Week 4 HW: Protein design part I
Part A. Conceptual Questions
1. How many molecules of amino acids do you take with a piece of 500 grams of meat? (on average an amino acid is ~100 Daltons)
Assumptions:
- Lean meat (e.g., beef, chicken, fish) contains approximately 20% protein by weight (the remainder is water, fat, minerals, and glycogen).
- 500 g meat × 0.20 = 100 g of protein.
- Proteins are digested into individual amino acid monomers.
- Average molar mass of an amino acid residue ≈ 100 g/mol (as stated).
Approximately 6 × 10²⁶ amino acid molecules are obtained from 500 g of meat.
2. Why do humans eat beef but do not become a cow, eat fish but do not become fish?
This is explained by the central dogma of molecular biology, digestion, and genetic isolation:
Digestion breaks down macromolecules:
- Dietary proteins are hydrolyzed by proteases (pepsin, trypsin, chymotrypsin, peptidases) into individual amino acids and small peptides.
- Nucleic acids (DNA/RNA) are degraded by nucleases into nucleotides and then further into nucleosides and bases.
- Intact macromolecular information does not survive digestion.
Absorption yields building blocks, not genetic information:
- Absorbed amino acids, sugars, nucleotides, and fatty acids enter a common metabolic pool.
- These are reassembled into human proteins according to instructions encoded in the human genome, transcribed into human mRNA, and translated by human ribosomes.
No horizontal gene transfer from diet:
- Eukaryotic cells lack mechanisms to incorporate foreign dietary DNA into the nuclear genome.
- Even if intact DNA fragments reached intestinal cells, they would be degraded by DNases or excluded by chromatin and membrane barriers.
3. Why are there only 20 natural amino acids?
The canonical set of 20 proteinogenic amino acids (plus selenocysteine and pyrrolysine in some organisms) reflects an evolutionary and biochemical optimum:
Chemical diversity and functional sufficiency:
- The 20 amino acids cover a wide range of chemical properties: hydrophobic, polar, charged (acidic/basic), aromatic, and conformationally constrained (proline).
- This diversity is sufficient to fold proteins into stable structures and catalyze essentially all cellular reactions.
Genetic code constraints:
- The genetic code maps 64 codons to amino acids and stop signals. Expanding the amino acid repertoire requires:
- New aminoacyl-tRNA synthetases
- Orthogonal tRNAs
- Unambiguous codon assignments
- Adding more amino acids increases translational errors and slows ribosomal elongation.
Historical contingency and error minimization:
- Early life likely used a smaller subset (~10 amino acids).
- The code expanded gradually and became “frozen” because the existing arrangement minimizes the phenotypic impact of point mutations (error minimization principle).
- Further expansion offers diminishing returns relative to the evolutionary cost of maintaining additional biosynthetic pathways and quality control.
4. Can you make other non-natural amino acids? Design some new amino acids.
Three Rationally Designed Non-Natural Amino Acids:
| Name | Structure/Modification | Functional Rationale |
|---|---|---|
| p-Azobenzene-phenylalanine | Phenyl ring replaced with azobenzene moiety | Photoswitchable: UV/visible light induces trans↔cis isomerization, enabling light-controlled conformational changes in engineered proteins (optogenetics, dynamic materials). |
| 2,2′-Bipyridyl-alanine | Chelating bipyridyl group on β-carbon | Metal coordination: High-affinity binding to Fe²⁺, Cu²⁺, Ru²⁺. Useful for installing synthetic metallocofactors, creating redox-active sites, or building metalloproteins with tunable properties. |
| ε-Azido-lysine | Lysine with azide (–N₃) on ε-amino group | Bioorthogonal chemistry: Azide is inert to biological functional groups but reacts rapidly via click chemistry (e.g., strain-promoted azide-alkyne cycloaddition), enabling site-specific labeling, crosslinking, and drug conjugation. |
ncAAs expand protein functionality for drug design, biosensors, biomaterials, mechanistic enzymology, and synthetic biology.
5. Where did amino acids come from before enzymes that make them, and before life started?
1. Atmospheric/oceanic synthesis (Miller–Urey type experiments):
- Reducing atmospheres (CH₄, NH₃, H₂, H₂O) subjected to energy sources (UV radiation, lightning, volcanic heat) produce amino acids.
- Modern models favor weakly reducing atmospheres (CO₂, N₂, H₂O + H₂) near hydrothermal vents, which still yield amino acids via formose-like and Strecker-like reactions.
