Week 5 HW: Protein design part II
Part A: SOD1 Binder Peptide Design (From Pranam)
Part 1: Generate Binders with PepMLM
the human SOD1 sequence (P00441): MATKAVCVLKGDGPVQGIINFEQKESNGPVKVWGSIKGLTEGLHGFHVHEFGDNTAGCTSAGPHFNPLSRKHGGPKDEERHVGDLGNVTADKDGVADVSIEDSVISLSGDHCIIGRTLVVHEKADDLGKGGNEESTKTGNAGSRLACGVIGIAQ
the A4V mutant SOD1 sequence: MATKVVCVLKGDGPVQGIINFEQKESNGPVKVWGSIKGLTEGLHGFHVHEFGDNTAGCTSAGPHFNPLSRKHGGPKDEERHVGDLGNVTADKDGVADVSIEDSVISLSGDHCIIGRTLVVHEKADDLGKGGNEESTKTGNAGSRLACGVIGIAQ
four peptides of length 12 amino acids conditioned on the mutant SOD1 sequence with the known SOD1-binding peptide FLYRWLPSRRGG for comparison:
| index | Binder | Pseudo Perplexity |
|---|---|---|
| 1 | WRYGPAAAAHWK | 9.318684 |
| 2 | WHYPAVVLRWKX | 16.435002 |
| 3 | WLYYPAAVRLWK | 16.527933 |
| 4 | WLYYVAVVALGE | 22.958134 |
| 5 | FLYRWLPSRRGG | 20.63523127283615 |
Conclusion
The model assigned the lowest perplexity (9.32) to peptide WRYGPAAAAHWK, indicating the highest sequence plausibility according to the language model.
The experimentally validated SOD1-binding peptide FLYRWLPSRRGG showed one of the higher perplexity (20.63), suggesting that the language model does not necessarily rank experimentally verified binders as the most probable sequences.
