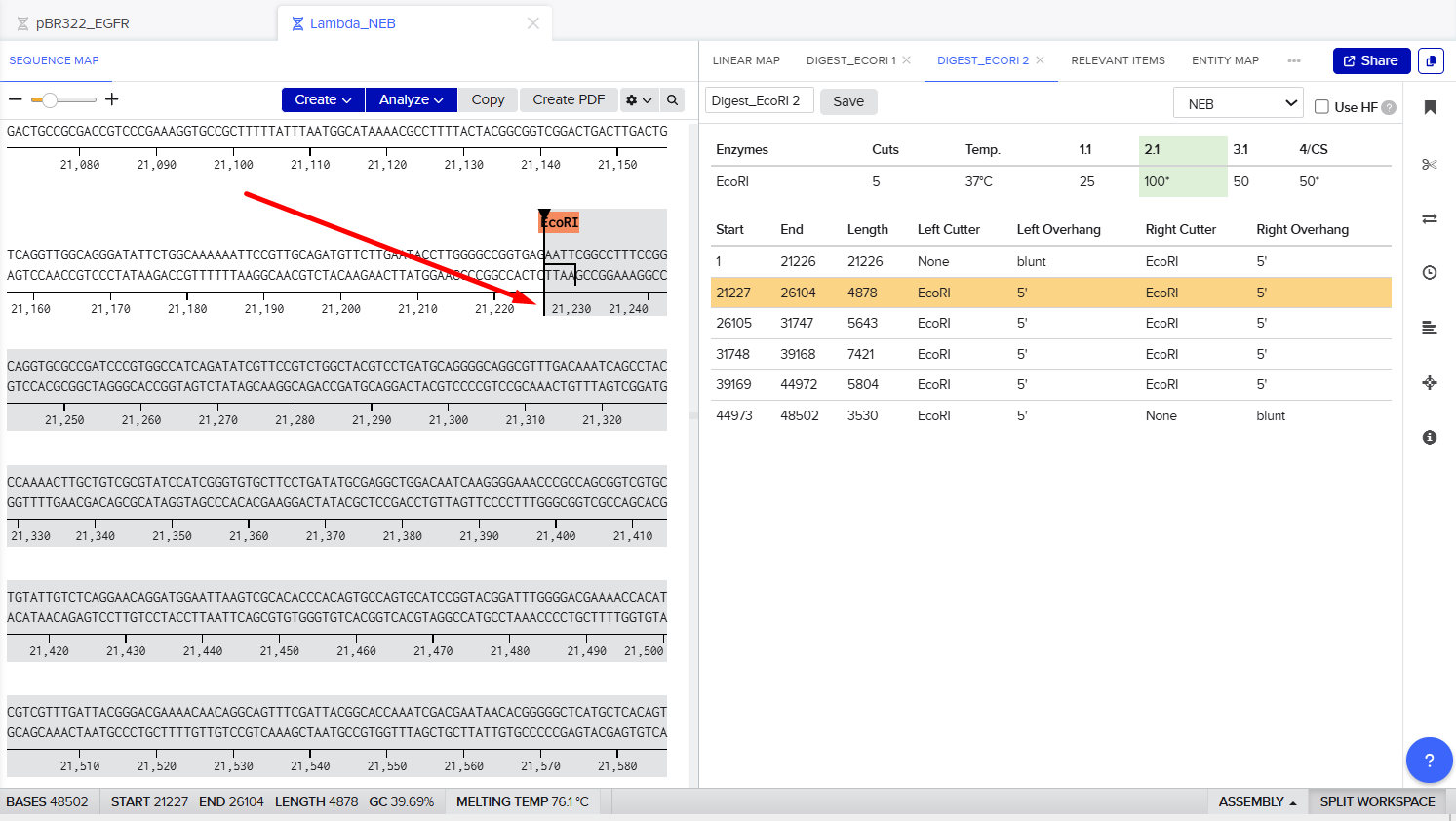
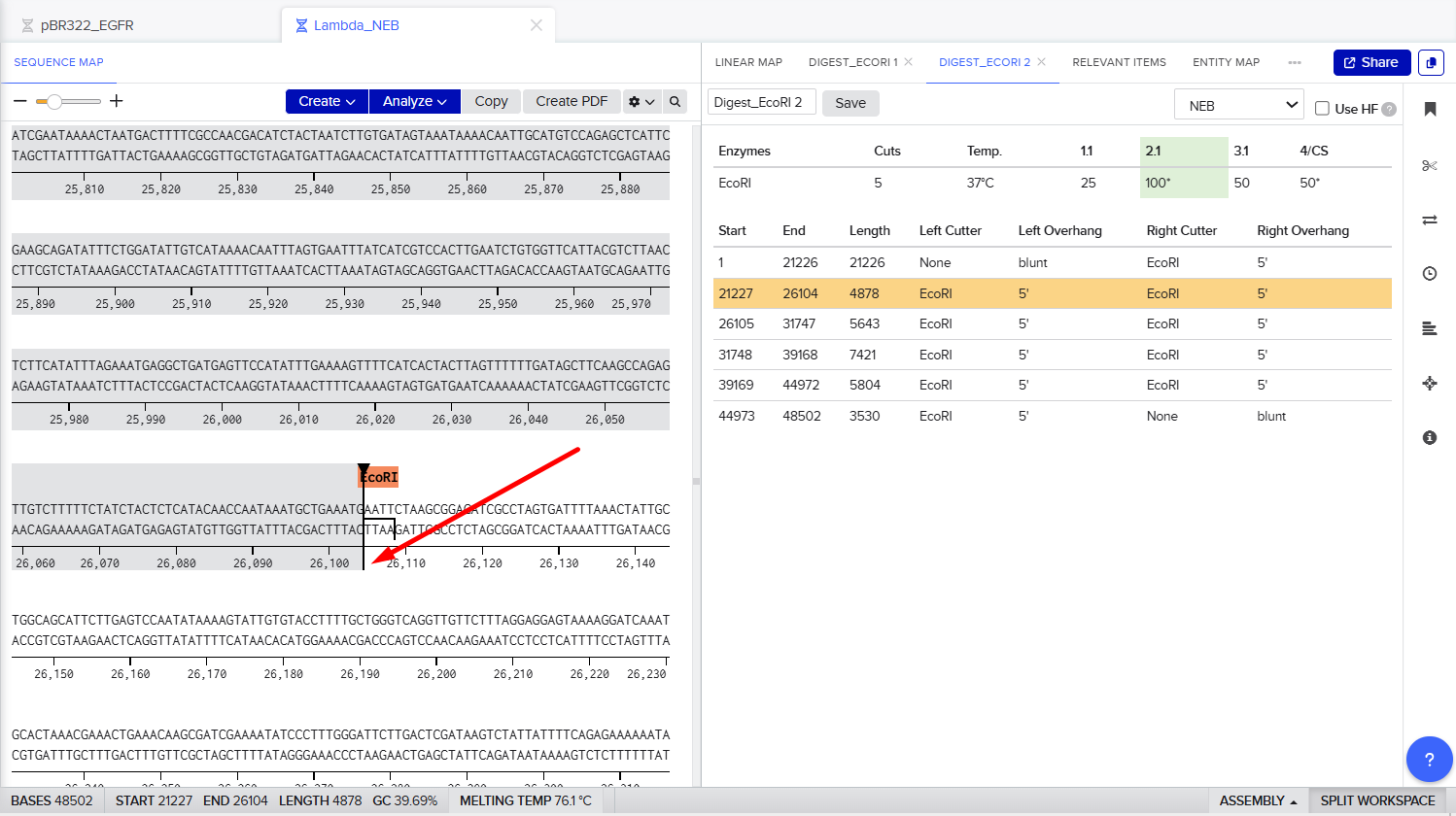
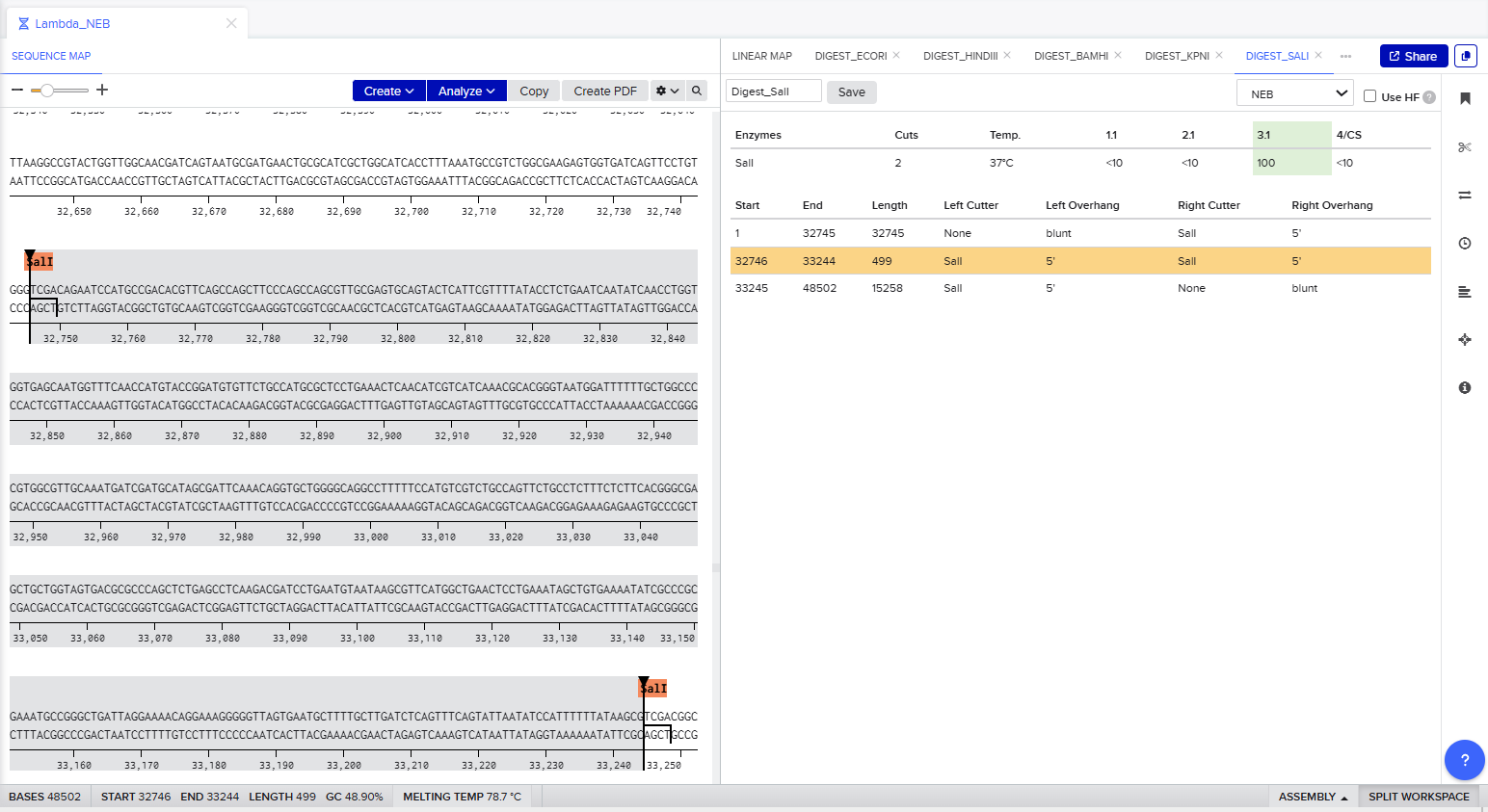
Final result:

For Gel art pattern I used KpnI, SacI and SalI Restriction Enzyme Digestion.