2. Strecker synthesis (aqueous chemistry):
- Aldehydes or ketones react with HCN and NH₃ to form α-aminonitriles, which hydrolyze to amino acids.
- This pathway occurs in alkaline hydrothermal vent systems and interstellar ice analogs.
3. Extraterrestrial delivery:
- Amino acids (>80 types, including non-biological ones) have been detected in carbonaceous chondrites (e.g., Murchison meteorite).
- Some samples show L-enantiomer excess, suggesting chiral symmetry-breaking in space.
- Comets and interstellar ices likely contributed to Earth’s prebiotic inventory.
6. If you make an α-helix using D-amino acids, what handedness (right or left) would you expect?
A polypeptide composed entirely of D-amino acids would form a left-handed α-helix. The reason because L-amino acids favor backbone dihedral angles φ ≈ –60°, ψ ≈ –45° (right-handed helical geometry). D-amino acids are mirror images (enantiomers) of L-amino acids, so their favorable φ/ψ angles are sign-inverted: φ ≈ +60°, ψ ≈ +45°. And these angles produce a left-handed helix with the same hydrogen-bonding pattern (i → i+4) but opposite helical sense.
7. Can you discover additional helices in proteins?
Yes. Beyond the canonical α-helix, 3₁₀-helix, and π-helix, new and rare helical motifs continue to be identified.
Known non-canonical helices:
- Polyproline II helix (PPII): left-handed, extended; common in collagen and disordered regions.
- γ-helix: rare; tighter than α-helix.
- α_L-helix: left-handed α-helix (rare in L-peptides; seen in certain contexts or D-residues).
- Hybrid/transitional helices: local 3₁₀/α blends or distorted segments.
Tools for discovery:
- High-resolution X-ray crystallography and cryo-EM: reveal short, rare, or distorted helices.
- NMR spectroscopy: detects transient helical states (e.g., via residual dipolar couplings, relaxation dispersion).
- AlphaFold2/3 and molecular dynamics (MD): predict metastable helical conformations and sample rare states via enhanced sampling (replica exchange, metadynamics).
Why new helices appear:
- Local sequence context, post-translational modifications, ligand binding, membrane environments, and pH shift φ/ψ distributions.
- Helices exist on a continuum; subtle energy differences stabilize non-canonical H-bond patterns.
8. Why are most molecular helices right-handed?
In biological polymers, right-handedness arises from monomer chirality and steric optimization.
For proteins (L-amino acids):
- L-amino acids favor φ/ψ angles (≈ –60°, –45°) that produce right-handed α-helices with minimal side-chain clashes and optimal backbone hydrogen bonding.
- Left-handed helices require φ/ψ values that place side chains in sterically unfavorable positions (forbidden regions of the Ramachandran plot).
General principle:
- Chirality of monomers dictates handedness.
- If you swap L ↔ D (e.g., D-amino acids), you get the mirror-image helix (left-handed).
In synthetic/achiral polymers:
- Handedness is determined by packing efficiency, torsional strain, and solvent interactions.
- Without chirality, helices may form as racemic mixtures or adopt the handedness that minimizes steric/electronic repulsion.
9. Why do β-sheets tend to aggregate? What is the driving force for β-sheet aggregation?
β-sheets are intrinsically prone to intermolecular association due to their geometry and chemical properties.
- β-strands often alternate polar and nonpolar residues on opposite faces. When β-sheets associate, hydrophobic side chains pack against each other, displacing high-energy ordered water molecules → favorable entropy gain.
- Aggregation is often initiated by partial unfolding, cleavage, or mutation, exposing β-strand-prone sequences.
- Some organisms use cross-β aggregates as structural materials (e.g., spider silk, curli fibers in biofilms).
Part B: Protein Analysis and Visualization
I selected the BglA (β-glucosidase) protein because my project focuses on horizontal gene transfer (HGT) in Escherichia coli using a bioprinting system. In this system, donor and recipient bacterial strains are printed onto agar plates in spatially defined layers. Successful conjugation events are visualized by a color change from pink to teal after recipient cells acquire the bglA gene from Enterococcus faecalis and begin producing β-glucosidase activity on chromogenic media.
There is limited structural information available for the E. faecalis BglA protein, so I used the structurally characterized homolog from E. coli. Although E. coli and E. faecalis are not from the same bacterial family, both proteins belong to the β-glucosidase enzyme group and perform similar functions.
The protein sequence for E. coli BglA was obtained from UniProt.