One generated peptide (WHYPAVVLRWKX) contains the residue “X”, which denotes an unknown or unspecified amino acid. This likely reflects a tokenization or sampling artifact of the language model. Because “X” does not correspond to a defined amino acid, this peptide should be interpreted cautiously when evaluating potential binding candidates which suggests that such a peptide may be invalid.
Part 2: Evaluate Binders with AlphaFold3
Each peptide was modeled in a separate AlphaFold 3 run using a two-chain setup. Chain A consisted of the SOD1 A4V mutant, while Chain B contained a single 12-residue peptide. All peptides were evaluated individually to compare ipTM scores and binding poses. Peptide 2 (WHYPAVVLRWKX) containing X was excluded from structural analysis because X denotes an unknown amino acid.
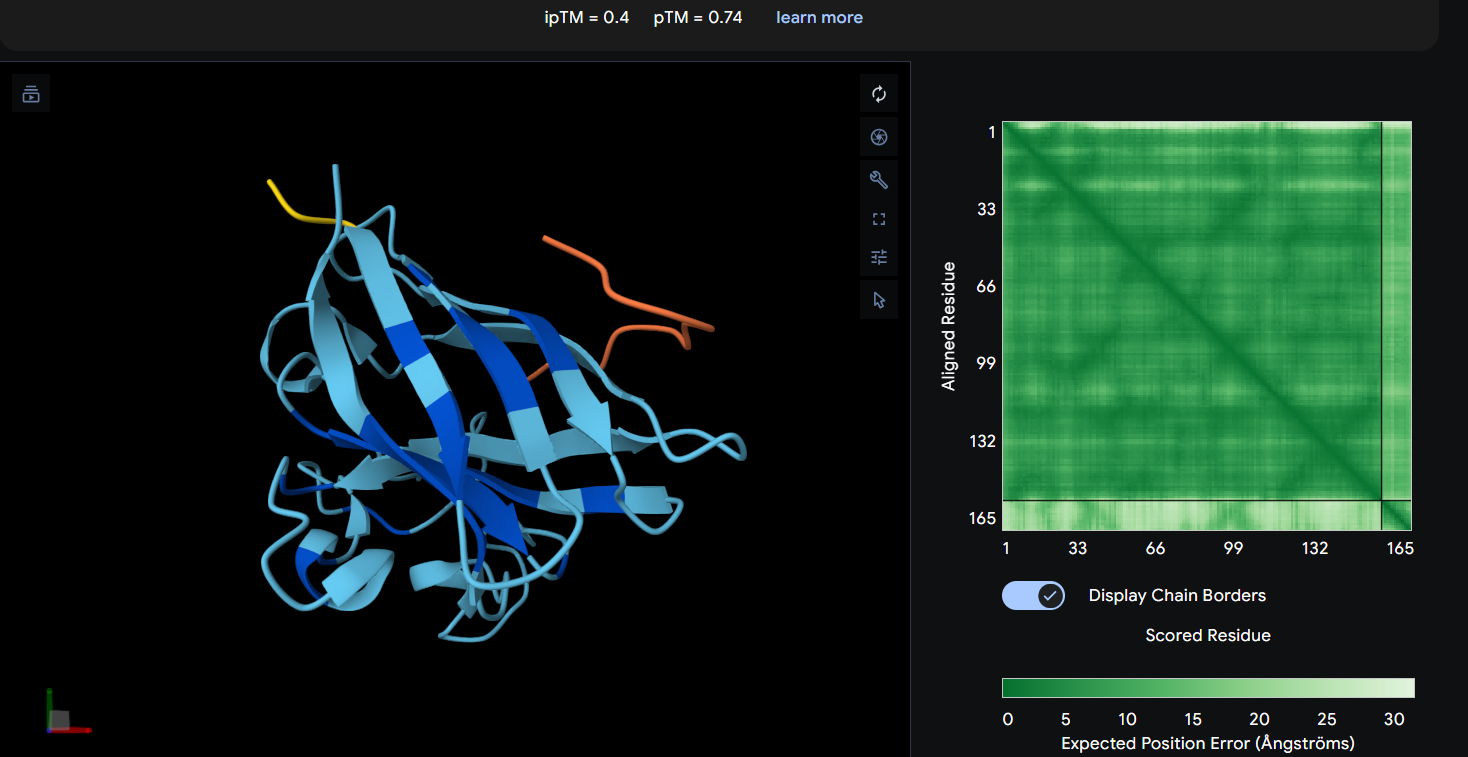
Peptide 1: WRYGPAAAAHWK (ipTM = 0.40)
Peptide 2: WHYPAVVLRWKX skipped (invalid X residue)
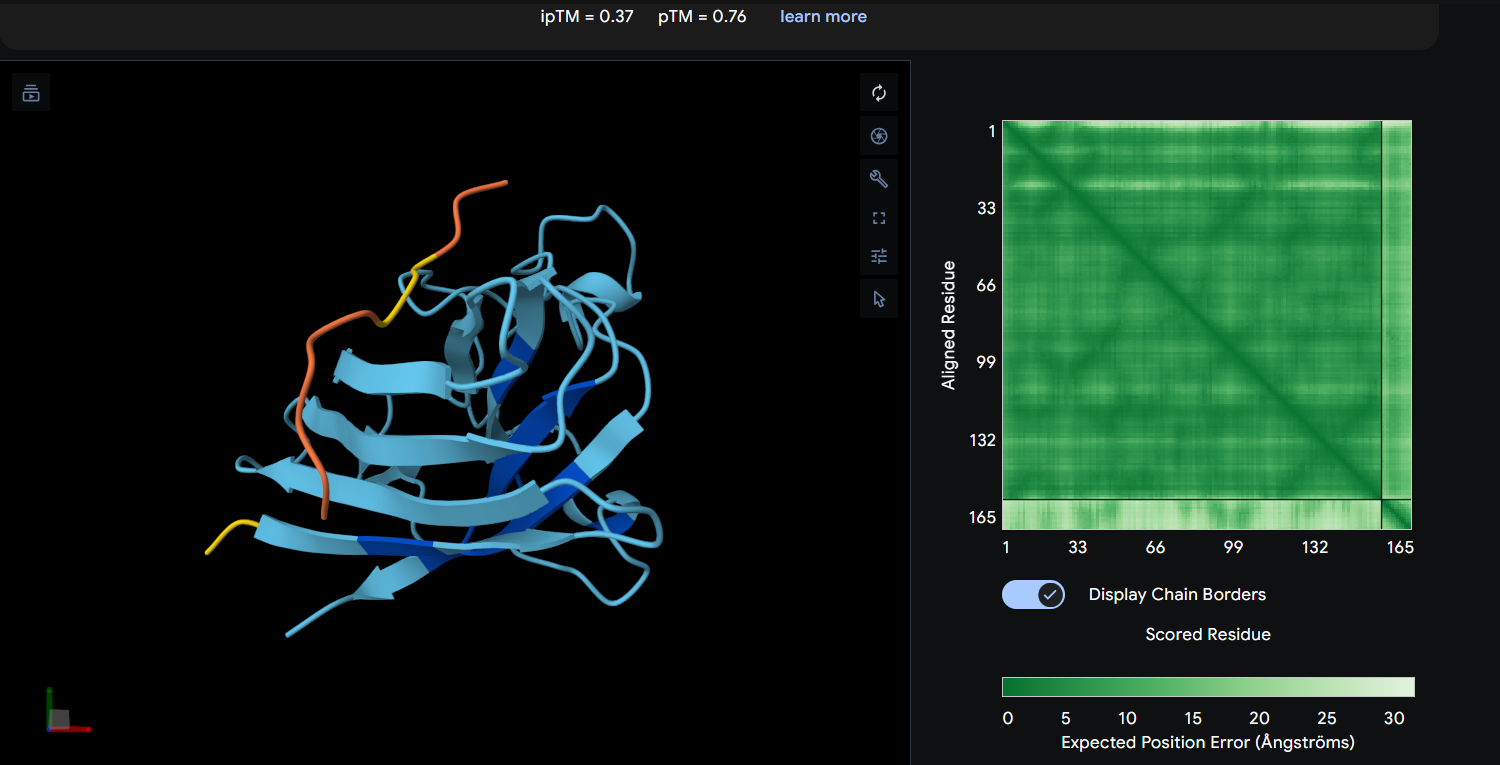
Peptide 3: WLYYPAAVRLWK (ipTM = 0.37)
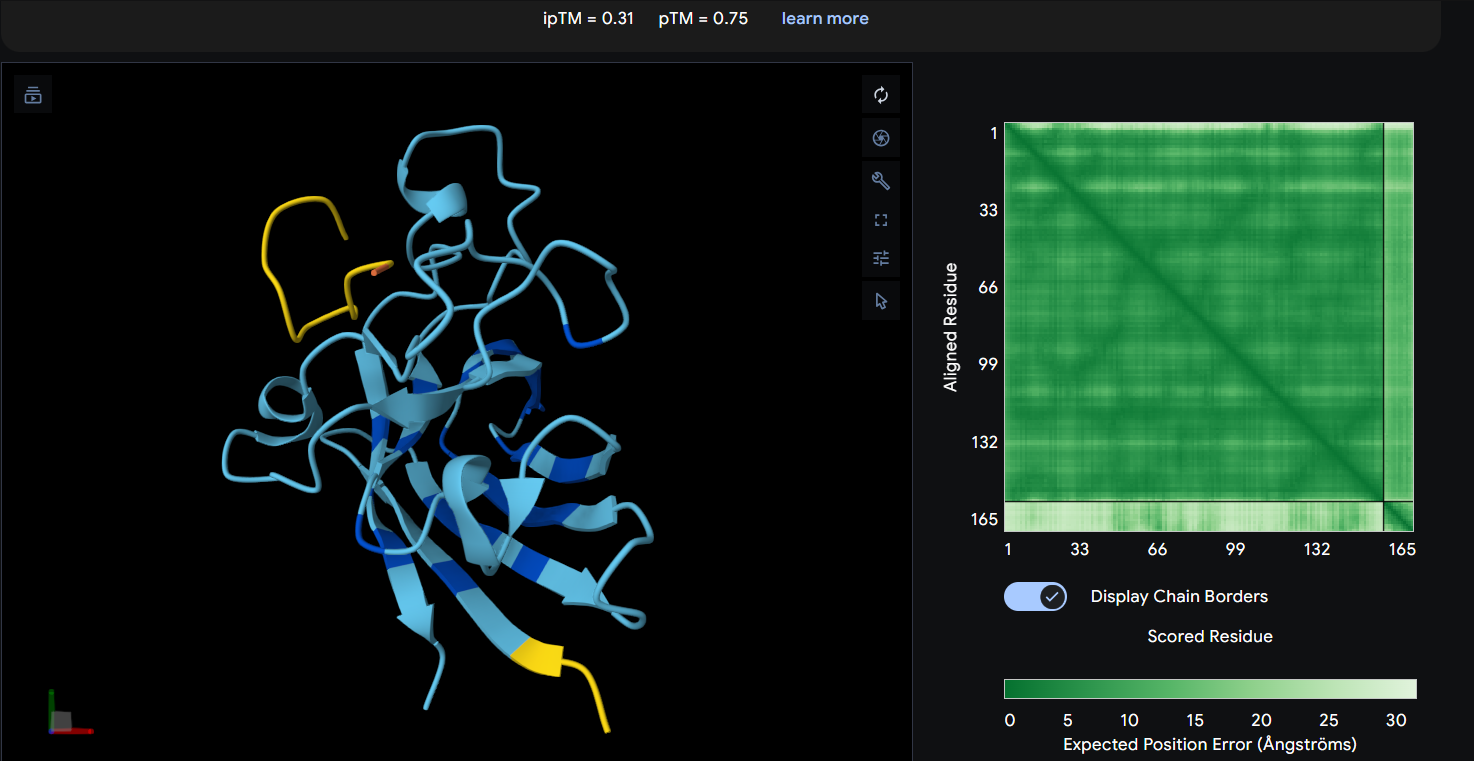
Peptide 4: WLYYVAVVALGE (ipTM = 0.31)