It is a pattern of wave.

Part 3: DNA Design Challenge
I chose the RecA protein from Enterococcus faecalis. I chose this protein because I am interested in horizontal gene transfer in bacteria. RecA is one of the key proteins involved in homologous recombination and DNA repair, which are important processes during horizontal gene transfer and bacterial adaptation. Studying RecA helps understand how bacteria exchange genetic material and acquire new traits such as antibiotic resistance.
3.1. Choose your protein
The protein sequence was obtained from UniProt. Protein: RecA from Enterococcus faecalis sp|P42444|RECA_ENTFA Protein RecA OS=Enterococcus faecalis (strain ATCC 700802 / V583) OX=226185 GN=recA PE=3 SV=2
MADDRKVALDAALKKIEKNFGKGSIMKLGEKADQKISTIPSGSLALDVALGVGGYPRGRI IEVYGPESSGKTTVSLHAIAEVQRNGGTAAFIDAEHALDPQYAEKLGVNIDELLLSQPDT GEQGLEIADALVSSGAIDIVVIDSVAALVPRAEIDGEMGASHVGLQARLMSQALRKLSGS INKTKTIAIFINQIREKVGVMFGNPETTPGGRALKFYATVRLEVRRAEQLKQGTDIVGNR TKIKVVKNKVAPPFKVAEVDIMYGQGISQEGELLDMAVEKDLISKSGAWYGYKEERIGQG RENAKQYMADHPEMMAEVSKLVRDAYGIGDGSTITEEAEGQEELPLDE
3.2. Reverse Translation: Protein Sequence to DNA Sequence
To obtain the nucleotide sequence corresponding to the RecA protein, I used the reverse translation tool from Sequence Manipulation Suite. Reverse translation converts an amino acid sequence into a possible DNA sequence based on the genetic code.
RecA protein DNA sequence
atggcggatgatcgcaaagtggcgctggatgcggcgctgaaaaaaattgaaaaaaactttggcaaaggcagcattatgaaactgggcgaaaaagcggatcagaaaattagcaccattccgagcggcagcctggcgctggatgtggcgctgggcgtgggcggctatccgcgcggccgcattattgaagtgtatggcccggaaagcagcggcaaaaccaccgtgagcctgcatgcgattgcggaagtgcagcgcaacggcggcaccgcggcgtttattgatgcggaacatgcgctggatccgcagtatgcggaaaaactgggcgtgaacattgatgaactgctgctgagccagccggataccggcgaacagggcctggaaattgcggatgcgctggtgagcagcggcgcgattgatattgtggtgattgatagcgtggcggcgctggtgccgcgcgcggaaattgatggcgaaatgggcgcgagccatgtgggcctgcaggcgcgcctgatgagccaggcgctgcgcaaactgagcggcagcattaacaaaaccaaaaccattgcgatttttattaaccagattcgcgaaaaagtgggcgtgatgtttggcaacccggaaaccaccccgggcggccgcgcgctgaaattttatgcgaccgtgcgcctggaagtgcgccgcgcggaacagctgaaacagggcaccgatattgtgggcaaccgcaccaaaattaaagtggtgaaaaacaaagtggcgccgccgtttaaagtggcggaagtggatattatgtatggccagggcattagccaggaaggcgaactgctggatatggcggtggaaaaagatctgattagcaaaagcggcgcgtggtatggctataaagaagaacgcattggccagggccgcgaaaacgcgaaacagtatatggcggatcatccggaaatgatggcggaagtgagcaaactggtgcgcgatgcgtatggcattggcgatggcagcaccattaccgaagaagcggaaggccaggaagaactgccgctggatgaa
3.3. Codon Optimization
Codon optimization is important because different organisms prefer different codons to encode the same amino acid. Although the genetic code is universal, some codons are used more frequently in certain species. Using preferred codons improves translation efficiency, increases protein expression, and can improve protein stability.