MIVKKLTLPKDFLWGGAVAAHQVEGGWNKGGKGPSICDVLTGGAHGVPREITKEVLPGKY YPNHEAVDFYGHYKEDIKLFAEMGFKCFRTSIAWTRIFPKGDEAQPNEEGLKFYDDMFDE LLKYNIEPVITLSHFEMPLHLVQQYGSWTNRKVVDFFVRFAEVVFERYKHKVKYWMTFNE INNQRNWRAPLFGYCCSGVVYTEHENPEETMYQVLHHQFVASALAVKAARRINPEMKVGC MLAMVPLYPYSCNPDDVMFAQESMRERYVFTDVQLRGYYPSYVLNEWERRGFNIKMEDGD LDVLREGTCDYLGFSYYMTNAVKAEGGTGDAISGFEGSVPNPYVKASDWGWQIDPVGLRY ALCELYERYQRPLFIVENGFGAYDKVEEDGSINDDYRIDYLRAHIEEMKKAVTYDGVDLM GYTPWGCIDCVSFTTGQYSKRYGFIYVNKHDDGTGDMSRSRKKSFNWYKEVIASNGEKL
The most frequent amino acids in the sequence is Glycine (G) as it appears in the sequence 42 times.
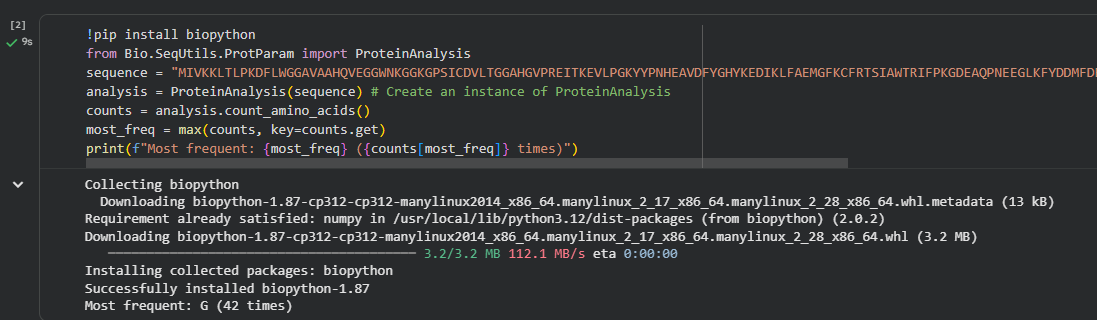
The BglA protein from E. coli contains 479 amino acids.
.png)
Using the UniProt BLAST tool, 250 results were identified as potential homologs of the BglA protein (UniProt ID: Q46829) with sequence identities ranging from 74.7% to 100%. Most homologs were found in bacteria (249 results), which is expected because β-glucosidases are common enzymes involved in bacterial carbohydrate metabolism. One homologous sequence was also identified in the eukaryotic organism Trichuris trichiura (1 result).
BglA belongs to the glycoside hydrolase family 1 (GH1) of β-glucosidases. These enzymes hydrolyze glycosidic bonds in carbohydrates and are involved in sugar metabolism.
The structure I used is Crystal Structure of E.coli BglA (2XHY).
The structure was solved using X-ray crystallography and released in 2011. The resolution of the structure is 2.30 Å, which is considered good quality for a protein crystal structure because lower resolution values indicate more precise atomic positions.
In addition to the protein, the structure contains sulfate ions (SO₄²⁻) and bromide ions. These molecules are commonly present from crystallization conditions and may stabilize the structure during crystal formation.
The protein belongs to the glycoside hydrolase structural family, which typically contains catalytic domains rich in α/β folds involved in carbohydrate hydrolysis.