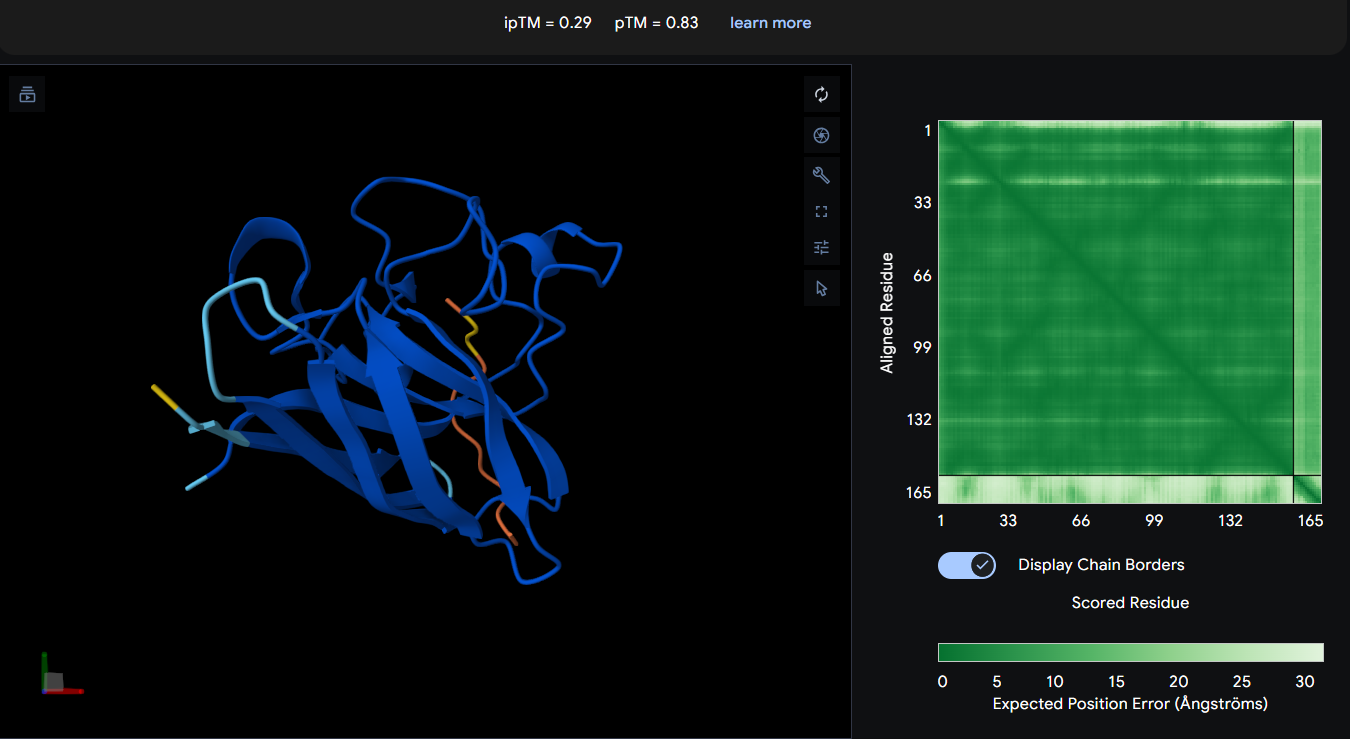
Known Binder: FLYRWLPSRRGG (ipTM = 0.29)
| Peptide | ipTM | pTM | Binding Interpretation |
|---|---|---|---|
| WRYGPAAAAHWK | 0.40 | 0.74 | Highest interface confidence among generated peptides, suggesting the most stable predicted interaction, although overall binding confidence remains low and primarily surface-associated. |
| WLYYPAAVRLWK | 0.37 | 0.76 | Weak and uncertain interaction with limited localized confidence; likely transient surface binding without a clearly defined interface. |
| WLYYVAVVALGE | 0.31 | 0.75 | Weakest predicted interface among generated peptides; interaction appears diffuse and poorly stabilized. |
| FLYRWLPSRRGG (known binder) | 0.29 | 0.83 | Despite being an experimentally known SOD1-binding peptide, AlphaFold3 predicted low interface confidence, suggesting that transient or flexible peptide interactions may not be captured reliably by structural prediction alone. |
Conclusion
AlphaFold3 analysis revealed generally low interface confidence across all peptide–SOD1 complexes, with all ipTM values remaining below 0.5. Among the PepMLM-generated peptides, WRYGPAAAAHWK achieved the highest ipTM score (0.40), indicating the strongest predicted interaction with the A4V mutant SOD1 structure, although the interaction still appeared weak and primarily surface-associated. WLYYPAAVRLWK and WLYYVAVVALGE showed progressively lower ipTM values (0.37 and 0.31, respectively), suggesting less stable peptide binding interfaces. None of the generated peptides localized clearly to the N-terminal region containing the A4V mutation, and no strongly buried or highly ordered binding mode was observed. Interestingly, the experimentally known SOD1-binding peptide FLYRWLPSRRGG produced the lowest ipTM score (0.29) despite its validated biological interaction, highlighting a limitation of AlphaFold3 in modeling transient or flexible peptide-mediated interactions. Overall, the results suggest that while some PepMLM-generated peptides may form weak surface interactions with mutant SOD1, none demonstrated highly confident or stable binding according to AlphaFold3 predictions.
Part 3: Evaluate Properties with PeptiVerse
Each peptide was evaluated against the A4V mutant SOD1 sequence using PeptiVerse for predicted binding affinity, solubility, hemolysis probability, net charge, and molecular weight.
