Week 1 HW: Principles and Practices

1. First, describe a biological engineering application or tool you want to develop and why. This could be inspired by an idea for your HTGAA class project and/or something for which you are already doing in your research, or something you are just curious about.
Looking at the work of Long, B., Li, Q., Hu, C. et al. (1), I am interested in the use of genetically modified algae for the purpose of aggregation and removal of microplastics in a manner that allows for the upcycling of the then microplastic-enriched cyanobacteria for plastic composites. The algae in the study referenced (Synechococcus elongatus UTEX 2973) were modified to produce limonene to increase cell hydrophobicity, facilitating both the aggregation of microplastics with the cyanobacteria and the self-aggregation of the cyanobacteria in water. I would be interested to see if changing the terpene produced by the cyanobcterium changes the efficacy of the microplastic removal in an aqeuous environment. Further studies could include exploring more uses or alternative processing methodologies for the plastic composite materials produced from the co-aggregated cyanobacteria and microplastics.
(1) Long, B., Li, Q., Hu, C. et al. Remediation and upcycling of microplastics by algae with wastewater nutrient removal and bioproduction potential. Nat Commun 16, 11570 (2025). https://doi.org/10.1038/s41467-025-67543-5
2. Next, describe one or more governance/policy goals related to ensuring that this application or tool contributes to an “ethical” future, like ensuring non-malfeasance (preventing harm). Break big goals down into two or more specific sub-goals. Below is one example framework (developed in the context of synthetic genomics) you can choose to use or adapt, or you can develop your own. The example was developed to consider policy goals of ensuring safety and security, alongside other goals, like promoting constructive uses, but you could propose other goals for example, those relating to equity or autonomy.
Policy goals may include:
A. Ensuring that the use of genetically modified cyanobacteria to remove microplastics from water does not harm the environment it is being deployed in, e.g.:
-Can this method be used for ocean or other waterway clean up without negatively impacting these environments?
-If this method is used by water treatment facilities, is there a way to ensure that the modified cyanobacteria does not escape into the environment?
-Is it necessary to prevent the release of this organism from the environment?
-To determine if the modified cyanobacteria is released into the environment from a water treatment facility, who is responsible for this monitoring?
-Who is responsible for removing the modified organism from the environment if a release is found (if it is determined that this should be actively prevented)?
-Should fines be levied towards companies that allow these organisms to escape into the environment? This alone will not be enough to ensure that this does not happen or that companies will be responsible for remediation (i.e. if there are only fines without specific regulation determining that companies must be responsible for remediation, then many companies may determine it is less expensive to pay fines than to fix the problem- already an issue with companies specifically as it relates to environmental impacts).
-May necessitate further study to determine what happens when the cyanobacteria-microplastic composite plastic itself decomposes. If it decomposes back into microplastics that are released into the environment, is this solution viable long-term?
B. Biosecurity: Determining who has or should have access to these genetically modified bacteria for the purpose of microplastic remediation. If the end result is using the cyanobacteria-microplastic aggregate to form plastic composites which can then be used in industry to replace traditional plastics, should the modified bacteria or the resulting plastic composites be considered commodities that should be protected as intellectual property? How available should strains be? Making them more widely available allows equity in ability to remove plastic from water (most likely application being removal from water supplies at water treatment facility plants, though it could be possible to use in natural environment depending on what impact this could have on broader ecosystems). Not a good idea to allow anyone to have these organisms, however, if the consequences of releasing these organisms into ecosystems is unknown at this time. Restrict to industry and researchers? What process should be used to vet? Who should have the authority and responsibility to vet industry and researchers?
C. Encouraging industry to either adopt this method for microplastic remediation (e.g. for water treatment plants) or to use the composite plastic material for producing plastic consumable goods. Right now, these are two separate industries. Who would be responsible for the treatment of the cynobacteria-microplastic aggregates that allow these to be used to produce plastic good, the water treatment plants generating the aggregates or the manufacturers that would be producing the plastic goods? A third party dedicated to this process? Manufacturers right now have manufacturing plants dedicated to producing items in regular plastics- would switching to the cyanobacteria-plastic composite require reworking the entire current processing process? This would be expensive, would need an incentive such as government stipends or grants to make this adoption realistic. Newer companies could form dedicated to using these composite materials, but this would be counter to the broader adoption required to make a an upcycling circular economy viable. Most water treatment facilities are run at the state or county level; who should pay for the increased cost of adding in this kind of remediation step? Would this require raising taxes in areas implementing this or would grant programs or stipends allow for this adoption? Early adopters could be at the private industry level (e.g. bottled wter companies) because they could charge higher prices for their product, but this could mean that these companies could lobby against broader adoption of this method because it would remove the novelty that allows them to charge higher prices for their products.
3. Next, describe at least three different potential governance “actions” by considering the four aspects below (Purpose, Design, Assumptions, Risks of Failure & “Success”). Try to outline a mix of actions (e.g. a new requirement/rule, incentive, or technical strategy) pursued by different “actors” (e.g. academic researchers, companies, federal regulators, law enforcement, etc). Draw upon your existing knowledge and a little additional digging, and feel free to use analogies to other domains (e.g. 3D printing, drones, financial systems, etc.).
Purpose: What is done now and what changes are you proposing?
Design: What is needed to make it “work”? (including the actor(s) involved - who must opt-in, fund, approve, or implement, etc)
Assumptions: What could you have wrong (incorrect assumptions, uncertainties)?
Risks of Failure & “Success”: How might this fail, including any unintended consequences of the “success” of your proposed actions?
A. Goverment/NGO Stipends/Grants: Likely needed for widespread industry adoption. Assumes that money is both available and that there is an appetite for this research to be implemented in the first place. NGOs may want to take up the mantle to support these efforts, but often have more limited funding (assuming that they even could fund these endeavors, which I am unsure about, espeically since water treatment is often a county or state effort). US federal funding would require approval by Congress at a time when environmental concerns are at a low. State funding may be easier to obtain, but could hinder more widespread adoption. Many companies may refuse to adopt if funding is not guaranteed for a certain number of years (no one wants to take on a project that could take 10 years with only 5 years of funding available). Government funding may increase distrust in communities since this affects their water supply.
B. Federal Monitoring for Release: Would need to determine who has authority (EPA, perhaps?). Federal agencies are already strained in terms of resources, both funding and personnel, and further requirements would strain this further. Would need a system in place to monitor water, requiring decisions as to where water should be collected and tested and what lab should be doing the testing. Would need to determine next steps (is the federal government only monitoring, or is there responsibility to also assist in remediation if a leak is found? If assisting with response, who pays the bill? American tax payers on the hook for assisting or fully the responsibility of the company? Depending on where the organism is found, what happens if it cannot be definitively determined which company is responsible?) If only monitoring, then this does not assist in response.