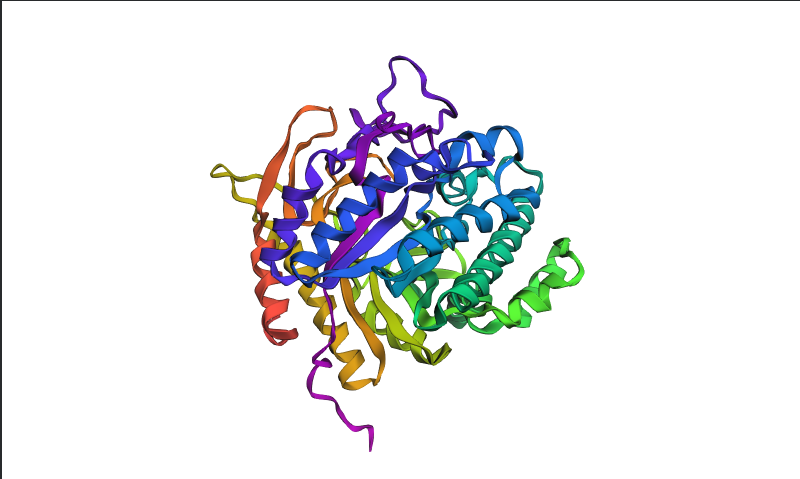
Visualized protein in PyMol using cartoon representations:
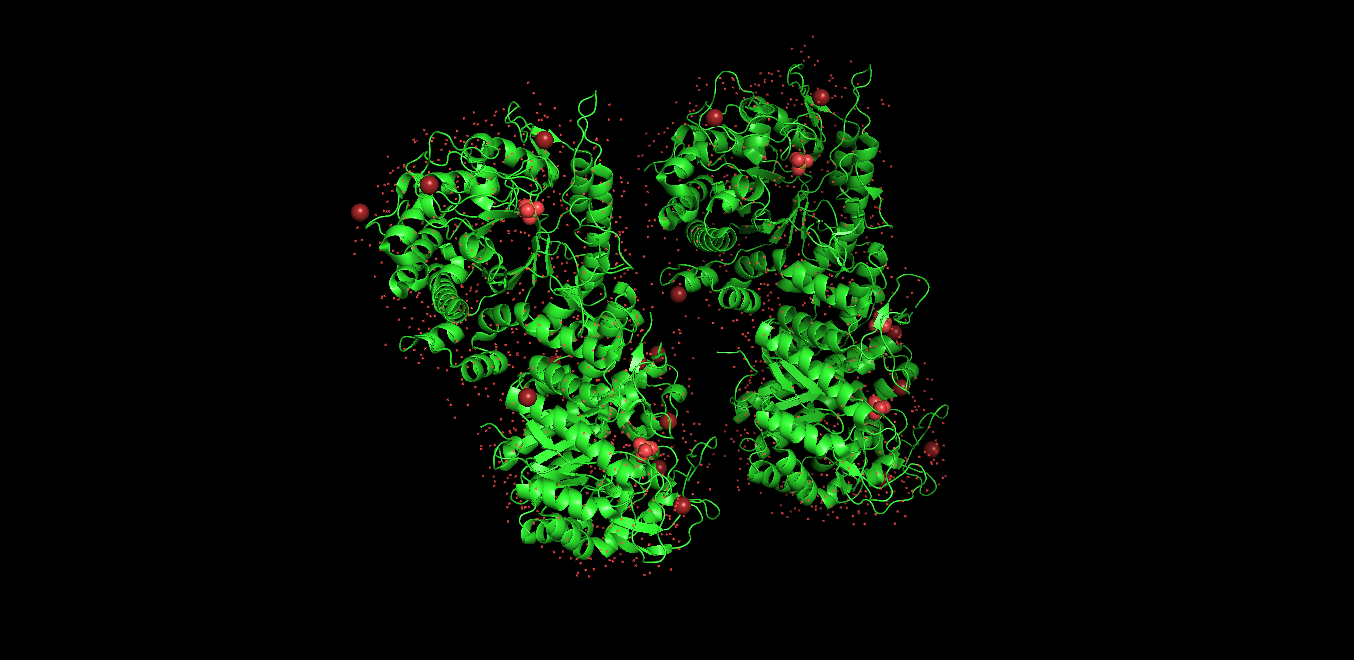
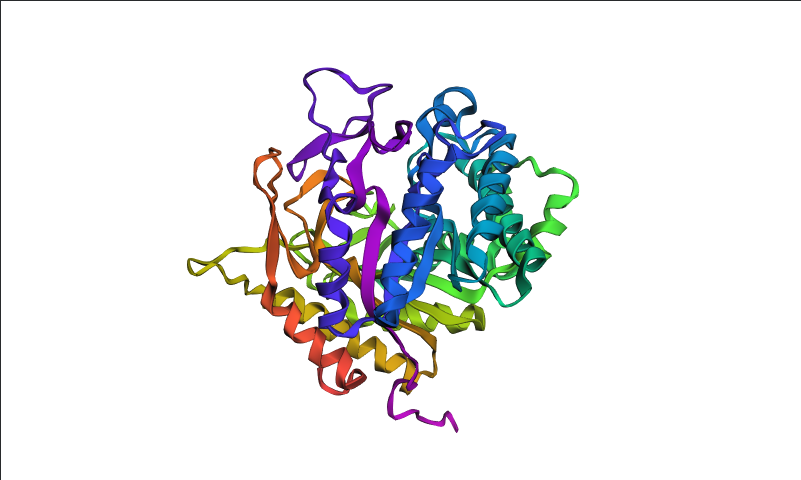
Visualized protein in PyMol using ribbon representations:
Visualized protein in PyMol using ball-and-stick representations:

Secondary structure
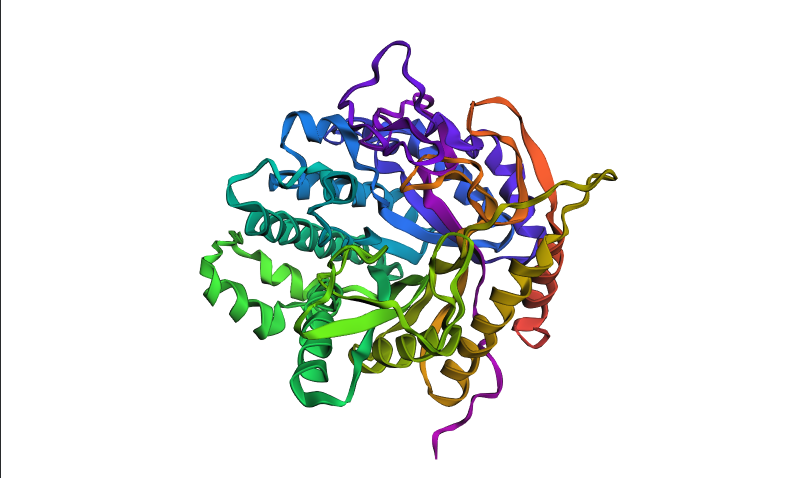
After coloring the protein by secondary structure in PyMol, the structure appeared to contain substantially more α-helices than β-sheets. Loops and turns were also distributed throughout the structure. The predominance of α-helices is consistent with the overall fold of many glycoside hydrolase enzymes.
.png)
Residue type distribution
After coloring the protein by residue type in PyMol, hydrophobic residues (yellow) appeared to cluster mainly in the internal regions of the protein structure, while hydrophilic residues (cyan) were more exposed on the protein surface. This organization is characteristic of soluble enzymes and contributes to protein stability and interactions with the aqueous environment.
.png)
Surface visualization
Surface representation was colored according to B-factor values using a blue-white-red spectrum. Regions with higher B-factors (red/pink) indicate increased atomic displacement or structural flexibility, while lower B-factor regions (blue) correspond to more rigid parts of the structure. The protein surface exhibits several grooves and cavities, consistent with the typical topology of glycoside hydrolases, which often possess an elongated substrate-binding cleft.
.png)