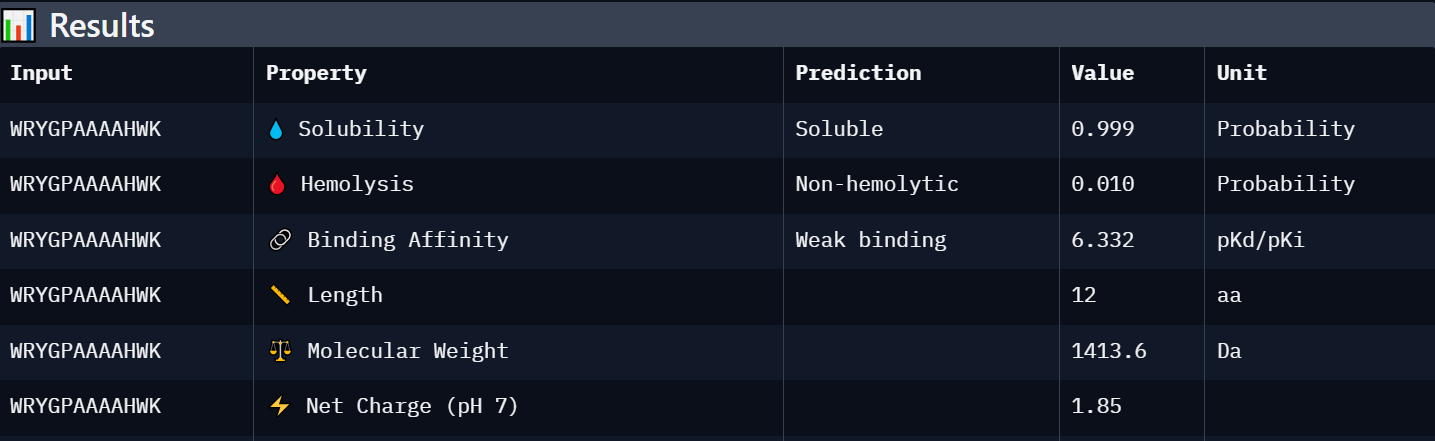
Peptide 1: WRYGPAAAAHWK
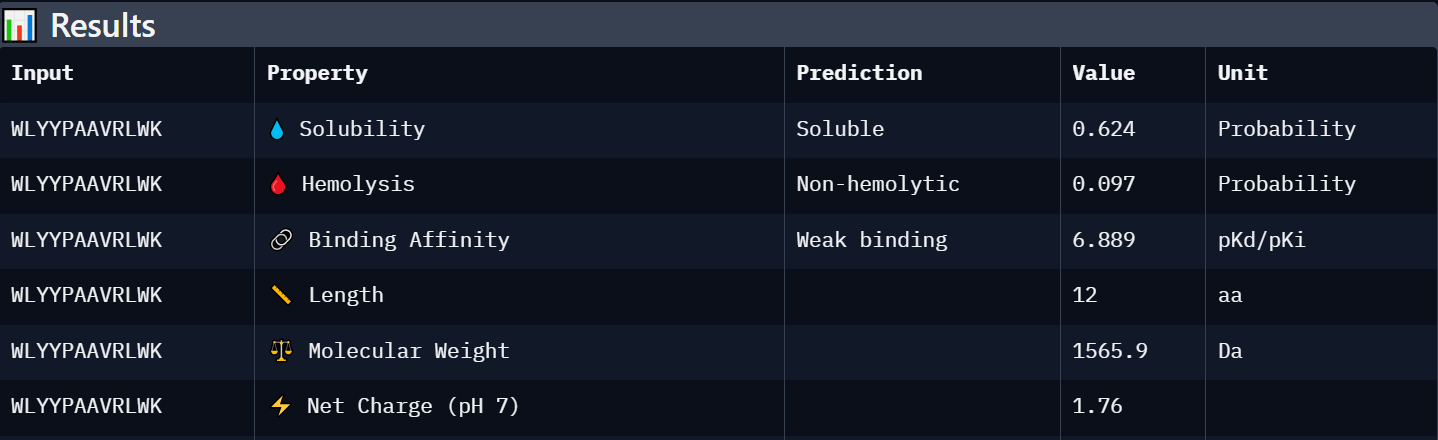
Peptide 3: WLYYPAAVRLWK
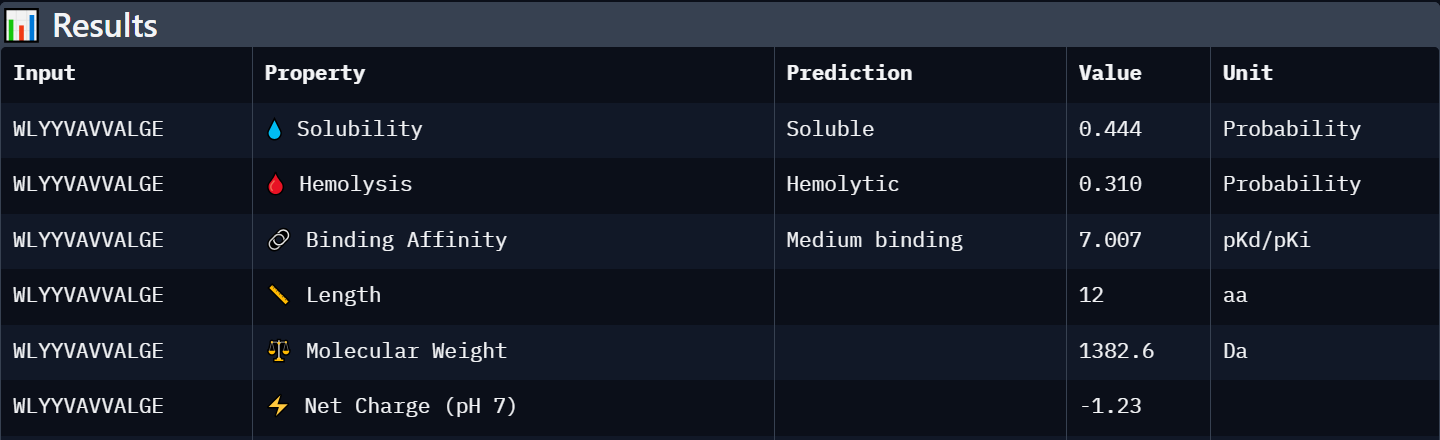
Peptide 4: WLYYVAVVALGE
Known Binder: FLYRWLPSRRGG
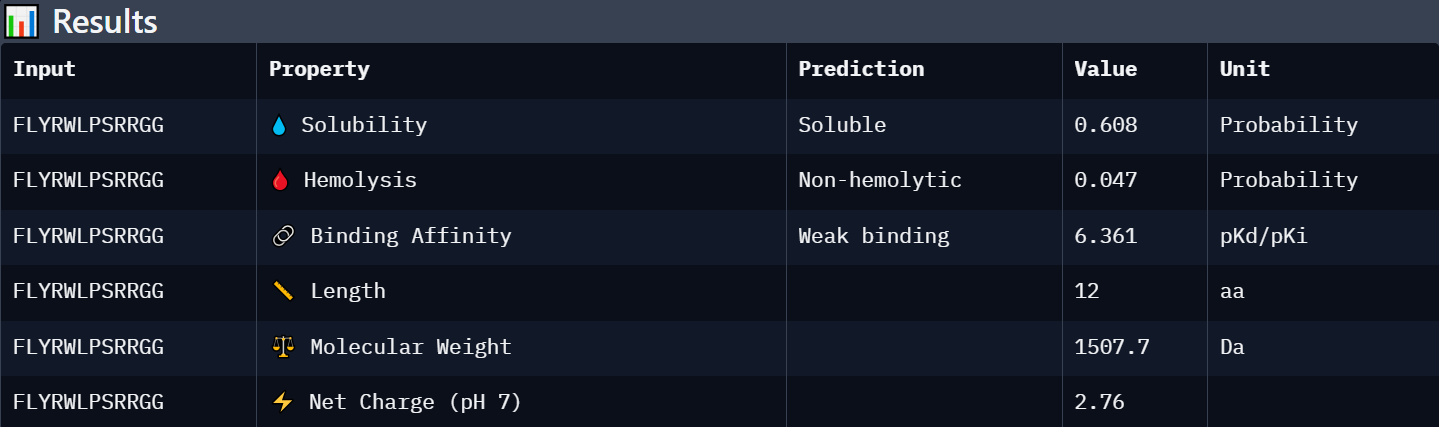
| Peptide | Predicted Binding Affinity | Solubility | Hemolysis Probability | Net Charge (pH 7) | Molecular Weight | Interpretation |
|---|---|---|---|---|---|---|
| FLYRWLPSRRGG | Weak binding (6.361) | Soluble (0.608) | Non-hemolytic (0.047) | +2.76 | 1507.7 | Known SOD1-binding reference peptide with moderate solubility, low toxicity, and acceptable predicted affinity. |
| WRYGPAAAAHWK | Weak binding (6.332) | Soluble (0.999) | Non-hemolytic (0.010) | +1.85 | 1413.6 | Best overall candidate with the strongest predicted affinity among generated peptides, excellent solubility, and minimal predicted toxicity. |
| WLYYPAAVRLWK | Weak binding (6.889) | Soluble (0.624) | Non-hemolytic (0.097) | +1.76 | 1565.9 | Intermediate candidate with acceptable solubility and low hemolysis risk, but weaker predicted affinity and structural confidence. |
| WLYYVAVVALGE | Weak binding (6.889) | Soluble (0.444) | Hemolytic (0.310) | -1.23 | 1382.6 | Least favorable candidate due to lower solubility, predicted hemolytic activity, and weak structural interaction confidence. |
Conclusion