I chose to optimize the RecA gene for Escherichia coli because E. coli is one of the most commonly used organisms for recombinant protein production. It grows quickly, is inexpensive to culture, and has many well-established molecular biology tools available. Since RecA is a bacterial protein and is not highly toxic, expression in E. coli is relatively straightforward.
For protein expression, I would use the plasmid pET-28a(+). To insert the optimized RecA gene into the plasmid, restriction enzymes can be used. A suitable pair of restriction enzymes would be NdeI and XhoI. These enzymes are commonly used with pET-28a(+) because their recognition sites are present in the plasmid multiple cloning site and allow directional cloning of the gene insert. Directional cloning ensures that the gene is inserted in the correct orientation for expression. I used Codon Optimization Tool from Vector Builder.
Improved DNA: GC=54.79%, CAI=0.94
ATGGCGGATGATCGCAAAGTGGCCCTTGATGCGGCGCTGAAAAAAATCGAAAAAAATTTTGGCAAAGGCAGCATTATGAAACTGGGCGAAAAAGCGGATCAGAAAATTAGCACCATTCCGTCGGGCAGCCTGGCCCTGGATGTGGCGCTGGGCGTGGGTGGCTATCCGCGTGGCCGCATTATTGAAGTGTATGGCCCGGAAAGCAGCGGCAAAACCACCGTGAGCCTGCATGCAATTGCGGAAGTCCAGCGCAACGGCGGCACCGCGGCCTTTATTGATGCGGAACATGCGCTGGATCCGCAGTATGCGGAAAAACTGGGTGTGAACATTGATGAACTGCTGCTGAGCCAGCCGGATACCGGCGAACAGGGCCTGGAAATTGCGGATGCCCTGGTGAGCTCAGGCGCGATTGATATTGTTGTTATTGACAGTGTGGCGGCCCTGGTGCCGCGCGCCGAGATTGATGGCGAAATGGGCGCAAGCCACGTGGGCCTGCAGGCGCGCCTGATGAGCCAGGCGCTGCGCAAACTGTCAGGCTCGATTAACAAAACCAAAACCATCGCAATTTTTATTAACCAGATTCGTGAAAAAGTGGGCGTGATGTTTGGCAATCCGGAAACCACCCCGGGCGGCCGCGCCCTGAAATTTTATGCGACCGTGCGTCTGGAAGTGCGCCGTGCGGAACAGCTGAAACAGGGCACCGATATTGTGGGCAATCGCACCAAAATTAAAGTGGTGAAAAACAAAGTTGCCCCGCCGTTTAAAGTGGCGGAAGTGGATATTATGTATGGCCAGGGCATTTCGCAGGAAGGCGAACTGCTGGATATGGCGGTGGAAAAAGATCTGATTTCGAAAAGCGGCGCGTGGTATGGCTACAAAGAAGAACGTATTGGCCAGGGCCGTGAAAACGCGAAACAGTACATGGCCGATCATCCGGAAATGATGGCCGAAGTGAGCAAACTGGTTCGTGATGCCTACGGTATCGGCGATGGCAGCACCATCACCGAAGAAGCGGAAGGCCAGGAAGAACTGCCGCTGGATGAA
3.4. You have a sequence! Now what?
After codon optimization, the DNA sequence encoding RecA can be synthesized and inserted into the pET-28a(+) plasmid. The recombinant plasmid is then introduced into Escherichia coli cells through transformation. Because the pET-28a(+) vector can add a His-tag to the protein, the expressed RecA protein could later be purified using nickel affinity chromatography.
Expression in E. coli is a practical approach because bacterial systems are simple, cost-effective, and most practical method because RecA is a non-toxic bacterial protein and bacterial expression systems are highly optimized for this type of protein production.