Part C. Using ML-Based Protein Design Tools
C1. Protein Language Modeling
- Deep Mutational Scans - Crystal Structure of E.coli BglA protein.
.png) Columns with more dark cells (the wild-type amino acid is strongly preferred) indicate more conserved residues, which are likely important for the structure and function of the protein.
Columns with more dark cells (the wild-type amino acid is strongly preferred) indicate more conserved residues, which are likely important for the structure and function of the protein.
- Latent Space Analysis
Neighboring proteins usually share the same SCOPe structural class and superfamily number, which indicates that the latent space captures structural and functional similarities between proteins.
C2. Protein Folding
Folding a protein
The ESMFold prediction for BglA produced a highly confident structure with a pTM score of 0.936 and an average pLDDT of 91.16. Most regions were colored blue or green, indicating high local confidence, while only a few loop regions showed lower confidence (yellow/red). The predicted fold closely matched the experimentally determined crystal structure, suggesting that ESMFold accurately reproduced the overall topology of the protein.
The E164A point mutation had minimal effect on the predicted structure of BglA. The pTM score remained nearly unchanged (0.935 vs. 0.936 for the wild type), and the average pLDDT stayed very high (91.05), indicating that the overall fold and structural confidence were preserved. This suggests that the BglA structure is resilient to single amino acid substitutions.
Point mutation (E164A)
MIVKKLTLPKDFLWGGAVAAHQVEGGWNKGGKGPSICDVLTGGAHGVPREITKEVLPGKYYPNHEAVDFYGHYKEDIKLFAEMGFKCFRTSIAWTRIFPKGDEAQPNEEGLKFYDDMFDELLKYNIEPVITLSHFAMPLHLVQQYGSWTNRKVVDFFVRFAEVVFERYKHKVKYWMTFNEINNQRNWRAPLFGYCCSGVVYTEHENPEETMYQVLHHQFVASALAVKAARRINPEMKVGCMLAMVPLYPYSCNPDDVMFAQESMRERYVFTDVQLRGYYPSYVLNEWERRGFNIKMEDGDLDVLREGTCDYLGFSYYMTNAVKAEGGTGDAISGFEGSVPNPYVKASDWGWQIDPVGLRYALCELYERYQRPLFIVENGFGAYDKVEEDGSINDDYRIDYLRAHIEEMKKAVTYDGVDLMGYTPWGCIDCVSFTTGQYSKRYGFIYVNKHDDGTGDMSRSRKKSFNWYKEVIASNGEKL
Deletion of a larger sequence segment resulted in a moderate decrease in prediction confidence (pLDDT 89.1 vs. 91.2 in the wild type), while the overall fold remained largely preserved. The predicted structure retained a similar topology and confidence-color distribution, suggesting that the deleted region is not essential for maintaining the global structural framework of BglA.
Large mutation (removed segment GYCCSGVVYTEHENPEETMY)
MIVKKLTLPKDFLWGGAVAAHQVEGGWNKGGKGPSICDVLTGGAHGVPREITKEVLPGKYYPNHEAVDFYGHYKEDIKLFAEMGFKCFRTSIAWTRIFPKGDEAQPNEEGLKFYDDMFDELLKYNIEPVITLSHFEMPLHLVQQYGSWTNRKVVDFFVRFAEVVFERYKHKVKYWMTFNEINNQRNWRAPLFQVLHHQFVASALAVKAARRINPEMKVGCMLAMVPLYPYSCNPDDVMFAQESMRERYVFTDVQLRGYYPSYVLNEWERRGFNIKMEDGDLDVLREGTCDYLGFSYYMTNAVKAEGGTGDAISGFEGSVPNPYVKASDWGWQIDPVGLRYALCELYERYQRPLFIVENGFGAYDKVEEDGSINDDYRIDYLRAHIEEMKKAVTYDGVDLMGYTPWGCIDCVSFTTGQYSKRYGFIYVNKHDDGTGDMSRSRKKSFNWYKEVIASNGEKL
Conclusion
ESMFold accurately reproduced the experimentally determined fold of BglA with high confidence scores (pTM 0.936; pLDDT 91.16). A single-point mutation (E164A) had almost no effect on the predicted structure, indicating high resilience to minor sequence changes. Deletion of a larger sequence segment caused a moderate reduction in confidence scores but did not significantly disrupt the overall fold, suggesting that BglA maintains a robust global structural architecture even after substantial sequence perturbation.
C3. Protein Generation
Inverse-Folding a protein
- Predicted sequence probabilities
I used ProteinMPNN (model v_48_020, sampling temperature 0.1) to perform inverse folding on the backbone structure of BglA (PDB: 2XHY). The designed sequence achieved a sequence recovery of 45.5%, indicating that nearly half of the original amino acids were reproduced by the model. This suggests that the BglA backbone imposes substantial structural constraints on sequence selection, particularly within the conserved catalytic core and structurally important regions. Probability maps showed several highly confident residue positions, while other regions tolerated greater sequence variability, especially likely in surface-exposed loops.
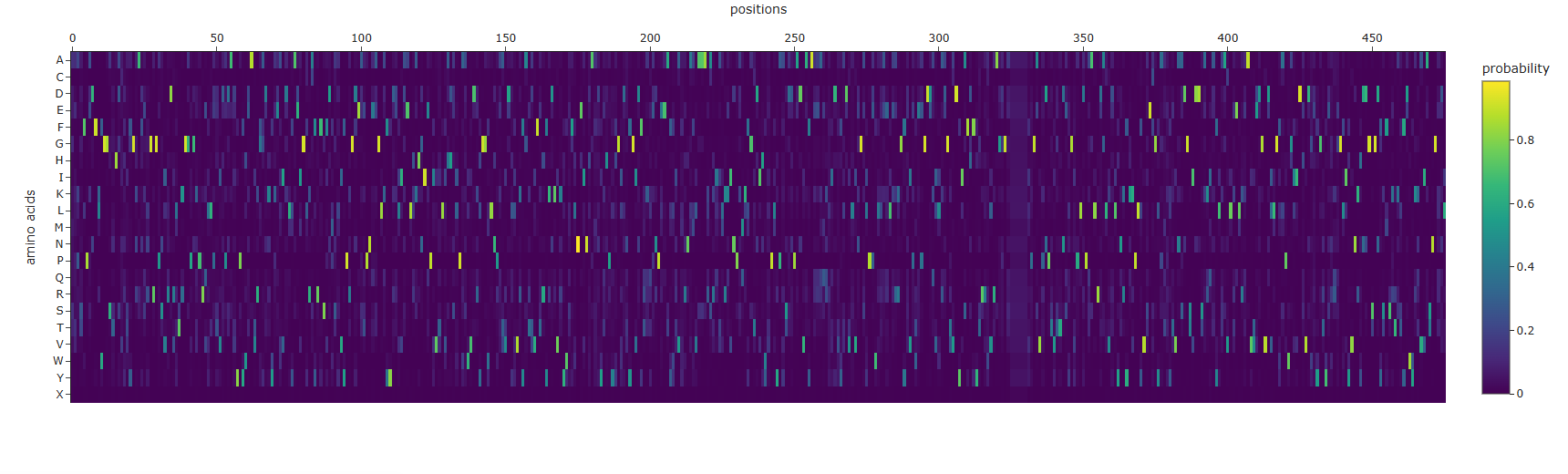 ProteinMPNN amino acid probabilities at each position. Bright spots indicate positions where the model is highly confident about which residue should occupy that position.
ProteinMPNN amino acid probabilities at each position. Bright spots indicate positions where the model is highly confident about which residue should occupy that position.
- Folding the designed sequence with ESMFold
I folded the ProteinMPNN-designed sequence of BglA with ESMFold and compared it to the original crystal structure.
| Structure | pTM | pLDDT |
|---|---|---|
| Original BglA | 0.936 | 91.16 |
| ProteinMPNN-designed sequence | 0.923 | 87.95 |
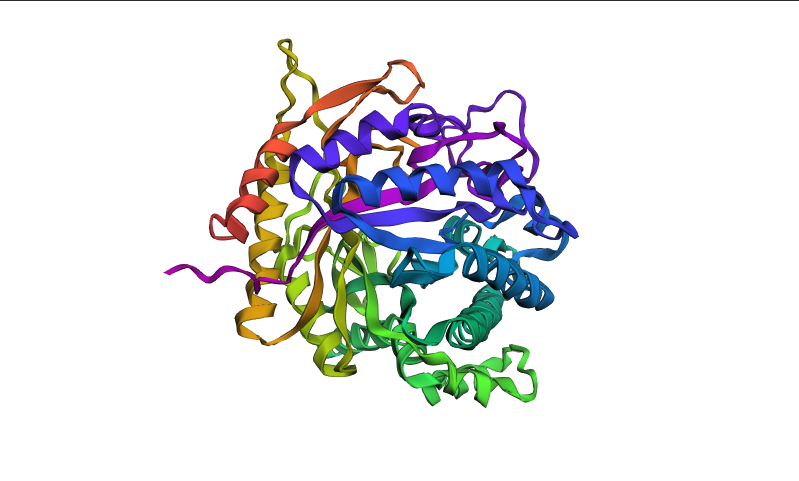
The ProteinMPNN-designed sequence retained a highly similar overall fold compared to the original BglA structure. Confidence coloring was also largely unchanged, with most regions remaining blue and green, indicating that the designed sequence still folds into a stable and well-defined globular structure.
Although the designed sequence recovered only ~45.5% of the original amino acids, the predicted structure remained highly similar to the native fold. This demonstrates that multiple different amino acid sequences can support the same overall protein architecture. In BglA, the structural constraints imposed by the conserved catalytic core and the α/β-fold appear to strongly determine the global topology even when substantial sequence variation is introduced.
BglA is a structured enzymatic protein with a robust and evolutionarily conserved fold. As a result, ProteinMPNN was able to design an alternative sequence that still folds with high confidence into a structure closely resembling the original enzyme.
Part D. Group Brainstorm on Bacteriophage Engineering
Main goal
Increased stability
Will focus on stabilizing the L protein computationally. By increasing its thermodynamic stability, there is aim to reduce its dependence on the bacterial chaperone DnaJ for proper folding and membrane insertion. A more stable L protein should maintain function even if E. coli mutates DnaJ, thereby helping the phage overcome host resistance mechanisms. This also indirectly supports higher toxicity by ensuring more functional lysis protein reaches the membrane.
Project Objective
Propose to computationally design stabilizing mutations in the MS2 L lysis protein to make it less reliant on the host chaperone DnaJ while preserving (or enhancing) its ability to form oligomeric pores in the bacterial membrane. The long-term goal is to create L protein variants that increase phage lysis efficiency and reduce the evolutionary window in which E. coli can develop resistance during phage therapy.
Proposed Tools and Computational Approaches
Will use a combination of modern protein design and structure prediction tools introduced in recitation:
- Protein Language Models (ESM-2 / ESMFold) for initial in silico mutagenesis and variant generation.
- AlphaFold3 (or AlphaFold-Multimer) to predict monomer structures and protein–protein/membrane complexes.
- FoldX and/or Rosetta (ddG protocol) to quantitatively evaluate mutational effects on folding stability (ΔΔG).
- ProteinMPNN for sequence redesign of surface residues to improve packing and reduce aggregation propensity.
- Molecular Dynamics (MD) simulations (short runs using GROMACS or OpenMM with a coarse-grained membrane model) to assess membrane insertion and oligomerization of top candidates.
Why These Tools Are Likely to Help
ESM-2 and ProteinMPNN
These models excel at proposing evolutionarily plausible and physically stable mutations without requiring an experimentally solved structure. Since the MS2 L protein is small, membrane-associated, and structurally under-characterized, sequence-based generative models provide an ideal starting point. They allow to explore sequence space while preserving foldability constraints learned from millions of natural protein sequences.
AlphaFold3 / AlphaFold-Multimer
AlphaFold enables structure prediction of the L protein monomer and potential oligomeric states. Using Multimer mode, can be modeled the L–DnaJ interaction to assess whether specific mutations preserve structural integrity while weakening dependence on the host chaperone. Recent AlphaFold versions handle small peptides and membrane proteins reasonably well, especially when modeled in a membrane-mimetic context.
FoldX / Rosetta (ddG calculations)
These physics-based tools estimate the change in folding free energy (ddG) upon mutation. Variants predicted to have ddG < -1.5 kcal/mol are likely more thermodynamically stable than wild type. This allows to prioritize mutations that may improve folding robustness and reduce chaperone dependence.
Molecular Dynamics (MD) Simulations
MD simulations provide dynamic validation. They allow to evaluate whether stabilized variants:
- Maintain structural integrity over time
- Insert more efficiently into lipid bilayers
- Form stable oligomeric pore-like assemblies
This connects computational stability predictions to functional outcomes such as faster lysis and potentially higher phage titers.
Overall Strategy
By integrating:
- Evolutionary information (protein language models)
- Deep learning-based structure prediction (AlphaFold)
- Physics-based stability estimation (Rosetta/FoldX)
- Membrane-context dynamic simulations (MD)
Potential Pitfalls
1. Limited Structural Confidence
The L protein is short (~75 amino acids), partially disordered, and membrane-associated. AlphaFold confidence scores (pLDDT) may be low in flexible or transmembrane regions. This uncertainty can propagate into inaccurate ddG predictions and false positives.
Mitigation strategy:
- Use consensus scoring (ESM + Rosetta + FoldX)
- Focus on mutations in higher-confidence structural regions
- Experimentally validate multiple top-ranked variants in parallel
2. Limited Training Data on Phage–Host Interactions
Most protein language models and structural predictors are trained primarily on soluble globular proteins and well-characterized complexes. The L–DnaJ interaction has limited structural and mutational data, which may reduce predictive accuracy for engineering chaperone independence.
Mitigation strategy:
- Prioritize global stability improvements before fine-tuning interface mutations
- Maintain diversity among selected candidates
- Use rapid experimental feedback (Stages 4–5) to iteratively refine computational predictions
Expected Outcome and Next Steps
This computational stage (Stage 1) will generate a prioritized list of 5–8 L protein variants predicted to exhibit improved thermodynamic stability and reduced reliance on host chaperones.
Selected variants will then:
- Be synthesized via Twist Bioscience
- Be cloned using Gibson Assembly
- Be structurally evaluated using the Nuclera system
- Be tested in E. coli for lysis efficiency and phage propagation
By focusing on stability, there is aim to engineer a more robust L protein that enhances MS2’s ability to lyse E. coli efficiently. This reduces the evolutionary window for bacterial resistance and strengthens the therapeutic potential of MS2-based phage therapy.
Pipeline Schematic
| Phase | Methods | Purpose |
|---|---|---|
| Sequence Design | ESM-2, ProteinMPNN | Generate stabilized variants |
| Structure Modeling | AlphaFold3, Multimer | Preserve fold, weaken DnaJ dependence |
| Stability Filtering | FoldX / Rosetta | Select low ddG mutants |
| Functional Validation | MD simulations | Evaluate membrane behavior |
| Experimental Transfer | Gene synthesis | Move to wet lab testing |