Each peptide was evaluated against the A4V mutant SOD1 sequence using PeptiVerse for predicted binding affinity, solubility, hemolysis probability, net charge, and molecular weight. Among the PepMLM-generated peptides, WRYGPAAAAHWK showed the strongest overall profile, combining the highest AlphaFold3 ipTM score (0.40) with the best predicted binding affinity (6.332), excellent solubility (0.999), and extremely low hemolysis probability (0.010). In contrast, WLYYVAVVALGE demonstrated weaker structural confidence (ipTM 0.31), lower solubility, and a higher predicted hemolysis probability (0.310), making it a less favorable therapeutic candidate despite similar predicted affinity values. WLYYPAAVRLWK showed intermediate behavior with moderate solubility and low hemolysis risk but weaker predicted binding and interface confidence than WRYGPAAAAHWK. Overall, peptides with higher ipTM values tended to display slightly stronger predicted binding affinity and more favorable therapeutic properties. Compared with the known binder FLYRWLPSRRGG, WRYGPAAAAHWK achieved a comparable affinity prediction while exhibiting superior solubility and lower predicted toxicity.
Among the evaluated candidates, WRYGPAAAAHWK I would select for further development. Although its predicted binding remains modest, it achieved the highest ipTM score among the generated peptides and demonstrated the most favorable therapeutic profile, including excellent solubility and minimal predicted hemolytic activity. These properties suggest a better balance between structural interaction potential and developability compared with the other candidates.