C. Laws for Industry Monitoring/Response Responsibility: Laws requiring industry using these organisms to monitor for and remediate an environmental release would be likely the only way to ensure that this happens. Traditionally, the use of fines for release of anything that may potentially harm the environment results in companies paying the fines without remediation, as this is generally cheaper than paying for the actual remediation. This also shifts the burden to the industry rather than forcing government agencies to pay for monitoring and response (although would likely still need independent government monitoring, otherwise you are relying fully on industry essentially self-reporting problems, a conflict of interest). Requirements such as these, however, my prevent adoption of these methods if the perceived industry risk is higher than potential reward.
D.IP Protections/Organism Use Restriction: Organism use restriction could prevent accidental release into the environment without proper determinination of how this would impact the environment, but does not entirely eliminate this risk. Restriction of use of organism would require some agency or company screening customers (unlikely that a DNA synthesis company would stop an order for limonene or other terpene-production gene, so would not get caught here if someone were to try to modify this organism themselves). IP protection for a modified organism via patent seems possible, but this is controversial and I am making assumptions about the feasbility here (unsure how feasible given how easy it generally is to modify these orgnanisms). Federal restriction of agents at the moment is limited primarily to the Select Agent program, which only covers highly pathogenic organisms, would need some other mechanism to restrict usage of an organism like this based upon industry usefulness/claims to it as intellectual property. Intellectual property protections would likely increase industry odds of adoption.
4. Next, score (from 1-3 with, 1 as the best, or n/a) each of your governance actions against your rubric of policy goals. The following is one framework but feel free to make your own:
RUMBA: Remediation and Upcycling of Microplastics by Algae
| Does the option: | Goverment/NGO Stipends/Grants | Federal Monitoring for Release | Laws for Industry Monitoring/Response Responsibility | IP Protections/Organism Use Restriction |
|---|
| Enhance Biosecurity | | | | |
| • By preventing incidents | n/a | 2 | 1 | 1 |
| • By helping respond | n/a | 3 | 1 | n/a |
| Protect the Environment | | | | |
| • By preventing incidents | n/a | 2 | 1 | 2 |
| • By helping respond | n/a | 3 | 1 | n/a |
| Industry Adoption | | | | |
| • Minimizing costs and burdens to stakeholders | 1 | 2 | 3 | n/a |
| • Feasibility? | 2 | 1 | 1 | 2 |
| • Promote constructive applications | 1 | n/a | 2 | 2 |
5.Last, drawing upon this scoring, describe which governance option, or combination of options, you would prioritize, and why. Outline any trade-offs you considered as well as assumptions and uncertainties.
Based upon the above layout of possible governance mechanisms, I would suggest that water treatment plants utilizing genetically engineered algae for the purpose of sequestering and removing microplastics have a legal requirement to self-monitor and report any possible leaks of the genetically modified organisms (assuming that there is a lack of large amounts of data over time demonstrating that these organisms would not cause ecosystem damage) with the ultimate responsibility of remediating any detected leaks. I recognize that this may dissuade industry from taking up this methodology, but to not have something in place like this would be irresponsible, as there is a possibility that an unintentional release could have broad, long-term impacts in the environment (cannot know without further testing). There would also be a need, then, to have independent monitoring and testing from a (likely federal) agency to ensure that there are no leaks and to ensure that we are not fully depending on industry self-reporting. I would also suggest that if there is a widespread desire for industry to take up this endeavor, there would need to be incentives, such as grants or stipends, to allow this. The best governance strategy, I think, would be a layered one that involves industry buy-in, federal oversight to ensure biosecurity and environmental protections are in place, and government financial support for this industry. There could also be information campaigns and industry outreach to educate consumers about the process to increase not only awareness, but drive demand either for this process to be put into place (in which case, constituents could, for example, write to members of Congress to voice support for the industry to increase odds of funding being made available to support these programs) or for the products produced through this method.
Ethical issues that arose during this thought experiment mainly were centered around equity versus biosecurity, i.e. restricting access to genetically modified organisms that could potentially have negative ecosystem impact would be a good biosecurity practice, but could prevent access to a potentially cost effective way to reduce microplastics in drinking water and produce composite plastics in areas of the globe (such as the global south) where it could do the most good. Additionally, the solution of allowing companies to patent either the modified organism itself or the production method for turning the aggregated cyanobacteria-microplastic into composite materials in order to protect intellectual property and incentivize the uptake of this idea could lead to one company holding a monopoly on this method. There is also the possibility that someone could purchase the patents in order to prevent them from being used by anyone, if they wanted to uphold the status quo for using conventional plastics.
Week 2 HW: DNA Read, Write, & Edit
Part 1: Benchling & In-silico Gel Art
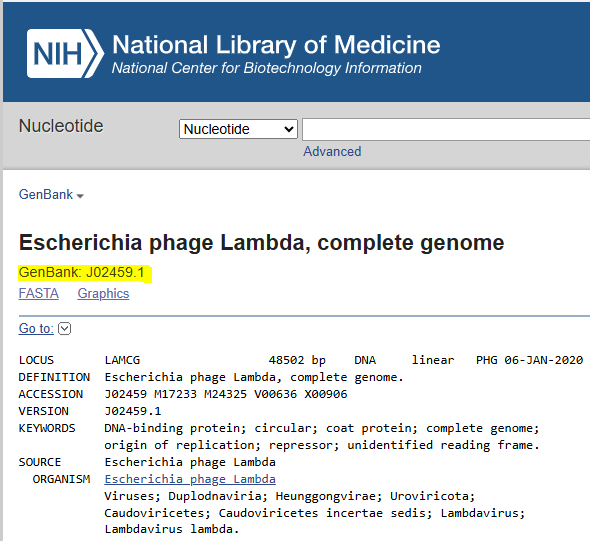
After locating the E. coli phage Lambda genome in the NCBI database, I loaded the sequence into my Benchling project by searching for the GenBank accession number
(GenBank: J02459.1):
I then went to the DNA Gel Art Interface (https://rcdonovan.com/gel-art) and went through the gallery for inspiration and to observe what bands looked like for the digestion enzymes listed both individually and in combination with one another, taking screenshots of these images to determine which of these I could use as “building blocks” for my image.