Part 4: Prepare a Twist DNA Synthesis Order
4.2. Build Your DNA Insert Sequence
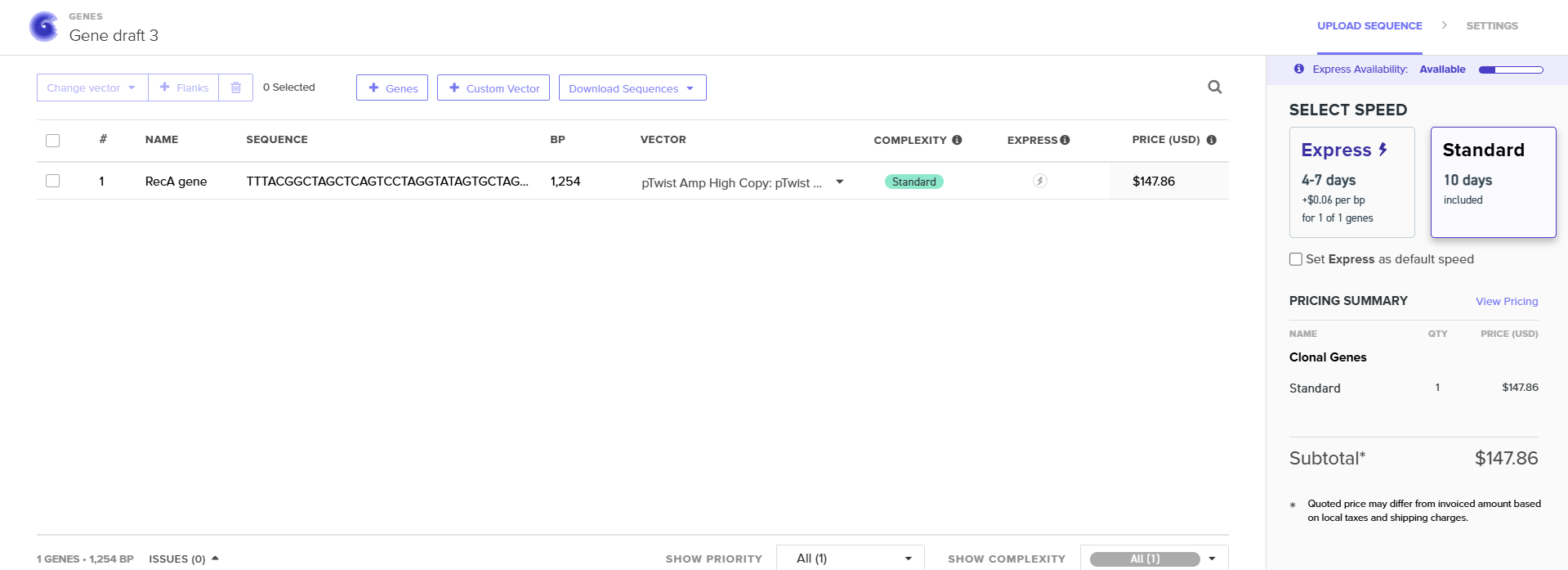
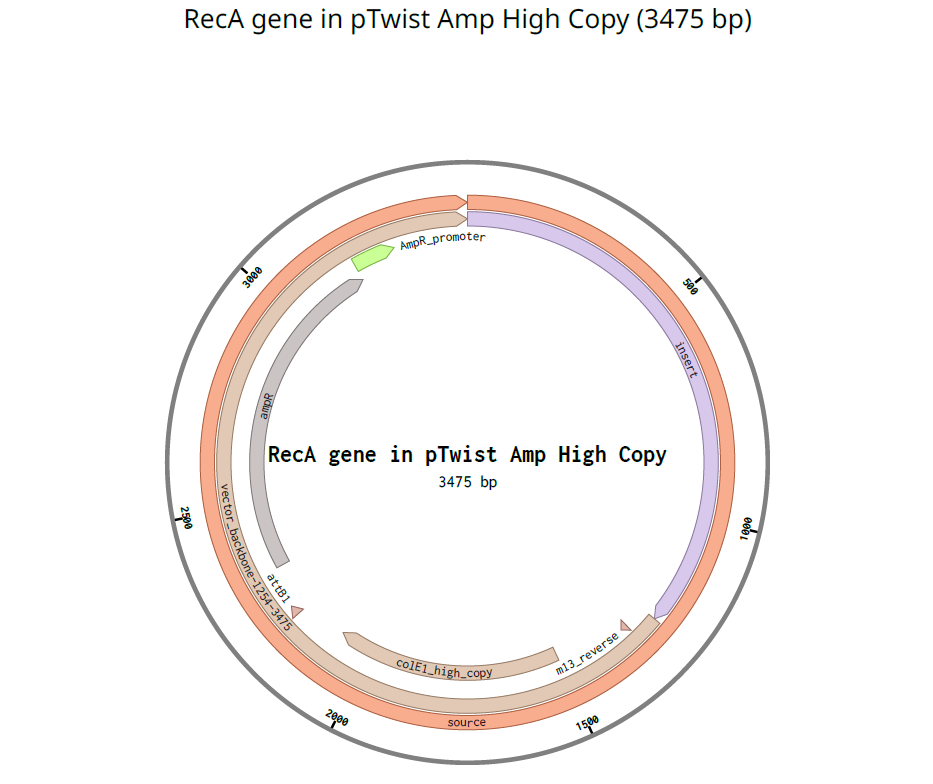
In Benchling I uploaded a sequence of RecA gene featured with Promoter, RBS, Start Codon, Coding Sequence, His Tag, Stop Codon, Terminator.