Part 4: Generate Optimized Peptides with moPPIt
moPPIt was run targeting motif positions 2–8 of A4V SOD1, with objectives set to Hemolysis Probability, Solubility, Predicted Affinity, and Motif (all weights = 1). Five 12-mer peptides were generated.
| Peptide | Hemolysis Probability | Solubility | Predicted Affinity | Motif | Interpretation |
|---|---|---|---|---|---|
| CTSGENVGAGVS | 0.0666 | 0.9999 | 6.0910 | 0.6461 | Highly soluble and low-risk peptide with moderate predicted affinity and acceptable motif targeting near the selected SOD1 region. |
| ANAPWPPAFSFH | 0.0155 | 1.0000 | 6.0548 | 0.6574 | Very low predicted hemolysis and excellent solubility, with moderate affinity and balanced therapeutic properties. |
| PSEKQCVKFHTT | 0.0481 | 1.0000 | 5.8624 | 0.8456 | Strong motif guidance score with improved predicted affinity and excellent solubility, making it one of the most balanced candidates. |
| MYAGIFEKNKQT | 0.0307 | 0.9999 | 5.6291 | 0.7912 | Best predicted affinity among generated peptides with low hemolysis probability and excellent solubility, representing the strongest overall candidate. |
| QPTCGSGQFNWF | 0.0334 | 1.0000 | 6.3863 | 0.8244 | Excellent solubility and favorable motif score, although predicted affinity is weaker compared with the other moPPIt peptides. |
Conclusion
moPPIt-generated peptides demonstrated consistently favorable therapeutic property predictions compared with the earlier PepMLM-generated candidates. All peptides showed near-perfect predicted solubility (~1.0) and very low hemolysis probabilities, indicating improved safety and developability profiles. In contrast to PepMLM, which generated peptides with mixed structural and therapeutic characteristics, moPPIt produced candidates optimized simultaneously for affinity, motif targeting, and physicochemical properties. Among the generated peptides, MYAGIFEKNKQT achieved the strongest predicted affinity score (5.6291) while maintaining excellent solubility and low predicted toxicity, making it the most promising overall candidate. PSEKQCVKFHTT also showed strong performance due to its high motif score (0.8456), suggesting improved targeting of the selected SOD1 A4V-adjacent region. Overall, the moPPIt peptides appeared more balanced and therapeutically favorable than the original PepMLM-generated peptides, highlighting the advantages of guided multi-objective peptide optimization.
Before advancing these peptides toward clinical development, additional validation would be required, including molecular docking, molecular dynamics simulations, biochemical binding assays, aggregation inhibition studies, cytotoxicity testing, and evaluation in cellular or animal ALS models to confirm both efficacy and safety.
Part C: Final Project: L-Protein Mutants
The goal of this project is to reduce the interaction between the MS2 lysis protein (L-protein) and the bacterial chaperone DnaJ. Since DnaJ is important for proper folding and processing of the L-protein, weakening this interaction may help the phage remain functional even if bacteria modify DnaJ. To study this interaction, co-folding predictions were performed using AlphaFold2 Multimer with both proteins entered together. In AlphaFold2, both sequences are inserted into a single input field separated by a colon :.
Mutations were introduced only in the soluble N-terminal domain of the L-protein and not in the transmembrane region.
Mutant 1
- R18A
- R19A
- R20A
- R30A
- R34A
Full Mutant L-Protein Sequence
METRFPQQSQQTPASTNAAAPFKHEDYPCRAQQASSTLYVLIFLAIFLSKFTNQLLLSLLEAVIRTVTTLQQLLT
AlphaFold2 Multimer Results
Among the generated AlphaFold2 multimer models, the top-ranked model (rank_001, model_4) was selected for analysis because it showed the highest predicted interaction confidence with an ipTM score of 0.165.