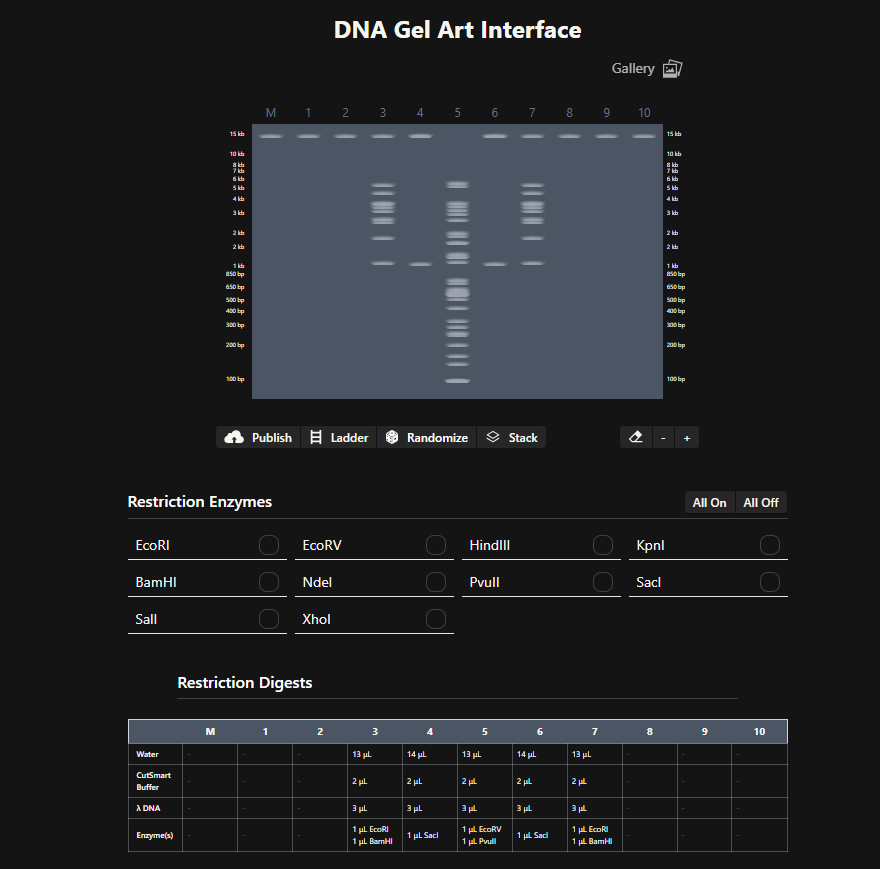
I took inspiration from the following design on the gel art website, thinking I could modify it to make “boxes” using three lanes that could look like eyes that could then be used for a “smiley face” design:
I used my notes/screenshots to determine what other bands could be used for the top parts of these boxes and for the parts of the smile, planning them out on paper first:

I then tested this design using the in-silico digest in Benchling:
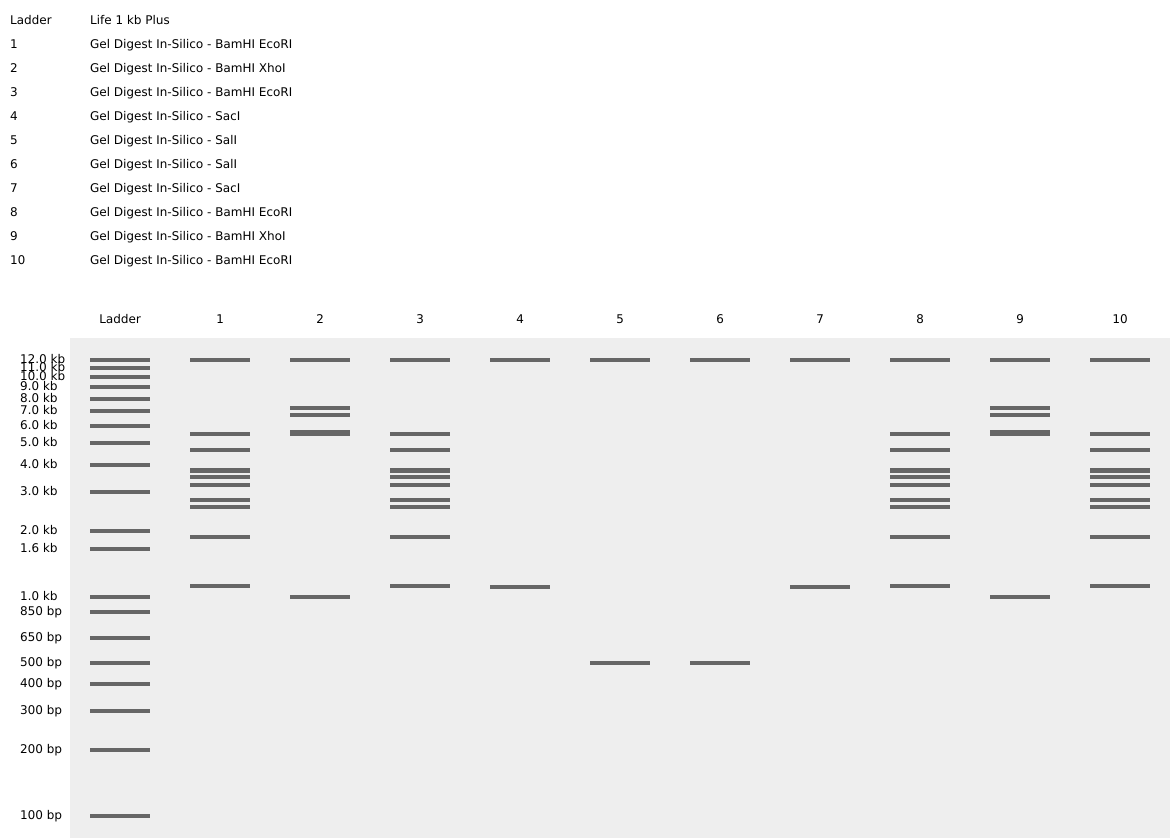
I did not like that the bands meant to represent the edges of the smile were level with the bottoms of the eyes, so I adjusted the design to make the smile start higher up in the lanes:
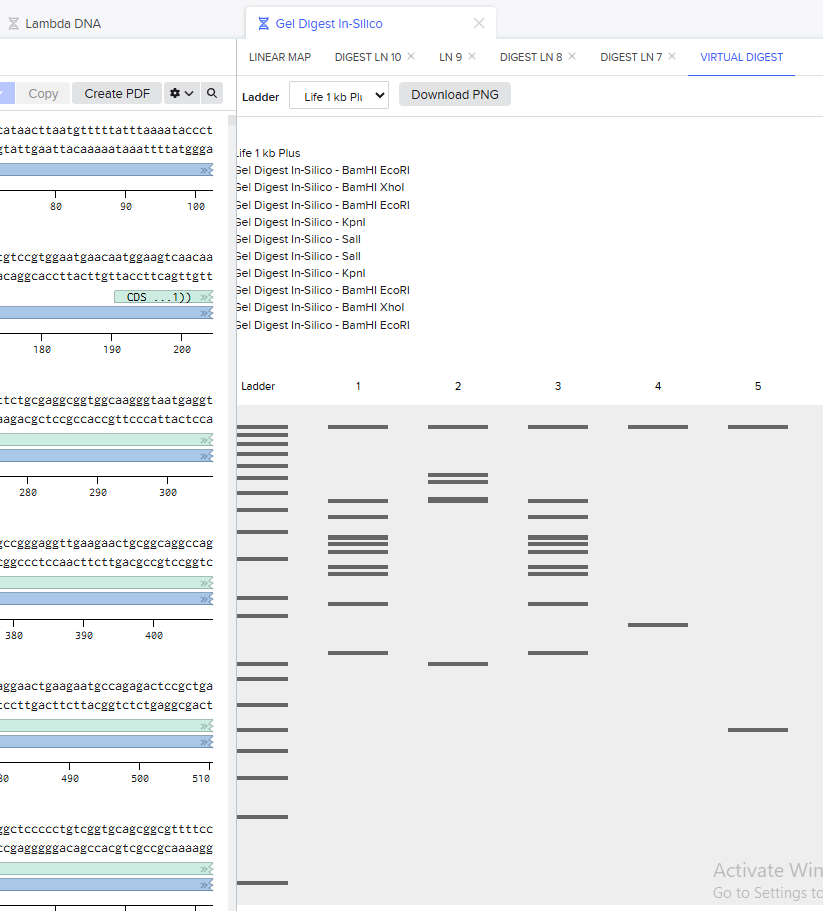
Final In-Silico Gel Design: https://benchling.com/s/seq-FfTcfmv79fwJMljaZij3?m=slm-Gjs2Z4pczSrUZzB76zUr
Part 2: Gel Art - Restriction Digests and Gel Electrophoresis Wet Lab
Note: I will not be able to make most wet labs due to EMT school causing scheduling conflicts but was able to attend this first in-person node lab and will participate in-person when possible.