TTTACGGCTAGCTCAGTCCTAGGTATAGTGCTAGCCATTAAAGAGGAGAAAGGTACCATATGGCGGATGATCGCAAAGTGGCCCTTGATGCGGCGCTGAAAAAAATCGAAAAAAATTTTGGCAAAGGCAGCATTATGAAACTGGGCGAAAAAGCGGATCAGAAAATTAGCACCATTCCGTCGGGCAGCCTGGCCCTGGATGTGGCGCTGGGCGTGGGTGGCTATCCGCGTGGCCGCATTATTGAAGTGTATGGCCCGGAAAGCAGCGGCAAAACCACCGTGAGCCTGCATGCAATTGCGGAAGTCCAGCGCAACGGCGGCACCGCGGCCTTTATTGATGCGGAACATGCGCTGGATCCGCAGTATGCGGAAAAACTGGGTGTGAACATTGATGAACTGCTGCTGAGCCAGCCGGATACCGGCGAACAGGGCCTGGAAATTGCGGATGCCCTGGTGAGCTCAGGCGCGATTGATATTGTTGTTATTGACAGTGTGGCGGCCCTGGTGCCGCGCGCCGAGATTGATGGCGAAATGGGCGCAAGCCACGTGGGCCTGCAGGCGCGCCTGATGAGCCAGGCGCTGCGCAAACTGTCAGGCTCGATTAACAAAACCAAAACCATCGCAATTTTTATTAACCAGATTCGTGAAAAAGTGGGCGTGATGTTTGGCAATCCGGAAACCACCCCGGGCGGCCGCGCCCTGAAATTTTATGCGACCGTGCGTCTGGAAGTGCGCCGTGCGGAACAGCTGAAACAGGGCACCGATATTGTGGGCAATCGCACCAAAATTAAAGTGGTGAAAAACAAAGTTGCCCCGCCGTTTAAAGTGGCGGAAGTGGATATTATGTATGGCCAGGGCATTTCGCAGGAAGGCGAACTGCTGGATATGGCGGTGGAAAAAGATCTGATTTCGAAAAGCGGCGCGTGGTATGGCTACAAAGAAGAACGTATTGGCCAGGGCCGTGAAAACGCGAAACAGTACATGGCCGATCATCCGGAAATGATGGCCGAAGTGAGCAAACTGGTTCGTGATGCCTACGGTATCGGCGATGGCAGCACCATCACCGAAGAAGCGGAAGGCCAGGAAGAACTGCCGCTGGATGAACATCACCATCACCATCATCACTAACCAGGCATCAAATAAAACGAAAGGCTCAGTCGAAAGACTGGGCCTTTCGTTTTATCTGTTGTTTGTCGGTGAACGCTCTCTACTAGAGTCACACTGGCTCACCTTCGGGTGGGCCTTTCTGCGTTTATA
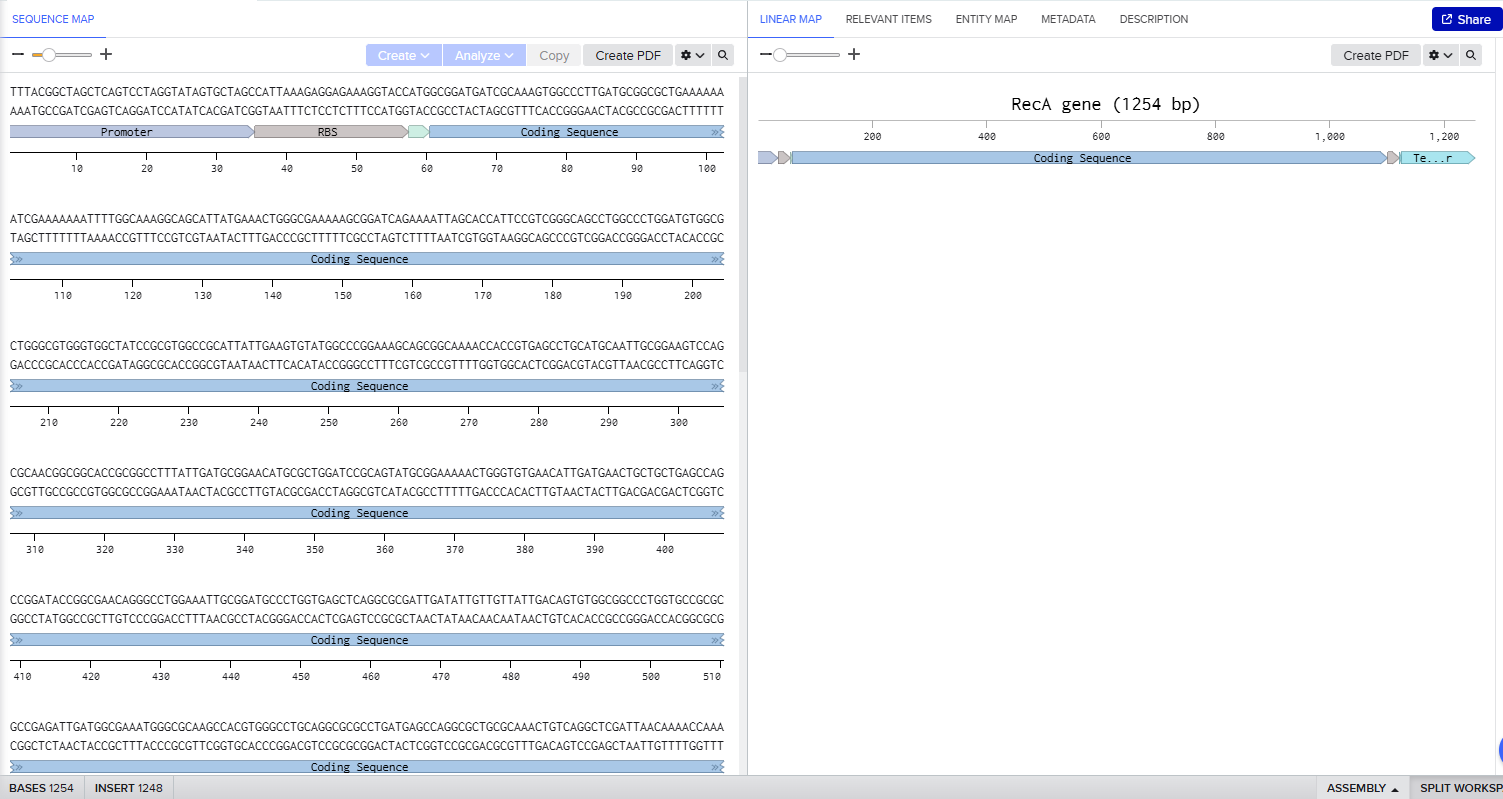
4.3.-4.6. Twist and Benchling
I used pTwist Amp High Copy as a Twist cloning vector because I require constant expression of the RecA gene for my experiment.