| Metric | Value |
|---|---|
| pLDDT | 74.8 |
| pTM | 0.528 |
| ipTM | 0.165 |
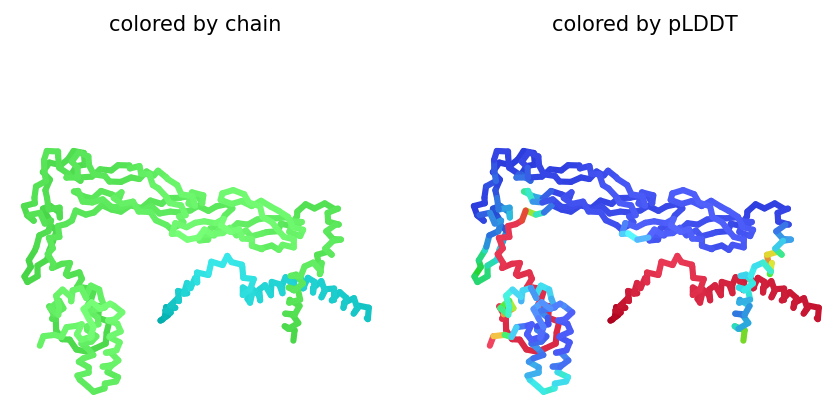
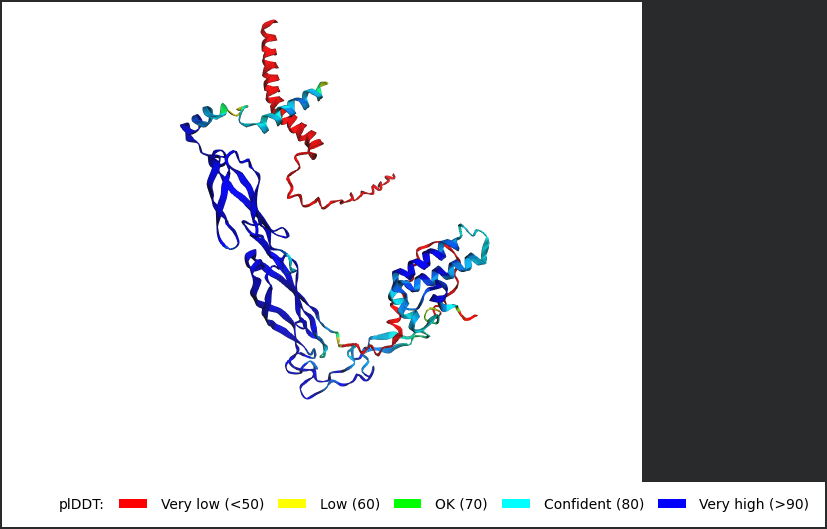 3D structure of top-ranked model (rank_001, model_4)
3D structure of top-ranked model (rank_001, model_4)
Explanation
This mutant was designed to weaken the interaction between the L-protein and DnaJ. The soluble N-terminal domain contains several positively charged arginine residues that may participate in electrostatic interactions with DnaJ. Replacing arginine with alanine removes positive charges and may reduce binding affinity to the chaperone.
Alanine substitutions were chosen because alanine is small and structurally non-disruptive, allowing the protein to maintain overall folding while reducing interaction surfaces.
The transmembrane domain was left unchanged because it is important for lysis activity.
Conclusion
Mutant 1 remained structurally stable after introducing alanine substitutions in the soluble domain. The pLDDT score indicates that the mutant protein still folds with moderate confidence.
However, the ipTM score was low (0.165), indicating a weak predicted interaction between DnaJ and the mutant L-protein. This suggests that the introduced arginine-to-alanine substitutions may successfully reduce DnaJ binding while preserving overall protein structure.
The structural model also showed limited contact between the two proteins, supporting the hypothesis that these mutations weaken the DnaJ–L-protein interaction.
Mutant 2
- Q7L
- Q10L
- Q11L
- Q32A
- Q33A
Full Mutant L-Protein Sequence
METRFPQLLSQLLTPASTNRRRPFKHEDYPCRRAAARSSTLYVLIFLAIFLSKFTNQLLLSLLEAVIRTVTTLQQLLT
AlphaFold2 Multimer Results
Among the generated AlphaFold2 multimer models, the top-ranked model (rank_001, model_4) was selected for analysis because it showed the highest predicted interaction confidence with an ipTM score of 0.149.
| Metric | Value |
|---|---|
| pLDDT | 74.4 |
| pTM | 0.523 |
| ipTM | 0.149 |
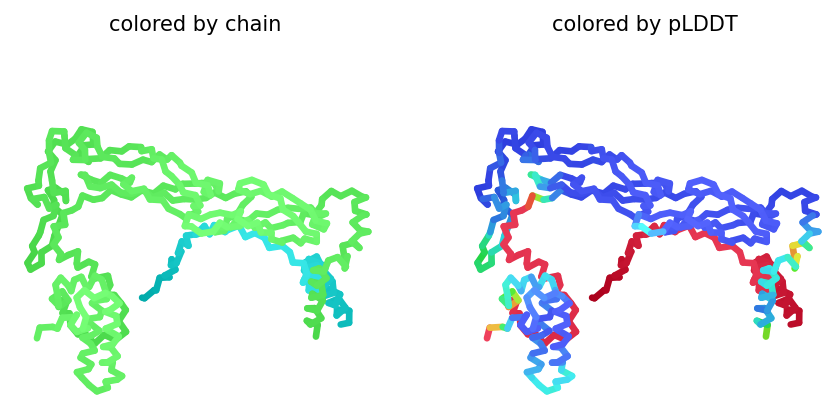
 3D structure of top-ranked model (rank_001, model_4)
3D structure of top-ranked model (rank_001, model_4)
Explanation
This mutant was designed to improve autonomous folding of the L-protein and reduce dependence on DnaJ. The soluble domain contains multiple glutamine residues that may contribute to structural flexibility and chaperone dependence. Replacing some glutamines with leucine increases hydrophobic stabilization, while replacing others with alanine reduces polar interactions. These mutations may stabilize local folding and decrease the need for DnaJ-assisted folding.
The transmembrane region was not modified to preserve lysis function.
Conclusion
Mutant 2 remained structurally stable after introducing glutamine-to-leucine and glutamine-to-alanine substitutions in the soluble domain. However, the low ipTM score (0.149) indicates a weak interaction with DnaJ. These results suggest that the mutations may reduce DnaJ dependence while maintaining overall folding of the L-protein.