Protocol
Part 1a: Preparing a 1% agarose electrophoresis gel
*Chemicals:
-Agarose powder
-1x TBE buffer
-SmartGlow Safe Green Pre Stain (https://www.labproservices.com/buy-smartglow-safe-green-pre-stain-1-0ml-ea-1-e4500-ps-ea.html?gad_source=1&gad_campaignid=20923765122&gbraid=0AAAAAqHTWoNkTzYIzDQi_alxD1vIk_2bP&gclid=CjwKCAiAncvMBhBEEiwA9GU_fvIAPFHPYWljiQUeFtU-1_CGZxCC4XhO-unP2Wn_t-_yl_bK6QYBLBoCFlwQAvD_BwE)
*Equipment
-Pipette Set
-Gel tray
-9 or 13-well comb
-Microwave
-Precision scale
*Consumables
-Pipette tips
-250 mL beaker
-Graduated cylinder (100mL)
TBE Buffer Preparation
The TBE buffer provided in our lab was already diluted to the proper 1X working solution.
Gel Preparation
To prepare an agarose gel for electrophoresis:
Added 0.4 g of agarose powder (exact weight came out to 0.3999g) and 40mL of 1x TBE buffer to a microwavable beaker (1% w/v)
Placed the flask in a microwave, heated in short pulses of 15–20 seconds, swirling gently between pulses, until the agarose was completely dissolved, watching to prevent any boil-overs.
Allowed the solution to cool until the beaker was warm but comfortable to touch.
Once cooled, added 2μL of SmartGlow DNA pre-stain to the solution and mixed gently.
Slowly poured the agarose into the tray to avoid forming bubbles, placed the comb into the gel tray to create wells. 9 or 13 well-combs were available; I selected the 13-well comb and adapted my design to this.
Allowed the gel to solidify for about 30 minutes at room temperature. Once set, carefully removed the comb, and the gel was ready for use.
Protocol
Part 1b: Restriction Digest
*Chemicals
-1X Lambda DNA
-The following enzymes: EcoRI-HF, BamHI-HF, Xhol, KpnI-HF, SacI-HF, SalI-HF (see calculated quantities below)
-Nuclease-free water
*Equipment and Consumables
-Thermocycler
-PCR tube rack
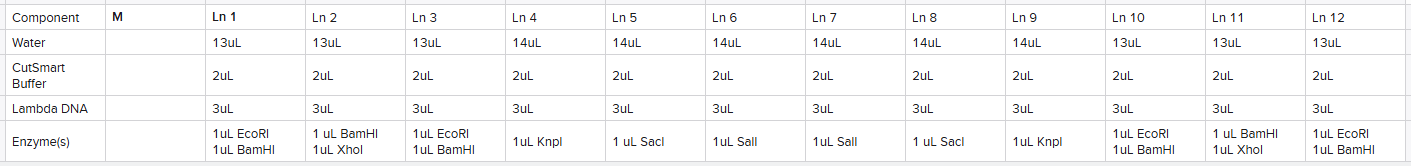
I planned out an adjusted design from what I had in-silico due to the availability of 13 lanes:
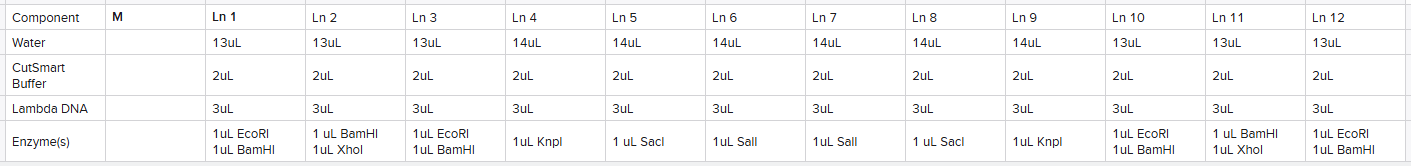
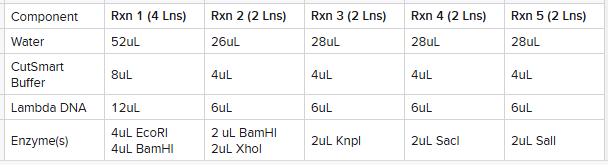
Based upon this plan, I made 5 reaction tubes as follows calculated based upon the number of lanes I planned on using the reaction for:
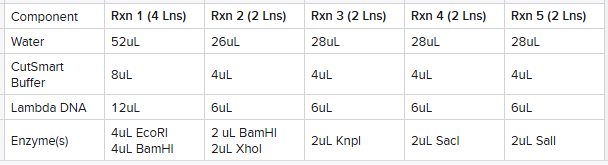
I then placed the rxn tubes in the thermocycler set to 37ºC and incubated for 30 minutes (while the agarose gel solidified).
Protocol
Part 2: Gel Run
*Chemicals
-6x loading dye
-1X TBE
-SmartGlow Safe Green Pre Stain
*Equipment
-Gel Box
-Power supply (ours did not require the addition of leads, everything was built into the gel box with just a power supply connected to an outlet, voltage already adjusted to what was required)
Gel Run
Comb was removed once the gel was set.
Gel box was filled with approximately 30-40mL 1x TBE (enough to ensure gel was covered entirely). 0.72uL of pre-stain was added to the 1X TBE.