Benchling
Part 5: DNA Read/Write/Edit
5.1 DNA Read
(i) What DNA would you want to sequence (e.g., read) and why?
In the context of my idea project, I would sequence DNA from putative transconjugant E. coli colonies that appear blue in the recipient-printed zones on chromogenic agar (Plate B + ampicillin).
Why:
- Confirm true horizontal gene transfer (conjugation): verify that recipient cells acquired the mobilizable plasmid carrying oriT_RK2–Ptrc–RBS–bglA, rather than the blue phenotype being caused by donor contamination, spontaneous mutations, or unexpected chromogenic substrate metabolism.
- Verify construct integrity after transfer: check whether bglA, Ptrc, BBa_B0034, and oriT_RK2 are intact (no deletions/rearrangements), since plasmids can mutate or recombine.
- Map spatial HGT patterns: sequence colonies sampled from different printed coordinates (e.g., overlap boundary vs center) to see whether transfer frequency or plasmid variants correlate with spatial proximity and incubation conditions.
(ii) What technology or technologies would you use to perform sequencing on your DNA and why?
I would use a two-tier approach:
Sanger sequencing (targeted validation)
- Fast and cost-effective for confirming a limited number of colonies and key regions (bglA + junctions).
- High per-base accuracy and straightforward interpretation.
Illumina short-read sequencing (scaled, high-throughput)
- Appropriate when sequencing many colonies across multiple printed locations.
- High accuracy and depth for detecting low-frequency variants.
Oxford Nanopore long-read sequencing
- Useful for reading the entire plasmid in one/few long reads and detecting structural rearrangements.
Is your method first-, second- or third-generation or other? How so?
- Sanger sequencing: first-generation (chain-termination; low throughput; ~700–900 bp reads with high accuracy).
- Illumina: second-generation (massively parallel sequencing-by-synthesis with cluster amplification; short reads).
- Oxford Nanopore: third-generation (single-molecule, real-time, long reads; PCR not mandatory).
What is your input? How do you prepare your input (e.g. fragmentation, adapter ligation, PCR)? List the essential steps.
Input:
- Colonies from chromogenic plates:
- Putative transconjugants: blue colonies in recipient zones on Plate B (+ ampicillin)
- Controls: donor strain, original recipient strain, and ideally a “no-helper” negative control mating
Essential preparation steps (by method):
A) Sanger (PCR + sequencing)
- Pick colony → short liquid culture (or colony PCR).
- DNA extraction (plasmid miniprep or crude lysate).
- PCR amplify:
- bglA internal region (presence/identity)
- junctions (e.g., oriT→Ptrc, Ptrc→RBS, RBS→bglA) to confirm correct order/integrity
- PCR cleanup.
- Sanger sequencing.
B) Illumina (amplicon-seq or plasmid-enriched sequencing)
- DNA extraction (preferably plasmid-enriched if focusing on the construct).
- Either:
- PCR amplify target regions (amplicon sequencing), or
- Fragment DNA for whole-plasmid/whole-genome sequencing.
- End repair / A-tailing (kit-dependent).
- Adapter ligation + indexing (barcodes).
- PCR enrichment (often used).
- Sequencing.
C) Nanopore (whole plasmid)
- Extract high-quality DNA with minimal shearing.
- End repair (kit-dependent).
- Ligate nanopore adapters with motor protein.
- Load onto flow cell.
What are the essential steps of your chosen sequencing technology, how does it decode the bases of your DNA sample (base calling)?
- Sanger: ddNTP incorporation terminates extension; fragments are separated by capillary electrophoresis; fluorescence color determines the terminal base.
- Illumina: sequencing-by-synthesis with fluorescent reversible terminators; imaging each cycle; base calling from image intensities.
- Nanopore: DNA passes through a pore and modulates ionic current; neural-network base calling converts current traces (“squiggles”) into bases.
What is the output of your chosen sequencing technology?
- Sanger: chromatograms (
.ab1) + base-called sequences for each amplicon. - Illumina:
FASTQshort reads (e.g., 2×150 bp) with Phred quality scores. - Nanopore:
FASTQlong reads (plus raw signal files such asPOD5/FAST5, workflow-dependent).
5.2 DNA Write
(i) What DNA would you want to synthesize (e.g., write) and why?
I would synthesize the mobilizable bglA expression cassette that generates the blue phenotype after conjugation:
- oriT_RK2 (mobilization origin; enables plasmid transfer using RK2 machinery supplied by pRK2013 in trans)
- Ptrc promoter (strong E. coli-compatible promoter for robust expression)
- BioBrick RBS BBa_B0034 (efficient translation initiation)
- bglA CDS from Enterococcus faecalis (β-glucosidase intended to produce a blue chromogenic phenotype)
Why: this DNA encodes the key “sensor output” of the system: recipient cells become blue only after acquiring bglA via HGT, enabling spatial visualization of conjugation.
(ii) What technology or technologies would you use to perform this DNA synthesis and why?
I would use:
- Phosphoramidite-based chemical DNA synthesis (commercial gene synthesis / oligo pools)
- Assembly methods such as Gibson Assembly or Golden Gate Assembly
Why:
- Phosphoramidite chemistry is the standard for accurate, scalable oligo/gene-fragment synthesis.
- Gibson/Golden Gate support modular assembly of oriT, promoter, RBS, and CDS, and enable rapid variant construction.
1) What are the essential steps of your chosen synthesis methods?
- In silico design (Benchling):
- verify ORF, start/stop, and reading frame of bglA
- codon-optimize for E. coli if needed
- check synthesis constraints (repeats, extreme GC, hairpins)
- add overlaps or Type IIS sites for assembly
- Oligo/gene fragment synthesis (phosphoramidite cycles).
- Assembly (Gibson/Golden Gate) into the plasmid backbone.
- Transformation into E. coli and clone picking.
- Sequence verification (Sanger/Illumina) of bglA and junctions.
- Functional validation on chromogenic agar.
2) What are the limitations of your synthesis method (if any) in terms of speed, accuracy, scalability?
- Length limits: single oligos are short; longer constructs require multi-fragment assembly.
- Error accumulation: synthesis errors accumulate with length; verification and clone screening are required.
- Sequence-dependent difficulty: repeats, strong secondary structures, and extreme GC can reduce synthesis/assembly success.
- Speed: outsourcing is fast but still usually days–weeks including cloning and verification.
- Scalability bottleneck: generating variants is feasible; the limiting step becomes screening + validation (though the color phenotype helps throughput).
5.3 DNA Edit
(i) What DNA would you want to edit and why?
To improve the robustness and interpretability of the HGT, I would edit DNA in the recipient strain and/or the plasmid system.
Examples of useful edits:
- Recipient genome edits to reduce background coloration or off-pathway metabolism on chromogenic media, making blue strictly dependent on bglA acquisition and expression.
- Regulatory tuning edits (promoter/RBS fine-tuning) to calibrate the onset and intensity of the blue signal and improve spatial resolution.
- Plasmid stability edits (e.g., removing recombinogenic sequences or adding stability features) to reduce plasmid loss and signal variability.
Why: the project’s readout is phenotypic (color), so improving specificity and stability directly strengthens conclusions about HGT frequency and spatial dynamics.
(ii) What technology or technologies would you use to perform these DNA edits and why?
I would use CRISPR-based editing in E. coli:
- CRISPR-Cas9 + recombineering (λ-Red) + donor DNA for precise insertions/replacements, and/or
- Base editors for targeted point mutations in promoters/RBS without introducing double-strand breaks.
Why:
- Cas9 + recombineering is well established for precise bacterial genome engineering.
- Base editing is ideal for fine-tuning regulatory elements controlling bglA expression.
How does your technology of choice edit DNA? What are the essential steps?
CRISPR-Cas9 + donor DNA (with recombineering):
- Design gRNA targeting the locus of interest (adjacent to a PAM).
- Deliver Cas9 + gRNA (typically via plasmid expression).
- Provide a donor DNA template with desired edits flanked by homology arms.
- Cas9 creates a double-strand break at the target site.
- λ-Red recombination integrates the donor sequence → precise edit.
- Cure editing plasmids if needed and verify.
Base editing:
- Design gRNA near the base(s) to be changed.
- Express nCas9/dCas9 fused to a deaminase.
- Targeted binding induces base conversion (C→T or A→G) within an editing window.
- Screen and sequence-verify edited clones.
What preparation do you need to do (e.g. design steps) and what is the input for the editing?
Preparation/design:
- Choose target loci (recipient genes affecting background; promoter/RBS tuning; plasmid stability elements).
- Design gRNAs and check off-targets against the recipient genome.
- Design donor templates (if using Cas9 + recombination) or plan the editing window (base editing).
- Define screening/selection strategy (markers, counterselection, phenotype).
Input:
- gRNA(s)
- Cas9 or base editor construct
- Donor DNA template (ssDNA/dsDNA) for precise edits
- Editing plasmids and recombineering functions (λ-Red)
- Competent E. coli cells (recipient/donor as appropriate)
What are the limitations of your editing methods (if any) in terms of efficiency or precision?
- Variable efficiency depending on locus, strain, and delivery; requires optimization.
- DSB toxicity/lethality if repair/recombination is inefficient.
- Off-target edits are possible and require sequencing verification.
- Base-editing constraints: depends on PAM availability and the editor’s window/context; not all bases are targetable.
- Plasmid/genome context issues: copy number, recombination, and selection dynamics can complicate stable outcomes.