For each rxn (per lane), I mixed 7uL of my digest from the rxn tube with 2uL of 6X loading dye on a sheet of parafilm prior to loading into wells, following the schema described above for all lanes.
Made sure the top was place on the gel box the correct way (negative to negative, positive to positive, gel positioned with loaded lanes at the negative end).
Ran the gel at 80V - 115V for around 45 minutes (originally checked after 30 minutes, let run about another 15 to allow for further separation of bands). Checked that there were bubbles in the buffer and that lanes were moving after about 10 minutes.
Protocol
Part 3: Imaging Your Results
*Equipment
-Blue light transilluminator (for our lab, this was actually built into the gel electrophoresis chamber/gel box)
-Phone camera
*Consumables and Safety
-Gloves
To view my results, I simply turned on the blue light built into our gel boxes, removing the need to transfer the gel to a separate transilluminator.
Put a light-blocking device around the gel box to block out ambient room light and allow for better photos to be taken.
Took a picture using my phone’s camera through an imaging hole on the top of the light-blocking device.
Disposed of the gel in the solid waste bin (burn box).
Final Results
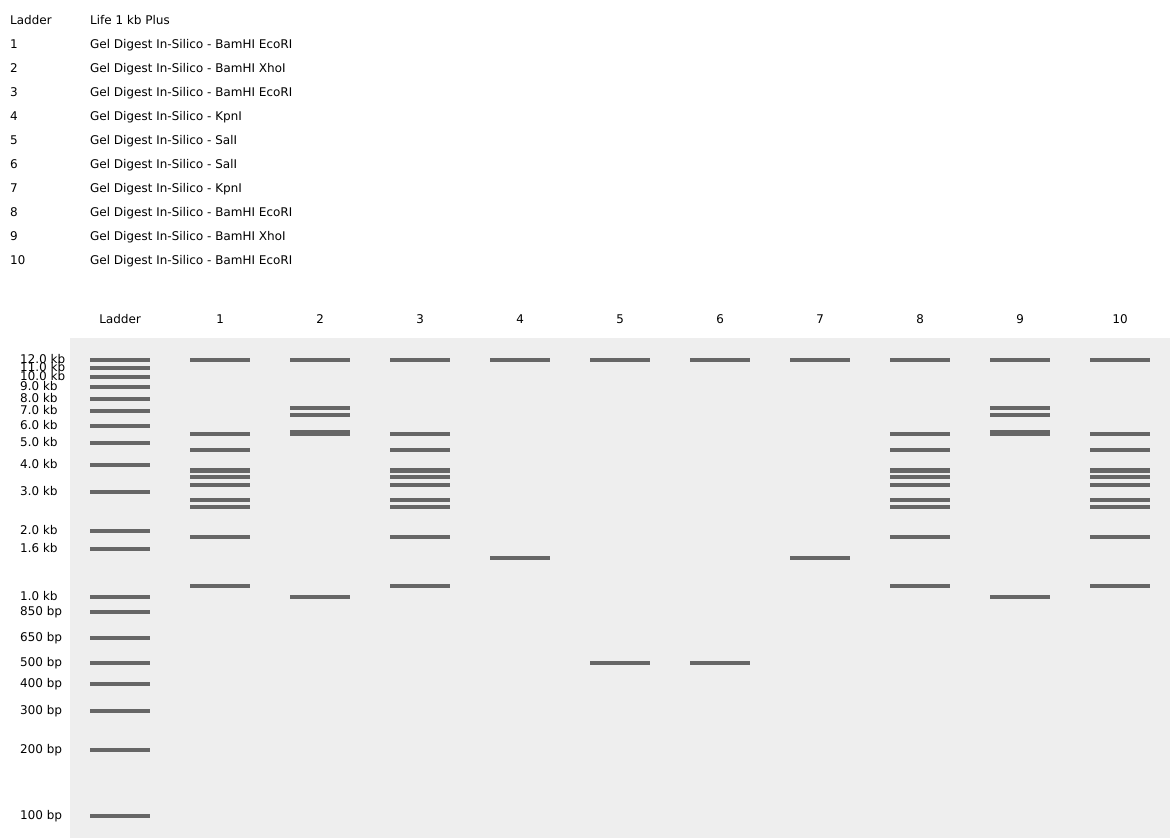
Here is a picture of the final results:

I forgot to account for the fact that some of the single bands created were rather small sections base-pair wise and thus would not be very visible. I also was hoping for better separation from my deisgn from the uncut sections at the top, making my smiley face a bit hard to make out (especially since the bottom of the smile is not actually visible). Someone commented that they think the design looked like a neat bridge, so if anyone asks…it was definitely a bridge I was trying to make here!
Part 3: DNA Design Challenge
3.1. Choose your protein.
The protein I have chosen is Chitin deacetylase 2 (CDA2), as I am interested in the usage of chitosan in hemostatic dressings to treat traumatic injury by stopping hemorrhage. Most chitosan produced for the purpose of hemostatic agents is produced chemically, but there is newer research being performed looking into using enzymes such as CDA2 for a “greener” method for the production of chitosan by removing N-acetyl groups from chitin. Chitin is naturally found in the exoskeleons of insects and crustaceans as well as fungi and certain algae. Chitosan produced from shellfish was used more heavily in the past and can still be found today in some hemostatic bandages, but this chitosan had to be irrigated out of wounds and posed danger to those with shellfish allergies.
The specific protein I found in UniProt was found in Saccharomyces cerevisiae (strain ATCC 204508 / S288c):
sp|Q06703|CDA2_YEAST Chitin deacetylase 2 OS=Saccharomyces cerevisiae (strain ATCC 204508 / S288c) OX=559292 GN=CDA2 PE=1 SV=1
MRIQLNTIDLQCIIALSCLGQFVHAEANREDLKQIDFQFPVLERAATKTPFPDWLSAFTG
LKEWPGLDPPYIPLDFIDFSQIPDYKEYDQNHCDSVPRDSCSFDCHHCTEHDDVYTCSKL
SQTFDDGPSASTTKLLDRLKHNSTFFNLGVNIVQHPDIYQRMQKEGHLIGSHTWSHVYLP
NVSNEKIIAQIEWSIWAMNATGNHTPKWFRPPYGGIDNRVRAITRQFGLQAVLWDHDTFD
WSLLLNDSVITEQEILQNVINWNKSGTGLILEHDSTEKTVDLAIKINKLIGDDQSTVSHC
VGGIDYIKEFLS
3.2. Reverse Translate: Protein (amino acid) sequence to DNA (nucleotide) sequence.
CDA2_YEAST Chitin deacetylase 2 OS=Saccharomyces cerevisiae (strain ATCC 204508 / S288c) AA sequence to nucleotides using the CUSABIO Protein to DNA Sequence Converter tool (including a start and stop codon):
ATGATGCGTA TTCAATTAAA TACTATTGAT TTACAATGTA TTATTGCTTT ATCTTGTTTA GGTCAATTTG TTCATGCTGA AGCTAATCGT GAAGATTTAA AACAAATTGA TTTTCAATTT CCTGTTTTAG AACGTGCTGC TACTAAAACT CCTTTTCCTG ATTGGTTATC TGCTTTTACT GGTTTAAAAG AATGGCCTGG TTTAGATCCT CCTTATATTC CTTTAGATTT TATTGATTTT TCTCAAATTC CTGATTATAA AGAATATGAT CAAAATCATT GTGATTCTGT TCCTCGTGAT TCTTGTTCTT TTGATTGTCA TCATTGTACT GAACATGATG ATGTTTATAC TTGTTCTAAA TTATCTCAAA CTTTTGATGA TGGTCCTTCT GCTTCTACTA CTAAATTATT AGATCGTTTA AAACATAATT CTACTTTTTT TAATTTAGGT GTTAATATTG TTCAACATCC TGATATTTAT CAACGTATGC AAAAAGAAGG TCATTTAATT GGTTCTCATA CTTGGTCTCA TGTTTATTTA CCTAATGTTT CTAATGAAAA AATTATTGCT CAAATTGAAT GGTCTATTTG GGCTATGAAT GCTACTGGTA ATCATACTCC TAAATGGTTT CGTCCTCCTT ATGGTGGTAT TGATAATCGT GTTCGTGCTA TTACTCGTCA ATTTGGTTTA CAAGCTGTTT TATGGGATCA TGATACTTTT GATTGGTCTT TATTATTAAA TGATTCTGTT ATTACTGAAC AAGAAATTTT ACAAAATGTT ATTAATTGGA ATAAATCTGG TACTGGTTTA ATTTTAGAAC ATGATTCTAC TGAAAAAACT GTTGATTTAG CTATTAAAAT TAATAAATTA ATTGGTGATG ATCAATCTAC TGTTTCTCAT TGTGTTGGTG GTATTGATTA TATTAAAGAA TTTTTATCTT AA
3.3. Codon optimization.
Codon optimization for the host organism that is intended to express the sequence is necessary as each organism may have different tRNAs available in different quantities, impacting the speed at which translation may take place or even be possible. I used Benchling’s back translate with codon optimization tool to optimize this sequence for the host organism E.coli K12 and avoid the BglI digest enzyme cut-site because it is non-pathogenic and would allow for selection of only transformed bacteria in an experiment if I use a plasmid vector that includes a gene that confers antibiotic resistance to an antibiotic that kills E. coli K12 wild type.
Signal Peptide:
ATGCGCATTCAGCTTAATACCATCGATTTACAATGCATCATTGCGTTGAGCTGTCTGGGCCAATTTGTTCATGCCGAAGCAAACCGTGAGGACCTGAAACAGATTGATTTCCAGTTTCCGGTGCTGGAACGC
Chain CDA2:
GCAGCGACCAAGACGCCGTTCCCGGATTGGCTGTCGGCTTTTACGGGCCTTAAAGAGTGGCCGGGCCTTGACCCGCCGTATATTCCCCTGGATTTTATCGATTTCAGCCAAATTCCAGATTACAAAGAATATGACCAGAACCATTGTGATAGTGTGCCACGCGATAGCTGCTCGTTCGACTGCCACCATTGCACCGAGCACGATGACGTCTACACTTGTTCCAAACTGAGTCAAACCTTTGACGATGGTCCTAGCGCCTCTACGACGAAACTGTTAGATCGCTTAAAACATAACTCAACCTTTTTTAATCTGGGCGTCAACATTGTTCAGCACCCCGACATTTATCAGCGTATGCAGAAAGAAGGCCATCTGATTGGAAGCCATACCTGGTCTCATGTATATCTGCCTAATGTGAGCAATGAGAAGATCATTGCGCAGATTGAATGGAGCATCTGGGCGATGAACGCCACCGGCAATCATACACCGAAATGGTTTCGTCCGCCGTACGGGGGCATTGACAATCGCGTTCGGGCCATTACCCGTCAATTCGGTCTGCAAGCGGTGTTGTGGGATCACGATACTTTCGATTGGTCGCTGTTGCTTAACGATTCCGTGATAACCGAACAGGAAATCCTGCAGAACGTAATTAATTGGAACAAAAGTGGCACAGGTTTGATTCTAGAACACGACAGCACGGAAAAGACTGTTGACTTAGCAATCAAGATTAATAAACTCATCGGTGATGATCAGTCGACCGTGTCACACTGTGTCGGAGGGATAGATTACATCAAAGAATTTCTCTCA
Together:
ATGCGCATTCAGCTTAATACCATCGATTTACAATGCATCATTGCGTTGAGCTGTCTGGGCCAATTTGTTCATGCCGAAGCAAACCGTGAGGACCTGAAACAGATTGATTTCCAGTTTCCGGTGCTGGAACGCGCAGCGACCAAGACGCCGTTCCCGGATTGGCTGTCGGCTTTTACGGGCCTTAAAGAGTGGCCGGGCCTTGACCCGCCGTATATTCCCCTGGATTTTATCGATTTCAGCCAAATTCCAGATTACAAAGAATATGACCAGAACCATTGTGATAGTGTGCCACGCGATAGCTGCTCGTTCGACTGCCACCATTGCACCGAGCACGATGACGTCTACACTTGTTCCAAACTGAGTCAAACCTTTGACGATGGTCCTAGCGCCTCTACGACGAAACTGTTAGATCGCTTAAAACATAACTCAACCTTTTTTAATCTGGGCGTCAACATTGTTCAGCACCCCGACATTTATCAGCGTATGCAGAAAGAAGGCCATCTGATTGGAAGCCATACCTGGTCTCATGTATATCTGCCTAATGTGAGCAATGAGAAGATCATTGCGCAGATTGAATGGAGCATCTGGGCGATGAACGCCACCGGCAATCATACACCGAAATGGTTTCGTCCGCCGTACGGGGGCATTGACAATCGCGTTCGGGCCATTACCCGTCAATTCGGTCTGCAAGCGGTGTTGTGGGATCACGATACTTTCGATTGGTCGCTGTTGCTTAACGATTCCGTGATAACCGAACAGGAAATCCTGCAGAACGTAATTAATTGGAACAAAAGTGGCACAGGTTTGATTCTAGAACACGACAGCACGGAAAAGACTGTTGACTTAGCAATCAAGATTAATAAACTCATCGGTGATGATCAGTCGACCGTGTCACACTGTGTCGGAGGGATAGATTACATCAAAGAATTTCTCTCA
3.4. You have a sequence! Now what?
My selected codon optimized sequence could be expressed in the host organism if this sequence were placed into a plasmid (preferably with an antibiotic resistance gene for an antibiotic that the E. coli K12 wild type is susceptible to) and the plasmid were uptaken by the bacteria, generally induced with heat-cold shock or electroporation. The presence of an antibiotic resistance gene in this plasmid that is lacking in the wild type would allow for me to select only for transformed bacteria by growing the bacteria in antibiotic-containing media.
Part 4: Prepare a Twist DNA Synthesis Order
Alas, I’m going to have to come back to this section and complete at another time. I have been attempting to make a Twist account for the past several days and have received an error message each time after attempting to access from different browsers:
I attempted to communicate with Twist through the chat system, but it is manned by actual people that are not working this long weekend. They should get back to me 17 February 2026, so I’ll come back to this for practice after this is resolved and I can make my account. In the meantime:
5.1 DNA Read
(i) What DNA would you want to sequence (e.g., read) and why? This could be DNA related to human health (e.g. genes related to disease research), environmental monitoring (e.g., sewage waste water, biodiversity analysis), and beyond (e.g. DNA data storage, biobank).
(ii) In lecture, a variety of sequencing technologies were mentioned. What technology or technologies would you use to perform sequencing on your DNA and why?
Also answer the following questions:
Is your method first-, second- or third-generation or other? How so?
What is your input? How do you prepare your input (e.g. fragmentation, adapter ligation, PCR)? List the essential steps.
What are the essential steps of your chosen sequencing technology, how does it decode the bases of your DNA sample (base calling)?
What is the output of your chosen sequencing technology?
Maybe this is a bit of a trick answer, but I work in biosurveillance/biodefense, so I would be interested in looking for sequences of novel pathogens in order to identify possible disease outbreaks before they spread into pandemics. The question then becomes, how do you look for a sequence that you don’t know?
- For biosurveillance of novel, potential pandemic-causing pathogens in a population, the preferred sample type would either be waste water (collected either from the municipal supplies or from specific large-gathering places such as airports) or nasal or oropharyngeal swabs in transport media taken from patients that, for example, shows respiratory illness symptoms but test negative on respiratory panels looking for common flu or cold-caueing viruses and bacteria. Swabs could also be taken of asymptomatic passengers at airports that decide to opt into such proposed programs.
- Nucleic acid from the samples of choice would then be extracted, typically using kits (e.g. Qiagen) for the specific sample type or other high-throughput methods (e.g. KingFisher or liquid handler-based extraction methods). This nucleic acid would then be quantified and checked for any contaminants, such as proteins, via methods such as NanoDrop or electrophoresis.
QC: Quantify DNA concentration (e.g., Qubit) and check for fragmentation or contamination (e.g., NanoDrop, electrophoresis).
- The extracted DNA would then need to be sheared into smaller, randomized fragments (sequencing fragmentation). A library prep step would then be performed for end repair, A-tailing, and ligation of adapters/indices for multiplexing, allowing multiple samples to be sequenced together.The sample would then be sequenced on a NextGenn platform such as Illumina (e.g., NovaSeq, NextSeq) to produce millions of short reads. Short read sequencing via sequencing by synthesis (SBS) is the most common method on the market, though long read methods like NanoPore are also used.
- Bioinformatics analysis, which can be read-centric or assembly-centric, then takes place and includes steps to: filter out low-quality reads and trim adapters (preprocessing); remove host reference genomes (e.g., human) as applicable;identify microbial species and their relative abundances (e.g., using Kraken2, MetaPhlAn) (taxonomic profiling); use assemblers (e.g., MEGAHIT, SPAdes) to stitch reads into longer contiguous sequences (contigs);predict genes (e.g., Prodigal) and annotate functions (e.g., Prokka, DIAMOND) to understand the metabolic potential; and cluster contigs into Metagenome-Assembled Genomes (MAGs) to identify novel organisms (e.g., MetaBAT2) (binning). Data can also be verified via methods such as qPCR and visualized by performing alpha/beta diversity analysis, and metabolic pathway mapping using tools like R, Python, or QIIME 2.
5.2 DNA Write
(i) What DNA would you want to synthesize (e.g., write) and why? These could be individual genes, clusters of genes or genetic circuits, whole genomes, and beyond. As described in class thus far, applications could range from therapeutics and drug discovery (e.g., mRNA vaccines and therapies) to novel biomaterials (e.g. structural proteins), to sensors (e.g., genetic circuits for sensing and responding to inflammation, environmental stimuli, etc.), to art (DNA origamis). If possible, include the specific genetic sequence(s) of what you would like to synthesize! You will have the opportunity to actually have Twist synthesize these DNA constructs! :)
(ii) What technology or technologies would you use to perform this DNA synthesis and why?
Also answer the following questions:
What are the essential steps of your chosen sequencing methods?
What are the limitations of your sequencing method (if any) in terms of speed, accuracy, scalability?
I would be interested in on-demand production of mRNA vaccines, an endeavor that is already being explored by the 100 days mission and by BARDA. One way that this could be done would be by sequencing a pathogen of interest and then selecting for a protein expressed on the outside of the bacteria or virus (one that would be interacting with the immune system directly) and construct an mRNA with the instructions for how your own ribosomes could produce the protein in order to train the immune system against the specific protein found on the pathogen (building up an immunity or resistance against the pathogen without using a killed or live-attenuated version of the pathogen). Using the SARS-CoV-2 mRNA vaccine as a reference, the genetic sequence for the SARS-CoV-2 spike protein is synthesized (e.g. through solid-phase phosphoramidite chemistry) and then inserted into a plasmid. From there, they separate the two strands of plasmid DNA. RNA polymerase, the molecule that transcribes RNA from DNA, then uses the spike protein gene to create a single mRNA molecule. Other molecules break down the rest of the plasmid to ensure that only the mRNA is packaged as a vaccine. This single-stranded mRNA is then encapsulated in a lipid bilayer to protect the fragile molecule. This method is limited by both the limitations previously discussed for phosphoramidite chemistry (losing efficiency and accuracy after a certain length, necessitating synthesis of smaller pieces to be stitched together) and by the fragility of the mRNA molecule (which requires protection via a lipid bilayer and, at the present, a continuous cold chain from production to end user). mRNA vaccines being made on demand should decrease the amount of time between novel pathogen identification/outbreak and a vaccine, but may of limited use in areas where a maintained (very) cold chain is unfeasible, such as areas in the global south. The hope is that these methods may be more accessible as the technology progresses, allowing for on-demand production at the point of the end user (reducing the need for the cold chain).
5.3 DNA Edit
(i) What DNA would you want to edit and why? In class, George shared a variety of ways to edit the genes and genomes of humans and other organisms. Such DNA editing technologies have profound implications for human health, development, and even human longevity and human augmentation. DNA editing is also already commonly leveraged for flora and fauna, for example in nature conservation efforts, (animal/plant restoration, de-extinction), or in agriculture (e.g. plant breeding, nitrogen fixation). What kinds of edits might you want to make to DNA (e.g., human genomes and beyond) and why?
(ii) What technology or technologies would you use to perform these DNA edits and why?
Also answer the following questions:
How does your technology of choice edit DNA? What are the essential steps?
What preparation do you need to do (e.g. design steps) and what is the input (e.g. DNA template, enzymes, plasmids, primers, guides, cells) for the editing?
What are the limitations of your editing methods (if any) in terms of efficiency or precision?
I think a good candidate for gene editing would be using the CRISPR/Cas9 technology to replace harmful/pathogenic variant BRCA1 and BRCA2 inherited gene mutations with functional BRCA1 and BRCA2 genes in individuals that have these harmful mutations. Many of these individuals know that they have these gene mutations due to a family history (generally confirmed during genetic testing/counseling) and have no recourse other than preventative mastectomies and other surgeries, certain medications, and increased cancer-screenings. While this gene does not run in my family, I think it would be a good candidate for clinical gene editing due to public awareness of this gene and the fact that many of the pathogenic variants of the BRCA gene are due to frame shift mutations caused by small insertions or deletions (indels). CRISPR/Cas9 gene-editing technology involves using a guide RNA (in our case, a synthetic single guide RNA) to match a desired target gene, and Cas9 (CRISPR-associated protein) endonuclease which causes a double-stranded DNA break, allowing modifications to the genome. In order to cut, a specific sequence of DNA of between 2 and 5 nucleotides (the exact sequence depends upon the bacteria which produces the Cas9) must lie at the 3’ end of the guide RNA, called the protospacer adjacent motif (PAM). Repair after the DNA cut may occur via two pathways: non-homologous end joining, typically leading to a random insertion/deletion of DNA; or homology directed repair where a homologous piece of DNA is used as a repair template (e.g. a syntheaized non-pathogenic gene variant), allowing for precise genome editing. The homologous section of DNA with the required sequence change may be delivered with the Cas9 nuclease and sgRNA, theoretically allowing changes as precise as a single base-pair. Based upon my limited knowledge of CRISPR/Cas9 technology, I would think targeted delivery would be one of the largest technical challenges in using this technology to gene edit in a way that produces non-pathogenic BRCA gene variants. If this method were to be used to treat children or adults, then we would be looking to make alterations in somatic cells where we would expect the gene variant to cause disease; this can differ between individuals, but generally impacts more than the breast tissue (individuals with BRCA mutations also have higher incidence of uterine, ovarian, and other reproductive cancers, as well as elevated risk of pancreatic cancers). Ensuring that all of the cells that could become cancerous are effectively treated would be extremely difficult (and I am doubtful there would be an effective method to screen or otherwise check this, clinically speaking). Even if all of the cells that could become cancerous due to this gene mutation were treated in an individual, this individual would still be able to pass on their pathogenic gene variant via germline, unless this were also treated. Treating the patient’s germline via CRISPR/Cas9 would ensure that offspring would inherit the “fixed” gene variant, but leads to ethical considerations regarding eugenics and “designer babies”.
Week 3 HW: Lab Automation
1. Find and describe a published paper that utilizes the Opentrons or an automation tool to achieve novel biological applications.
I came across the following paper, Brown DM, Phillips DA, Garcia DC, Arce A, Lucci T, Davies JP Jr, Mangini JT, Rhea KA, Bernhards CB, Biondo JR, Blum SM, Cole SD, Lee JA, Lee MS, McDonald ND, Wang B, Perdue DL, Bower XS, Thavarajah W, Karim AS, Lux MW, Jewett MC, Miklos AE, Lucks JB. Semiautomated Production of Cell-Free Biosensors. ACS Synth Biol. 2025 Mar 21;14(3):979-986. doi: 10.1021/acssynbio.4c00703. Epub 2025 Mar 12. PMID: 40073441., that compares the semi-automated production of cell-feee biosensors against manual production, utilizing the Opentron OT-2 liquid handler. I hope to use this and similar papers (I believe Opentron has prepared Python scripts for applications like this that I could modify as needed) as a basis for my final project.
2. Write a description about what you intend to do with automation tools for your final project. You may include example pseudocode, Python scripts, 3D printed holders, a plan for how to use Ginkgo Nebula, and more. You may reference this week’s recitation slide deck for lab automation details.
While your description/project idea doesn’t need to be set in stone, we would like to see core details of what you would automate. This is due at the start of lecture and does not need to be tested on the Opentrons yet
I would be interested in utilizing a cloud laboratory to screen biosensor constructs for biological or chemical warfare agents that I design, synthesize, and express using cell-free protein synthesis.
I did find a paper (Park Y.J., Choi S., Lee K.W., Park S.Y., Song D.Y., Yoo T.H., Kim D.M. A Cell-Free Biosensor for Multiplexed and Sensitive Detection of Biological Warfare Agents. Biosens. Bioelectron. 2024;257:116331. doi: 10.1016/j.bios.2024.116331.) in which the researchers converted detection of 16S rRNA for traditional biothreat agents into functional proteins for detection and I’d be interested to see if this could be done for other sections of genes that may allow for differentiation of wild-type pathogenic bacteria and attentuated strains.
I need to further research precisely how to work design, synthesize, and express cell-free biosensor constructs (I have no prior experience here), but taken from the example provided:
Echo transfer biosensor constructs and any required cofactors into specified wells.
Bravo stamp in CPFS reagent master mix into all wells of a 96-well / 384-well plate.
Multiflo dispense the CFPS lysate to all wells to start protein expression.
PlateLoc seal the plate.
Inheco incubate the plate at 37°C while the biosensor proteins are synthesized.
XPeel remove the seal.
PHERAstar measure fluorescence to compare biosensor responses.
This example does assume that I have my biosensor constructs already designed and synthesized; I would likely design this in Benchling or another online tool and then request that it be synthesized by Twist Bioscience.
As I am not physically located at MIT/Harvard, I could discuss the possibility of using the Opentron OT-2 at my local node or MIT/Harvard’s cloud lab to work on my final project, though it would require having someone follow whatever deck layout I have in mind and aliquot out my reagents to place on the deck where I have designated. I have used a couple different liquid handlers (Hamilton STAR, Biomek i7) in different labs, but have never used them as part of a cloud lab.
Ideas for a final project:
(in progress)
Idea: Biothreat or chemical warfare agent cell-free biosensors
a. Level 1:
b. Level 2:
c. Level 3:
Idea: In-place/close to use production of nutrient and calorie-dense food stuffs that can be used for rations for warfighters or for space travel (could utilize yeast, bacteria, algae, etc.)
a. Level 1:
b. Level 2:
c. Level 3:
Idea: Venom-induced consumption coagulopathy (VICC): some way to impregnate cloth or gauze for wound-packing with synthetically produced venom (e.g. from certain species of snakes or catepillars) to cause localized coagulation and prevent major hemorrhaging
a. Level 1:
b. Level 2:
c. Level 3: