Hi everyone! I’m Violeta, a senior year of biomedical engineering in Peru. I’m passionate about synthetic biology, tissue engineering, and molecular diagnostics. My work explores biodesign as a tool for innovation in regenerative medicine and neuroengineering.
PRE-LECTURE ABOUT CLASS 2 SLIDES ABOUT PROFESSOR JACOBSON
-Question 1 When biological polymerase copies DNA, it makes about 1 mistake per million base pairs (1:10^6).Since the human genome has around 3.2 billion base pairs, that error rate would mean every time one of my cells divides, it would introduce over 3,000 mistakes if there weren’t any correction mechanisms. There’s a 3’-5’ exonuclease that catches and removes errors during DNA synthesis, and then the MutS repair system acts as a backup to fix any mismatches that slipped through afterward. Together, these mechanisms keep my genetic information stable across cell divisions.
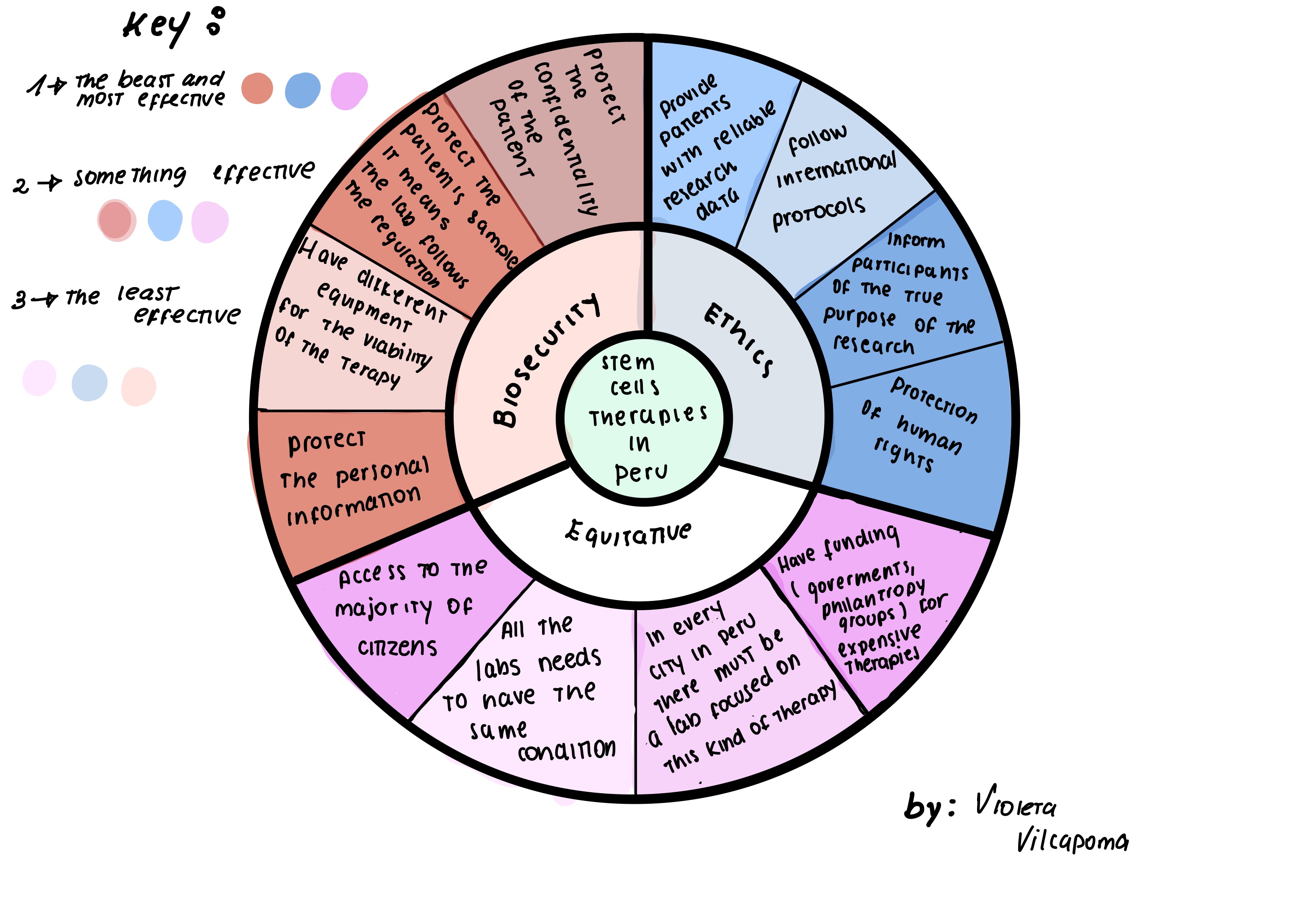
Biological engineering application: The development of various therapies in which they employ stem cells in Peru, to treat neurodegenerative diseases and chronic diseases. Along with this the appropriate regulations for this type of therapy. -> Why I chose this application: Because in Peru there is an increase in the development of various clinical therapies for diseases that have been very expensive to access, so the emergence of stem cell applications are recent. This is also due to a problem, this is more than anything focused on the little regulation on this type of treatment, which limits the creation of more trained centers, generating a delay in the access of new therapies and research.
Homework 3: Python Script for Opentrons Artwork Overview The task is to create a Python script that runs on an Opentrons OT-2 liquid handling robot and draws an artistic design on a 96-well plate using dye transfer. The design chosen for this project is a mercury droplet, representing the core element of the MeR (Mercury Bioremediation) project. Adapted to reflect a droplet / letter “M” for Mercurio.
Protein Design Part 1 Conceptual Questions Q1: How many molecules of amino acids do you take with a piece of 500 grams of meat? (on average an amino acid is ~100 Daltons)
First, we need to calculate the number of moles and multiply by Avogadro’s number (NA=6.022×1023 mol−1). An amino acid has an average mass of ~100 Daltons (Da), which is roughly equivalent to 100 g/mol. Meat is mostly protein (~20% of its weight is protein). → 500 g of meat contains approximately 100 g of protein. Since 1 mole of amino acids weighs ~100 g, there are ~1 mole of amino acids in 100 g of protein. → 1 mole is equivalent to 6.022 × 1023 molecules (Avogadro’s number). So you consume approximately 6 × 1023 amino acids in 500 g of meat.
What are some components in the Phusion High-Fidelity PCR Master Mix and what is their purpose? The Phusion Master Mix is basically a ready-to-use mix that makes setting up PCR way easier since everything is already in it. The main component is the Phusion High-Fidelity DNA Polymerase, which is the enzyme that actually copies the DNA. What makes it special is that it catches and fixes mistakes as it goes, giving you really accurate amplification. It also has the four dNTPs which are the building blocks the polymerase uses to build new DNA strands. There’s also a reaction buffer that keeps the pH and salt conditions stable so the enzyme works properly.
Genetic Circuits II 1.What advantages do IANNs have over traditional genetic circuits, whose input/output behaviors are Boolean functions? Traditional genetic circuits function as digital logic gates, processing inputs in a strictly binary manner — a signal is either present or absent, ON or OFF. While this approach is sufficient for simple regulatory tasks, it is inherently limited in its capacity to handle the complexity characteristic of many biological environments. Intracellular Analog Neural Networks (IANNs) offer several notable advantages over this paradigm. For example, IANNs operate on continuous, graded signals which is typically the concentration of transcription factors, proteins, or regulatory RNAs, it could rather than discrete binary states. This allows them to perform sophisticated pattern recognition and non-linear classification that Boolean logic gates are fundamentally incapable of. Also, IANNs can assign differential weights to distinct inputs, enabling the circuit to be more responsive to certain signals than others, which more accurately reflects the nuanced regulatory logic observed in natural biological systems.
Advantages of CFPS The main advantage is that CFPS is an open system. Unlike in vivo methods, there is no cell membrane, allowing direct access to the reaction.
Flexibility: You can adjust $Mg^{2+}$ levels, add chaperones, or use non-natural amino acids easily. Toxic Proteins: You can produce proteins that would normally kill a living host cell. Speed: It enables “benchtop” production in hours rather than days of cell culture. 2. Main Components Cell Extract (Lysate): The “machinery” (ribosomes, tRNAs, enzymes). DNA Template: The “instructions” for the protein. Energy System: ATP/GTP and a regeneration substrate (e.g., PEP). Amino Acids: The “building blocks.” Salts/Cofactors: Specifically $Mg^{2+}$ and $K^{+}$ for ribosome function. 3. Energy Provision Why it’s critical: Protein synthesis is energy-expensive. Without a regeneration system, ATP is depleted in minutes by side reactions, stopping production.
HTGAA 2026 — Week 11 Homework Cell-Free Protein Synthesis & Collaborative BioArt What I Liked About the Project The experiment beautifully demonstrated that biological creativity and collective computation can be bridged through a structured, yet open-ended collaborative format. What stood out most was the emergent complexity: each individual’s single-pixel decision was locally simple, but globally the artwork encoded recognizable biological imagery. This mirrors how distributed cellular systems encode complex phenotypes from individual gene expression events. The fact that the “canvas” was limited to 1,536 pixels (a deliberate constraint) also made each contribution weighty and meaningful — a great lesson in resource allocation at biological scale.
DNA Design Challenge
Protein: GFP (Green Fluorescent Protein) Reason: Because GFP is commonly used as a biological marker to visualize various cellular processes due to its green fluorescence. sp|P42212|GFP_AEQVI Green fluorescent protein OS=Aequorea victoria OX=6100 GN=GFP PE=1 SV=1 MSKGEELFTGVVPILVELDGDVNGHKFSVSGEGEGDATYGKLTLKFICTTGKLPVPWPTL VTTFSYGVQCFSRYPDHMKQHDFFKSAMPEGYVQERTIFFKDDGNYKTRAEVKFEGDTLV NRIELKGIDFKEDGNILGHKLEYNYNSHNVYIMADKQKNGIKVNFKIRHNIEDGSVQLAD HYQQNTPIGDGPVLLPDNHYLSTQSALSKDPNEKRDHMVLLEFVTAAGITHGMDELYK
Reverse Translate: Protein (amino acid) sequence to DNA (nucleotide) sequence GFP DNA >ATGTCCAAGGGTGAGGAGCTGTTTACCGGCGTGGTTCCGATTCTTGTGGAATTAGACGGCGATGTCAACGGCCACTTCTCCGTTTCT GGCGAGGGCGAGGGAGGCGACGCCACGTATGGCAAATTGACCCTGAAGTTTATTTGCACGACCGGAAAATTGCCTGTACCGTGGCCCACACTTTGGT CACTACCGTTATCAATGTTTCTCGCTATCCGGACCACATGAAGCAGCATGACTTCTTTAAAAGTGCAATGCCCGAGGGTTATGTTCAAGAGCGGACCA TCTTTTTTAAAAGACGACGGCAACTACAAGACGCGCGAGGTGAAGTTCGAGGGCGACACGCTGGTGAATCGGATTGAGTTAAAAGGAATTGACTTTAA AAGATGACGGCAACATCCTTGGACATAAGTTAGAGTACAATTATAATTCAAACCACGTGTACATCATGGCCGACAAACAAAAAAACGGCATCAAGGTA AACTTTAAAATTAGACATAATATCGAGGATGGCAGTGTTCAATTAGCCGACCATTACCAACAGAACACCGATAGGCGGACGGTCCTGTATTACCTGAC AACCATTACCTTAGCACGCAGTCTGCACTGTCCAAGGACCCAAATGAGAAACGAGGACCATATGGTGTTGCTAGAGTTCGTTACCGCAGCAGGAATAAC
Codon optimization The selected organism: Escherichia coli (E. coli) Reason: This is because E. coli is a common model organism for the production of recombinant proteins due to its speed, low cost and ease of manipulation.
HTGAA 2026 — Week 5 Homework PROTEIN DESIGN PART II Part 1: Generating Peptides with PepMLM SOD1 A4V Sequence (Human):
MATVAVCVLKGDGPVQGIINFEQKESNGPVKVWGSIKGLTEGLHGFHVHEFGDNTAGCTSAGPHFNPLSRKHGGPKDEERHVGDLGNVTADKDGVADVSIEDSVISLSGDHCIIGRTLVVHEKADDLGKGGNEESTKTGNAGSRLACGVIGIAQ Generated Peptides: We obtained approximately 4 peptides plus one control peptide, each showing a perplexity score. The sequence modification made was Alanine to Valine substitution at position 4.
Subsections of Homework
Week 2 Lecture Prep
PRE-LECTURE ABOUT CLASS 2
SLIDES ABOUT PROFESSOR JACOBSON
-Question 1
When biological polymerase copies DNA, it makes about 1 mistake per million base pairs (1:10^6).Since the human genome has around 3.2 billion base pairs, that error rate would mean every time one of my cells divides, it would introduce over 3,000 mistakes if there weren’t any correction mechanisms. There’s a 3’-5’ exonuclease that catches and removes errors during DNA synthesis, and then the MutS repair system acts as a backup to fix any mismatches that slipped through afterward. Together, these mechanisms keep my genetic information stable across cell divisions.
-Question 2
The average human protein is about 345 amino acids long (roughly 1036 base pairs of DNA). Because the genetic code is redundant, there are approximately 3^345 different DNA sequences that could produce the same protein—an astronomically large number. However, most of these sequences don’t actually work well in practice. Some fold into secondary structures like hairpins that block the copying machinery.
SLIDES ABOUT DR.LEPROUST-Question 1
The method that’s most commonly used today is called the phosphoramidite method, or phosphoramidite chemistry. From what I learned, this technique was developed back in 1981 by a researcher named Caruthers, and it’s still the gold standard in the industry. It’s used for both the traditional column-based synthesis approach and the newer silicon-based platforms that companies are working with now.
-Question 2
Even though each individual base might attach with really high efficiency, small errors happen at every single step of the process. By the time you reach 200 nucleotides, the percentage of molecules that are perfect and full-length has fallen to around 37% or even lower.
-Question 3
When I think about the cumulative error rate logic, the probability of building a perfect 2000 base pair molecule one base at a time is essentially zero. That’s why in practice, when scientists need to manufacture long genes, they don’t try to synthesize them directly. Instead, they make shorter verified fragments (oligos) and then assemble those pieces together to build the full-length gene.
SLIDES ABOUT DR.CHURCH
There are 10 amino acids that humans can’t make on their own and have to get from food ->
Phenylalanine (F), Valine (V), Threonine (T), Tryptophan (W), Isoleucine (I), Methionine (M), Histidine (H), Arginine (R), Leucine (L), and Lysine (K).
The “Lysine Contingency” is a weak biocontainment strategy because lysine is already essential to all animals.
Week 1 HW: Principles and Practices
1. Biological engineering application:
The development of various therapies in which they employ stem cells in Peru, to treat neurodegenerative diseases and chronic diseases. Along with this the appropriate regulations for this type of therapy.
-> Why I chose this application:
Because in Peru there is an increase in the development of various clinical therapies for diseases that have been very expensive to access, so the emergence of stem cell applications are recent.
This is also due to a problem, this is more than anything focused on the little regulation on this type of treatment, which limits the creation of more trained centers, generating a delay in the access of new therapies and research.
2. Ethical future:
Governance and policy objectives in PeruOverall objective:
To establish a comprehensive framework that ensures stem cell therapies in Peru are safe, effective, and equitably accessible across all health sectors—both private and public—while fostering sustainable development of regenerative medicine in the country.
In addition, a specific and clear regulation must also be provided, which must be supervised by one or more public entities. Also to have control over the misleading commercial use of stem cells without clinical evidence, in order to provide health security for patients in the face of medical fraudulence. Finally, the approach of accessibility and equity, since what is expected to be achieved through the creation of centers with stem cell therapies is to facilitate access to these therapies in public and private hospitals, avoiding that they are only for sectors of high purchasing power those who have access to these therapies, this would help to reduce a gap in the health system that every year continues to be larger.
3. Governance Actions
Governance actions
Purpose
Design
Assumptions
Risks of Failure & “Success”
Option 1: Creation and implementation of a specific and explicit regulation focused on stem cell therapies.
Regulate the development and application of therapies, through various regulations and standards in which clinical evidence is provided prior to their use in humans, avoiding exposing patients to treatments that are not viable.
The Ministry of Health should approve regulations and require researchers to conduct clinical trials for new therapies.
It should be assumed that the Ministry of Health has sufficient budget to create a supervisory and oversight body. There may be resistance from clinics or private entities offering unregulated therapies.
If it “fails”, treatments without evidence will continue to exist, harming the lives of patients. If it “succeeds”, it could increase the cost and delay promising therapies, especially in terms of reducing the health care gap.
Option 2: Sanctions for clinics that offer treatments without scientific support or violate patient confidentiality.
Preventing medical and scientific fraud, seeking to ensure that patients receive safe treatments, without affecting their integrity.
Creating a supervisory unit within the Ministry of Health focused primarily on inspecting clinics and hospitals that provide such treatment. Instead of failing to comply with what is necessary, fines should be established and establishments closed.
It is assumed that patients are aware of the regulations that will be established and can report any type of infraction.
If it “fails”, medical fraud will continue, affecting many more patients, generating large-scale economic loss. If it is “successful”, some clinics could operate clandestinely, but it would be quicker to recognize whether they are fraud or not.
Option 3: Development of a program for equitable access therapies in public hospitals.
Ensure that most patients with chronic diseases have access to advanced therapies without high costs, especially in the public health system.
The Ministry of Health should finance local research in each region of Peru, along with allowing national hospitals to offer therapies regulated by the regulations that are sought to be established. Obtain joint cooperation, universities, research centers and hospitals to promote research in this sector.
It is assumed that there is sufficient infrastructure and personnel who can be trained to carry out these therapies safely. In any case, an investment in the private sector in conjunction with research centers at the national level would be assumed.
If it “fails”, only the wealthy will be able to access these treatments. If it “succeeds”, demand may exceed the capacity of the health system, but it would be sought to channel it to make it accessible to the majority of citizens.
4. Evaluation of Governance Actions
(Score from 1-3, with 1 as the best and 3 the least effective)
Governance actions (options)
Ethics
Accessibility
Equitative
Biosecurity
Promote investigation
Reduce breach in health system
Creation and implementation of a specific and explicit regulation
1
1
2
1
1
1
Sanctions for clinics that offer treatments without scientific support
1
1
2
1
2
3
Development of a program for equitable access therapies
2
1
1
1
2
1
5. DRAW AND THE BEST OPTION
The best option→ Creation and implementation of a specific and explicit regulation focused on stem cell therapies.
Justification: It is essential to establish clear and precise regulations, in order to avoid risks for patients
Second option→ Development of a program for equitable access therapies in public hospitals.
Justification: Since stem cell therapies are expensive, there is a need to generate and promote greater investment and infrastructure development.
Third option → Sanctions for clinics that offer treatments without scientific support or violate patient confidentiality.
Justification: Seeking to prevent medical fraud and ensure that only viable and safe therapies are offered to patients across the country.
AUDIENCE :
This proposal is addressed to the Ministry of Health, which is responsible for implementing regulations, standards and properly supervising the correct application of cell therapies in Peru.
REFLECTION
When I think back on our class conversations about ethics and safety, one ethical issue really stuck with me: the idea of “Therapeutic Misconception” and how desperate patients’ hope can be exploited. Honestly, before doing this assignment, I always thought stem cell regulations were just red tape—like bureaucratic obstacles or technical checkboxes researchers had to deal with. But now I understand there’s something much deeper going on ethically. When people are vulnerable and desperately searching for treatments, especially when they’re running out of options, they can end up being sold therapies that haven’t actually been proven to work.
PRE-LECTURE ABOUT CLASS 2
SLIDES ABOUT PROFESSOR JACOBSON
-Question 1
When biological polymerase copies DNA, it makes about 1 mistake per million base pairs (1:10^6).Since the human genome has around 3.2 billion base pairs, that error rate would mean every time one of my cells divides, it would introduce over 3,000 mistakes if there weren’t any correction mechanisms. There’s a 3’-5’ exonuclease that catches and removes errors during DNA synthesis, and then the MutS repair system acts as a backup to fix any mismatches that slipped through afterward. Together, these mechanisms keep my genetic information stable across cell divisions.
-Question 2
The average human protein is about 345 amino acids long (roughly 1036 base pairs of DNA). Because the genetic code is redundant, there are approximately 3^345 different DNA sequences that could produce the same protein—an astronomically large number. However, most of these sequences don’t actually work well in practice. Some fold into secondary structures like hairpins that block the copying machinery.
SLIDES ABOUT DR.LEPROUST-Question 1
The method that’s most commonly used today is called the phosphoramidite method, or phosphoramidite chemistry. From what I learned, this technique was developed back in 1981 by a researcher named Caruthers, and it’s still the gold standard in the industry. It’s used for both the traditional column-based synthesis approach and the newer silicon-based platforms that companies are working with now.
-Question 2
Even though each individual base might attach with really high efficiency, small errors happen at every single step of the process. By the time you reach 200 nucleotides, the percentage of molecules that are perfect and full-length has fallen to around 37% or even lower.
-Question 3
When I think about the cumulative error rate logic, the probability of building a perfect 2000 base pair molecule one base at a time is essentially zero. That’s why in practice, when scientists need to manufacture long genes, they don’t try to synthesize them directly. Instead, they make shorter verified fragments (oligos) and then assemble those pieces together to build the full-length gene.
SLIDES ABOUT DR.CHURCH
There are 10 amino acids that humans can’t make on their own and have to get from food ->
Phenylalanine (F), Valine (V), Threonine (T), Tryptophan (W), Isoleucine (I), Methionine (M), Histidine (H), Arginine (R), Leucine (L), and Lysine (K).
The “Lysine Contingency” is a weak biocontainment strategy because lysine is already essential to all animals.
Week 3 HW: OPENTRONS
Homework 3:
Python Script for Opentrons Artwork
Overview
The task is to create a Python script that runs on an Opentrons OT-2 liquid handling robot and draws an artistic design on a 96-well plate using dye transfer. The design chosen for this project is a mercury droplet, representing the core element of the MeR (Mercury Bioremediation) project. Adapted to reflect a droplet / letter “M” for Mercurio.
Labware
Slot
Labware
1
USA Scientific 96-well deep plate (2.4 mL)
2
Opentrons 96-tip rack (300 µL)
3
NEST 12-well reservoir (15 mL)
Python Script — MeR_art.py
fromopentronsimportprotocol_apimetadata={'protocolName':'MeR Art — Mercury Bioremediation','author':'Violeta Vilcapoma Torres','description':'Mercury droplet drawing via dye transfer on a 96-well plate','apiLevel':'2.13'}defrun(protocol:protocol_api.ProtocolContext):# --- Load labware ---plate=protocol.load_labware('usascientific_96_wellplate_2.4ml_deep','1')tiprack=protocol.load_labware('opentrons_96_tiprack_300ul','2')reservoir=protocol.load_labware('nest_12_reservoir_15ml','3')# --- Load pipette ---p300=protocol.load_instrument('p300_single_gen2','left',tip_racks=[tiprack])# --- Well coordinates: mercury droplet / "M" shape ---# Generated via opentrons-art.rcdonovan.com GUImercury_drop=['D4','E4','F5','G6','F7','E8','D8','C7','B6','C5']silver_dye=reservoir.wells_by_name()['A1']# Silver/gray dye in reservoir well A1# --- Drawing routine ---p300.pick_up_tip()forwellinmercury_drop:p300.distribute(20,# 20 µL per wellsilver_dye,plate.wells_by_name()[well],new_tip='never'# single tip for the full drawing pass)p300.drop_tip()
POST LAB QUESTIONS
Paper: Accelerated high-throughput imaging and phenotyping system for small organisms
This paper details the creation of a high-throughput experimentation (HTE) platform built around duckweeds — specifically Lemna minor, a tiny aquatic plant with applications in bioremediation and biofuel research. To run large-scale evolutionary ecology experiments, the team combined an Opentrons OT-2 liquid handling robot with a custom autonomous imaging system, creating a pipeline capable of operating at a scale that would be practically impossible by hand.
The central engineering challenge was that standard liquid handling robots are designed to work with, unsurprisingly, liquids. Duckweeds are solid floating plant fronds, which meant the OT-2 needed to be rethought for a very different kind of material. The researchers solved this by replacing the standard pipette tips on the OT-2’s P300 pipette heads with commercial inoculation loops. These loops exploit capillary action to gently lift individual fronds from the water’s surface, allowing the robot to pick and place solid biological matter with the same reliability it would otherwise bring to liquid transfers.
This seemingly simple hardware modification had enormous practical consequences. By enabling automated handling of the plants, the team was able to design an experiment encompassing 6,000 individual microcosms spread across 2,000 distinct combinations of nutrients and microbes — a scale of experimental complexity that manual pipetting and plant placement could never realistically achieve, given how tedious and error-prone working with tiny floating organisms at high volume would be for human researchers.
Question 2 — Describe what you intend to automate for your final project
Project: High-throughput screening of stabilized MerA enzyme variants for mercury bioremediation.
For my final project, I would like to automate the screening of different computationally designed MerA enzyme variants to identify which one shows the best stability and activity under mercury stress conditions.
The main idea is to test several MerA variants in parallel, instead of doing each assay manually. This would help reduce pipetting errors, save time, and allow the comparison of many conditions within a single working day.
Planned Automation Workflow
Step 1 — Sample preparation
First, an automated liquid-handling robot, such as the Opentrons OT-2, would prepare the samples. The robot would mix different concentrations of HgCl₂ with cell lysates containing each MerA variant. This step is important because it allows the same mercury stress conditions to be applied consistently across all samples.
Step 2 — Thermal stability screening
Then, the samples would be exposed to controlled temperature conditions using a temperature module, such as an Inheco module. This would allow me to evaluate how stable each MerA variant remains after thermal stress. During this stage, the robot could also add the required cofactors, such as NADPH and FAD, at defined time points.
Step 3 — Activity readout
After incubation, the samples would be transferred to a 384-well plate and analyzed using a plate reader, such as the PHERAstar. The readout would be based on absorbance changes related to the reduction of Hg²⁺ to Hg⁰, which would indicate the activity level of each MerA variant.
Q1: How many molecules of amino acids do you take with a piece of 500 grams of meat? (on average an amino acid is ~100 Daltons)
First, we need to calculate the number of moles and multiply by Avogadro’s number (NA=6.022×1023 mol−1).
An amino acid has an average mass of ~100 Daltons (Da), which is roughly equivalent to 100 g/mol.
Meat is mostly protein (~20% of its weight is protein).
→ 500 g of meat contains approximately 100 g of protein.
Since 1 mole of amino acids weighs ~100 g, there are ~1 mole of amino acids in 100 g of protein.
→ 1 mole is equivalent to 6.022 × 1023 molecules (Avogadro’s number).
So you consume approximately 6 × 1023 amino acids in 500 g of meat.
Q2: Why do humans eat beef but do not become a cow, eat fish but do not become fish?
When you consume animal proteins — whether from beef, fish, or any other source — your body
does not simply absorb and repurpose those proteins wholesale. Instead, the digestive system
dismantles them through a carefully orchestrated process involving enzymes such as pepsin and
various proteases, which hydrolyze the peptide bonds holding amino acids together, reducing
complex foreign proteins into their simplest building blocks: individual amino acids and small peptides.
These generic molecular units are then absorbed through the intestinal lining into the bloodstream,
where they are transported to cells throughout the body. Once inside the cell, the process shifts from
digestion to construction, guided by the Central Dogma of molecular biology — the principle that genetic
information flows from DNA to RNA to protein. Using the instructions encoded in human DNA, the cell’s
ribosomes read messenger RNA transcripts and reassemble those same basic amino acids into entirely new,
distinctly human proteins. This elegant process explains why eating cow or fish protein does not make you
more “cow-like” or “fish-like” — because by the time those amino acids are rebuilt into proteins,
they are following your body’s own genetic blueprint, not the animal’s.
Q3: Why are there only 20 natural amino acids?
The 20 protein amino acids were chosen during evolution for their chemical stability,
functionality and compatibility with the ribosomal machinery. Furthermore,
their structure allows them to perform a wide variety of enzymatic and structural
functions without being excessively complex.
Q5: Where did amino acids come from before enzymes that make them, and before life started?
Long before life existed on Earth and biological enzymes evolved to synthesize proteins,
amino acids were being formed through entirely abiotic — or non-living — chemical processes.
The landmark Miller-Urey experiment provided the first compelling laboratory evidence for this,
demonstrating that when the conditions of early Earth are simulated — a reducing atmosphere composed
of methane, ammonia, hydrogen, and water vapor — and electrical discharges are introduced to mimic
lightning strikes, amino acids spontaneously emerge from inorganic chemistry alone.
This groundbreaking result suggested that the fundamental building blocks of life were not the
product of biology, but rather an inevitable outcome of basic chemistry under the right environmental
conditions. Further supporting this idea, astrobiological research has revealed that amino acids are
not exclusive to Earth — they have been discovered on carbonaceous chondrite meteorites, most notably
the Murchison meteorite, which was found to contain a diverse array of extraterrestrial amino acids.
Q6: If you make an α-helix using D-amino acids, what handedness (right or left) would you expect?
If a protein were constructed entirely from D-amino acids rather than the L-amino acids found in nature,
you would expect it to fold into a left-handed α-helix. In natural proteins, L-amino acids preferentially
adopt right-handed α-helical conformations because this geometry minimizes steric clashes between the side
chains and the peptide backbone, making it the energetically favorable configuration. D-amino acids, however,
are the exact mirror images — or enantiomers — of their L counterparts, meaning their stereochemical constraints
are precisely reversed. As a result, a polymer built entirely from D-amino acids would experience the same
stabilizing forces and steric considerations as a natural protein, but reflected in the opposite direction,
driving the chain to fold into a left-handed helix instead. This mirror-image relationship illustrates just
how profoundly the chirality of individual amino acids influences the three-dimensional architecture of the
proteins they compose, and highlights why the near-universal selection of L-amino acids in biological systems
is so fundamental to the structural consistency of life as we know it.
Q7 Can you discover additional helices in proteins?
Beyond the well-known α-helix, proteins and engineered peptides can adopt a surprising variety of helical
conformations, and scientists have developed several creative strategies to discover and design them.
While naturally occurring proteins predominantly utilize the standard α-helix, the 3-helix, and the rarer π-helix,
the conformational space available to polypeptide chains is far broader than what evolution has sampled.
One powerful approach involves the use of unnatural amino acids — such as β- or γ-amino acids, which
carry extra carbon atoms in their backbone — to build synthetic peptides called foldamers that fold
into entirely novel helical geometries, such as the 14-helix or 12-helix, structures with no natural
counterpart. Another strategy exploits stereochemistry by alternating D- and L-amino acids within a
single chain, producing unusual structures like the hollow tubular helix found in the antibiotic Gramicidin A,
which behaves fundamentally differently from the solid cylinder of a conventional α-helix.
Finally, computational de novo design tools such as Rosetta allow structural biologists to
mathematically explore novel hydrogen-bonding networks and backbone torsion angles, effectively
engineering stable helical architectures that nature never arrived at through evolution.
Together, these approaches reveal that the helices found in natural proteins represent only
a small fraction of what is geometrically and chemically possible.
Q8 Why are most molecular helices right-handed?
Most molecular α-helices are right-handed because biological systems exclusively use L-amino acids.
In a right-handed helix made of L-amino acids, the side chains (R-groups) point outward and downward,
which minimizes steric hindrance (spatial crowding) with the carbonyl oxygen atoms of the polypeptide
backbone. If L-amino acids were forced into a left-handed α-helix, the side chains would structurally
clash with the backbone, creating a high-energy, thermodynamically unstable state.
Q9 Why do β-sheets tend to aggregate? What is the driving force?
The tendency of β-sheets to aggregate is driven by two primary forces that act in concert to pull
individual sheets together into larger, often insoluble structures. The first is the presence of
unsatisfied hydrogen bonds along the exposed edges of each β-sheet, where unpaired amide donors
and carbonyl acceptors remain highly reactive and eagerly seek out complementary partners on neighboring
sheets, effectively stitching them together through intermolecular hydrogen bonding. The second driving
force is the hydrophobic effect, which arises from the amphipathic nature of many β-sheets — one face is
hydrophilic while the opposing face is hydrophobic. In an aqueous environment, the hydrophobic faces are
thermodynamically compelled to shield themselves from surrounding water molecules, driving them to pack
tightly against one another and stack into larger fibrillar assemblies. Together, these two forces create
a powerful thermodynamic pull toward aggregation, which has significant biological consequences — this very
mechanism underlies the formation of amyloid fibrils, the insoluble protein aggregates associated with devastating
neurodegenerative diseases such as Alzheimer’s and Parkinson’s, where normally soluble proteins misfold and their
exposed β-sheet edges and hydrophobic faces drive runaway aggregation in the cell.
Q10 Why do many amyloid diseases form β-sheets? Can you use amyloid β-sheets as materials?
The prevalence of β-sheet misfolding in diseases like Alzheimer’s is rooted in the extraordinary
thermodynamic stability of a structure known as the cross-β spine, in which tightly interdigitated
β-strands form a rigid, highly ordered fibril that represents a deep low-energy state. Once a protein
misfolds into this conformation, it does not simply remain an isolated aberration — it acts as a template,
recruiting and converting neighboring normal proteins into the same misfolded state in a self-propagating
chain reaction that progressively builds insoluble amyloid plaques. This prion-like mechanism makes amyloid
formation particularly insidious, as the thermodynamic favorability of the fibril structure means the process
is essentially irreversible under physiological conditions. However, the same properties that make amyloid
fibrils so destructive in disease also make them remarkably attractive as engineering materials.
Their mechanical strength is exceptional, rivaling that of spider silk and steel at the nanoscale,
and their resistance to biological degradation gives them a durability that few natural materials
can match. Recognizing this potential, engineers and materials scientists have begun harnessing modified,
non-toxic amyloid sequences to design a wide range of advanced materials, including nanomaterials,
hydrogels, biosensors, and scaffolds for tissue engineering — effectively repurposing one of biology’s
most feared structural motifs into a platform for cutting-edge biotechnology.
Q11. Design a β-sheet motif that forms a well-ordered structure.
I would design a short β-hairpin motif, because it is one of the simplest ways to form a well-ordered β-sheet structure.
Proposed β-sheet motif
TVTVTVT-GNG-KVKVKVK
PART B
For this part, I selected MerA (mercuric reductase) from Pseudomonas aeruginosa.
MerA was chosen because it is the central enzyme of my final project. It catalyzes the reduction of toxic ionic mercury (Hg²⁺) into inert elemental mercury (Hg⁰), making it the key biological tool for detoxifying mercury-contaminated environments.
2. Amino Acid Sequence
The sequence corresponds to MerA / Mercuric reductase retrieved from UniProt.
Alanine which is a small, nonpolar amino acid known for its role in structural stability. Its high representation in MerA likely contributes to the protein’s overall fold stability — which is particularly relevant to this project, as one goal is to engineer MerA variants with enhanced thermostability.
4. Protein Sequence Homologs
A BLAST search via UniProt reveals numerous homologous sequences in other bacterial species, particularly in organisms carrying mercury resistance systems.
This is expected: MerA is a core component of the mer operon, a conserved genetic module that grants microorganisms the ability to survive in mercury-contaminated environments. Its widespread occurrence across bacteria reflects strong evolutionary pressure to maintain mercury detoxification capacity.
5. Protein Family
MerA belongs to the mercuric reductase family.
It is also classified within the broader superfamily of FAD/NADPH-dependent oxidoreductases, as it requires both FAD and NADPH as cofactors to perform the two-electron reduction of Hg²⁺ to Hg⁰.
6. RCSB Structure Page
Field
Value
PDB ID
1ZK7
Protein
Mercuric reductase MerA
Organism
Pseudomonas aeruginosa
Method
X-ray diffraction
Resolution
1.60 Å
The structure was solved by X-ray crystallography at 1.60 Å resolution. Since lower resolution values indicate higher structural detail, this represents a high-quality structure — well above the 2.70 Å reference threshold given in the assignment.
7. Other Molecules in the Solved Structure
Yes, the structure contains molecules beyond the protein chain itself.
Molecule
Role
FAD
Redox cofactor required for Hg²⁺ → Hg⁰ reduction
Sulfate ions
Small molecules present in the crystal lattice
The presence of FAD in the crystal structure is significant: it confirms that the solved conformation represents the catalytically relevant, cofactor-bound state of the enzyme — not just the apo form.
8. Structure Classification
In RCSB, MerA is classified as an oxidoreductase.
This classification directly reflects its biological function: MerA performs a redox reaction, accepting electrons from NADPH via FAD to reduce mercury from its toxic ionic form (Hg²⁺) to inert elemental mercury (Hg⁰).
Week 6 HW: GENETIC CIRCUITS PART I: ASSEMBLY TECHNOLOGIES
1. What are some components in the Phusion High-Fidelity PCR Master Mix and what is their purpose?
The Phusion Master Mix is basically a ready-to-use mix that makes setting up PCR way easier since everything is already in it. The main component is the Phusion High-Fidelity DNA Polymerase, which is the enzyme that actually copies the DNA. What makes it special is that it catches and fixes mistakes as it goes, giving you really accurate amplification. It also has the four dNTPs which are the building blocks the polymerase uses to build new DNA strands. There’s also a reaction buffer that keeps the pH and salt conditions stable so the enzyme works properly.
2.What are some factors that determine primer annealing temperature during PCR?
The annealing temperature matters a lot because if it’s too low, your primers bind nonspecifically and you get messy results, and if it’s too high, they won’t bind at all. The most important factor is the melting temperature (Tm) of your primers — the annealing temperature is usually set about 3–5°C below the lower Tm of the two primers. Primer length plays into this too since longer primers have higher Tm values. GC content is another big one — G-C pairs have three hydrogen bonds instead of two, so GC-rich primers are more stable and need higher temperatures to melt.
3.There are two methods from this class that create linear fragments of DNA: PCR, and restriction enzyme digests. Compare and contrast these two methods, both in terms of protocol as well as when one may be preferable to use over the other.
Both methods can give you linear DNA fragments, but they work pretty differently. PCR uses primers, a polymerase, and thermal cycling to amplify a specific region. The big advantage is flexibility — you can design primers to add any sequence you want to the ends of your fragment, like overlaps for Gibson assembly. The downside is there’s some risk of mutations, though high-fidelity polymerases like Phusion make this pretty minimal. Restriction digestion, on the other hand, cuts DNA at specific recognition sequences using enzymes, and it’s done at a constant temperature.
4.How can you ensure that the DNA sequences that you have digested and PCR-ed will be appropriate for Gibson cloning?
Gibson assembly works by joining fragments that share overlapping sequences at their ends (usually around 20–40 bp), so you have to make sure those overlaps are designed correctly. For PCR fragments, you add the overlap sequences directly into the 5’ tails of your primers — so the forward primer of one fragment carries the end of the previous fragment, and so on. For restriction-digested fragments, you need to check that the ends left by the enzyme line up with the overlap region of the adjacent fragment. The best way to check everything is to model the assembly in software like Benchling before you even start — you can simulate how all the fragments will come together and catch any design errors early. Basically, if the end of fragment A perfectly matches the beginning of fragment B, you’re good.
5.How does the plasmid DNA enter the E. coli cells during transformation?
E. coli cells don’t naturally take up DNA, so you have to make them “competent” first. With chemically competent cells, they’re treated with CaCl₂, which neutralizes the negative charges on both the DNA and the cell membrane. Then you do a heat shock which temporarily disrupts the membrane and lets the plasmid get in. With electrocompetent cells, it’s a bit different: a brief electroporation creates tiny pores in the membrane that the DNA can pass through. Both methods are essentially creating a temporary opening in the membrane for the DNA to enter.
6.Describe another assembly method in detail (such as Golden Gate Assembly)
Explain the other method in 5 - 7 sentences plus diagrams (either handmade or online).
Golden Gate Assembly is a really elegant method that lets you assemble multiple DNA fragments in a single reaction. It uses Type IIS restriction enzymes — like BsaI — which are special because they cut outside of their recognition sequence. So you can engineer your fragments so that after the enzyme cuts, it leaves behind a specific 4-bp overhang that you designed, and the recognition site itself gets removed. When you run the digestion and ligation at the same time in one tube, the correctly assembled fragments stick together through their matching overhangs and get ligated. Any fragments that aren’t assembled correctly still have the recognition site, so they just get cut again.
Week 07 HW: Genetic Circuits II
Genetic Circuits II
1.What advantages do IANNs have over traditional genetic circuits, whose input/output behaviors are Boolean functions?
Traditional genetic circuits function as digital logic gates, processing inputs in a strictly binary manner — a signal is either present or absent, ON or OFF. While this approach is sufficient for simple regulatory tasks, it is inherently limited in its capacity to handle the complexity characteristic of many biological environments. Intracellular Analog Neural Networks (IANNs) offer several notable advantages over this paradigm.
For example, IANNs operate on continuous, graded signals which is typically the concentration of transcription factors, proteins, or regulatory RNAs, it could rather than discrete binary states. This allows them to perform sophisticated pattern recognition and non-linear classification that Boolean logic gates are fundamentally incapable of. Also, IANNs can assign differential weights to distinct inputs, enabling the circuit to be more responsive to certain signals than others, which more accurately reflects the nuanced regulatory logic observed in natural biological systems.
2.Describe a useful application for an IANN; include a detailed description of input/output behavior, as well as any limitations an IANN might face to achieve your goal.
It has many application for an IANN, and it is the highly specific detection of cancerous cells coupled with selective activation of a pro-drug therapy.
In this system, we have different inputs such as X₁, X₂, X₃… that it would correspond to the intracellular concentrations of several cancer-associated microRNAs (miRNAs) or protein biomarkers. Crucially, while any one of these biomarkers may be present at low levels in healthy tissue, their specific combinatorial pattern and relative concentrations constitute a signature unique to the target cancer cell type. The IANN would process these inputs through weighted connections across its layers, integrating the signals in a manner analogous to a perceptron. If the combined weighted output surpasses a defined threshold which is the indicating that the cancer signature has been recognized, the for the final output (Y) would be the expression of an enzyme such as nitroreductase, which converts a systemically administered, non-toxic pro-drug into its cytotoxic active form, selectively eliminating the cancerous cell.
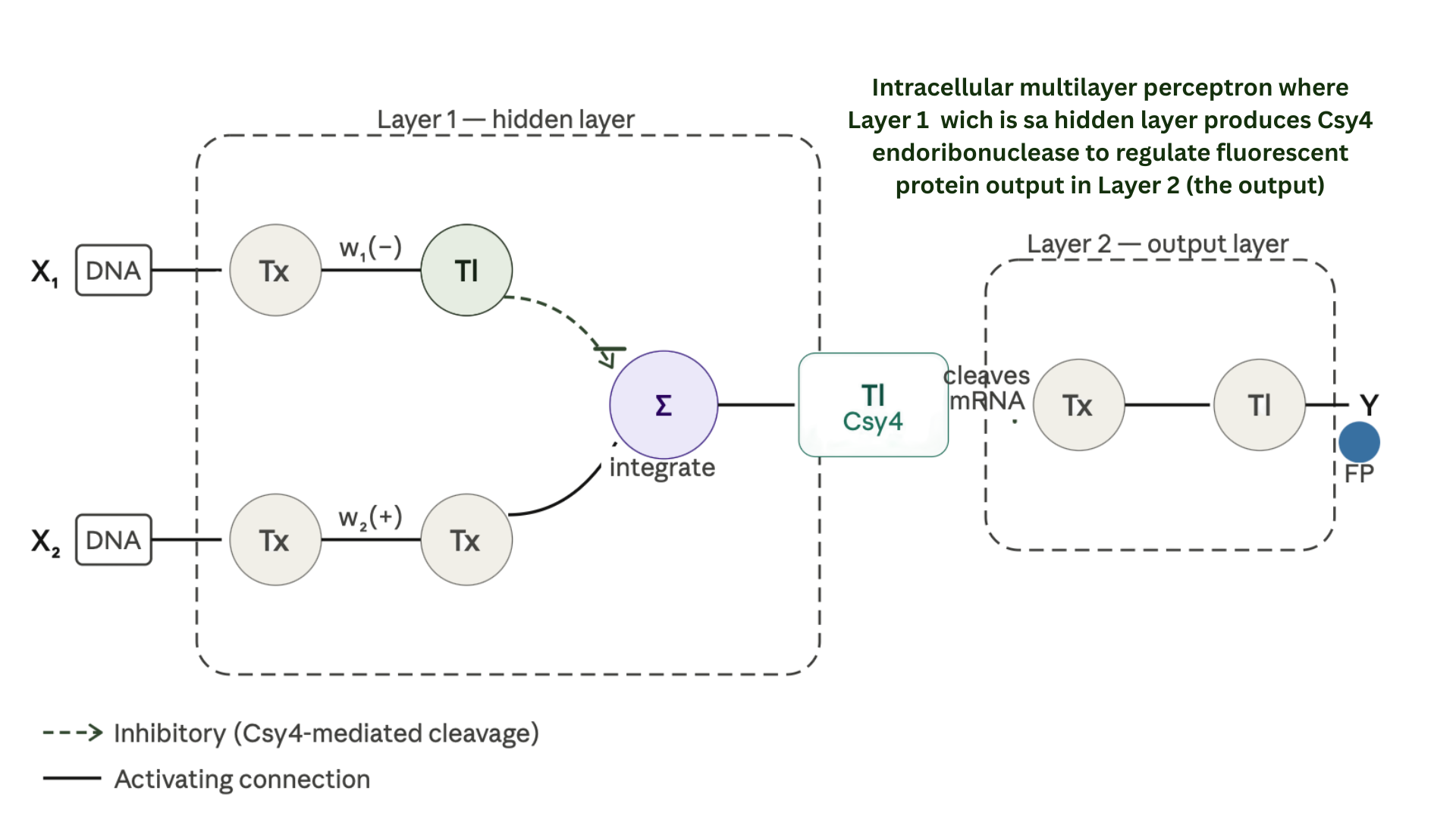
3.Below is a diagram depicting an intracellular single-layer perceptron where the X1 input is DNA encoding for the Csy4 endoribonuclease and the X2 input is DNA encoding for a fluorescent protein output whose mRNA is regulated by Csy4. Tx: transcription; Tl: translation.
SECOND PART
a. What are some examples of existing fungal materials and what are they used for? What are their advantages and disadvantages over traditional counterparts?
Existing fungal materials include mycelium-based biofilters, myco-bricks, and fungal leather. Mycelium biofilters can be used to filter pollutants from industrial runoff, acting as reactive matrices capable of absorbing metals instead of using only activated carbon. Myco-bricks are grown from agricultural waste and fungi, and can be used in construction for thermal and acoustic insulation. Fungal leather, such as Mylo or Reishi, is being developed as a sustainable alternative to animal or synthetic leather in fashion.
Their main advantages are that they are biodegradable, can be produced from organic waste, and have a lower environmental impact than conventional materials. However, they also have some limitations: they are less durable than traditional materials, can be sensitive to extreme humidity if they are not sealed, and in some cases still need further development to reach the same mechanical resistance or large-scale production capacity.
b. What might you want to genetically engineer fungi to do and why? What are the advantages of doing synthetic biology in fungi as opposed to bacteria?
This project proposes the genetic modification of fungi to secrete optimized MerA/MerB enzymes and express metallothioneins capable of sequestering mercury within fungal vacuoles. As eukaryotic organisms, fungi possess chaperones and advanced protein-folding machinery compared to bacteria, which may enhance the stability and functionality of complex proteins such as MerA, reducing the formation of inactive intracellular aggregates.
The use of fungi offers several advantages over bacteria. Their filamentous structure, composed of hyphae, allows the mycelium to anchor itself to contaminated soil, whereas bacteria can be easily washed away by water flow. This enables fungi to form a persistent physical barrier in polluted environments. Furthermore, fungi generally show greater tolerance to acidic and toxic conditions and are capable of secreting large amounts of proteins, making them promising platforms for mercury bioremediation.
Week 09 HW: Cell-Free Systems
1. Advantages of CFPS
The main advantage is that CFPS is an open system. Unlike in vivo methods, there is no cell membrane, allowing direct access to the reaction.
Flexibility: You can adjust $Mg^{2+}$ levels, add chaperones, or use non-natural amino acids easily.
Toxic Proteins: You can produce proteins that would normally kill a living host cell.
Speed: It enables “benchtop” production in hours rather than days of cell culture.
2. Main Components
Cell Extract (Lysate): The “machinery” (ribosomes, tRNAs, enzymes).
DNA Template: The “instructions” for the protein.
Energy System: ATP/GTP and a regeneration substrate (e.g., PEP).
Amino Acids: The “building blocks.”
Salts/Cofactors: Specifically $Mg^{2+}$ and $K^{+}$ for ribosome function.
3. Energy Provision
Why it’s critical: Protein synthesis is energy-expensive. Without a regeneration system, ATP is depleted in minutes by side reactions, stopping production.
Method: Use Creatine Phosphate and Creatine Kinase. This pair re-phosphorylates ADP back into ATP continuously during the reaction.
4. System Comparison: Prokaryotic vs. Eukaryotic
System
Host Example
Advantages
Best For
Prokaryotic
E. coli
High yield, fast, cost-effective.
Simple proteins (e.g., GFP).
Eukaryotic
Wheat Germ
Complex folding, post-translational mods.
Human proteins (e.g., EPO).
5. Membrane Protein Design
Challenge: These proteins are hydrophobic and aggregate in water-based lysates.
Solution: Add Nanodiscs or Liposomes to the reaction. These provide a lipid scaffold for the protein to insert into as it is being built.
6. Troubleshooting Low Yields
Template Degradation: Add RNase inhibitors to protect the mRNA/DNA.
Magnesium Levels: Perform a $Mg^{2+}$ titration; ribosomes are extremely sensitive to salt concentrations.
Codon Bias: Use a specialized lysate supplemented with rare tRNAs if the gene source and lysate source are different organisms.
Part 2: Synthetic Minimal Cell Design
An encapsulated arsenic biosensor for water quality monitoring.
Chassis->E. coli-based Tx/Tl system encapsulated in a POPC/POPG phospholipid membrane.
Communication->Alpha-hemolysin (αHL) pores allow arsenic ions to permeate the membrane.
Genetic Circuit-> An arsR repressor linked to a GFP reporter gene.
Mechanism-> Arsenic binding to ArsR triggers the expression of GFP.
Safety-> The system is non-replicative and biocontained, making it safe for environmental deployment.
Part 3: BioBits® in Space - Muscle Atrophy Diagnostic
Monitoring astronaut health via freeze-dried synthetic biology.
Challenge: Microgravity-induced muscle atrophy using the Myostatin (MSTN) gene sequence, a regulator of muscle growth.
Hypothesis: Freeze-dried BioBits® pellets can be rehydrated with astronaut samples to provide a visible fluorescent readout of MSTN levels.
Implementation Plan:
Deploy freeze-dried BioBits® containing an MSTN-responsive circuit.
Rehydrate with sample DNA/RNA.
Quantify results using the P51 Fluorescence Viewer.
Utilize non-target DNA as a negative control to ensure specificity.
BL21 (DE3) Star Lysate (T7 RNA Polymerase-expressing) Provides the endogenous transcription–translation machinery, including ribosomes, tRNAs, and metabolic enzymes. T7 RNA polymerase enables high-efficiency transcription from T7 promoters.
Salts and Buffer System
Potassium Glutamate Try to maintains intracellular-like ionic conditions and stabilizes ribosomal complexes.
HEPES-KOH (pH 7.5) These buffers work on the system to maintain optimal enzymatic activity.
Magnesium Glutamate Critical cofactor for ribosome assembly and ATP-dependent processes; directly influences translation efficiency.
Potassium Phosphate (Monobasic / Dibasic) Provides buffering and supplies phosphate for metabolic and nucleotide regeneration processes.
Energy and Nucleotide System
Ribose Precursor for nucleotide biosynthesis via salvage pathways.
Glucose Supports ATP regeneration through glycolysis.
AMP, CMP, GMP, UMP Nucleotide precursors that are phosphorylated into NTPs for transcription.
Guanine Salvageable base converted into GMP/GTP to sustain transcription.
Translation Substrate System
17 Amino Acid Mix Supplies most amino acids required for translation.
Tyrosine Supplemented separately due to solubility constraints.
Cysteine Added independently due to oxidation sensitivity; critical for folding.
Additives
Nicotinamide Enhances NAD⁺ regeneration and supports metabolic activity.
Backfill
Nuclease-Free Water Adjusts reaction volume while preserving nucleic acid integrity.
System Comparison
1-Hour PEP-NTP System
Rapid protein expression
High initial yield
Limited by substrate depletion and inhibitory byproducts
20-Hour NMP-Ribose-Glucose System
Slower expression rate
Sustained metabolic regeneration
Improved stability and long-term output
Nucleotide Salvage Mechanism
Even without exogenous GMP, transcription proceeds via salvage pathways:
Guanine → GMP → GTP This maintains the nucleotide pool required for RNA synthesis.
Fluorescent Protein Considerations
Protein
Key Property
Impact on CFPS
sfGFP
Fast folding
Early fluorescence readout
mRFP1
Slow maturation
Delayed fluorescence
mKO2
pH sensitivity
Performance depends on environment
mTurquoise2
High quantum yield
Requires efficient folding
mScarlet-I
High brightness
Improved but slower maturation
Electra2
Engineered variant
Dependent on folding and oxygen
Design Hypothesis (36-Hour Optimization)
Target Protein: mRFP1
Strategy:
Increase glucose and ribose concentrations
Optimize Mg²⁺ levels
We enhanced energy regeneration and ribosome stability will support prolonged translation and improved folding, increasing fluorescence output over extended incubation.
Applications
We can use all of these to the cell-free synthetic biology and a rapid prototyping of genetic circuits specially focus on a low-cost diagnostic platforms (for low-income countries) and for the fluorescent protein screening and optimization
Week 11 HW: Cell-Free Protein Synthesis
HTGAA 2026 — Week 11 Homework
Cell-Free Protein Synthesis & Collaborative BioArt
What I Liked About the Project
The experiment beautifully demonstrated that biological creativity and collective computation can be bridged through a structured, yet open-ended collaborative format. What stood out most was the emergent complexity: each individual’s single-pixel decision was locally simple, but globally the artwork encoded recognizable biological imagery. This mirrors how distributed cellular systems encode complex phenotypes from individual gene expression events. The fact that the “canvas” was limited to 1,536 pixels (a deliberate constraint) also made each contribution weighty and meaningful — a great lesson in resource allocation at biological scale.
What Could Be Made Better
Future iterations could benefit from a live fluorescence visualization layer — rather than a static pixel grid, students could watch their pixel “express” as a simulated fluorescence signal over time, giving a tangible connection between the bioart assignment and the cell-free expression lab that follows. Additionally, providing each participant with a brief protocol preview of how fluorescent protein expression maps to pixel intensity would reinforce the cross-lab connection and motivate deeper engagement.
Part B: Cell-Free Protein Synthesis — Cell-Free Reagents
B1. Component Roles in the Cell-Free Reaction
E. coli Lysate — BL21 (DE3) Star Lysate (includes T7 RNA Polymerase)
The lysate is the core “cellular machinery” of the reaction: a clarified extract of disrupted E. coli BL21(DE3) cells that retains the ribosomes, translation factors, aminoacyl-tRNA synthetases, chaperones, and metabolic enzymes required for coupled transcription and translation. BL21(DE3) cells are specifically used because they harbor a chromosomally integrated T7 RNA polymerase gene under an IPTG-inducible promoter; the T7 RNAP is induced prior to lysis, so the extract natively drives transcription from T7-promoter-containing DNA templates without the need for exogenous polymerase supplementation (Pardee et al., 2016; Sun et al., 2013). The “Star” designation refers to a recA-minus, RNase E mutant background that improves RNA stability and protein yield in cell-free reactions.
Reference: Sun, Z. Z. et al. (2013). Protocols for Implementing an Escherichia coli Based TX-TL Cell-Free Expression System for Synthetic Biology. JoVE, 79, e50762.
Salts / Buffer
Potassium Glutamate
Potassium glutamate serves two functions simultaneously: it provides potassium ions (K⁺) at physiologically relevant concentrations, which are critical for ribosome function and RNA polymerase activity, and it provides the glutamate counter-ion, which can act as a secondary carbon/energy source via oxidative metabolism in the cell extract. Mimicking the intracellular ionic environment of E. coli (where K⁺ is the dominant cation) is essential for maximal translation efficiency (Jewett & Swartz, 2004; Cai et al., 2015).
Reference: Jewett, M. C. & Swartz, J. R. (2004). Substrate replenishment extends protein synthesis with an in vitro translation system designed to mimic the cytoplasm. Biotechnol. Bioeng., 87(4), 465–472.
HEPES-KOH pH 7.5
HEPES (4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid) is a zwitterionic buffering agent that maintains the reaction pH near 7.5, mimicking the cytoplasmic pH of E. coli. Maintaining stable pH is critical because acidification (common when glucose or glycolytic intermediates are used as energy sources) can inhibit ribosome activity and denature enzymes. HEPES is preferred over phosphate buffers in certain formulations because it does not chelate magnesium ions, preserving Mg²⁺ availability for ribosomes (Calhoun & Swartz, 2005).
Reference: Calhoun, K. A. & Swartz, J. R. (2005). Energizing cell-free protein synthesis with glucose metabolism. Biotechnol. Bioeng., 90(5), 606–613.
Magnesium Glutamate
Magnesium is an indispensable cofactor for ribosome assembly and stability, for all nucleotide-utilizing enzymes (polymerases, kinases), and for numerous metalloenzymes involved in energy metabolism. The glutamate salt form is preferred over magnesium acetate because it mimics the ionic composition of the E. coli cytoplasm and avoids introduction of extraneous acetate ions. Mg²⁺ concentration must be carefully optimized — too low inhibits ribosomes, too high inhibits transcription — and is consistently identified as one of the most influential variables in CFPS performance (Caschera & Noireaux, 2015).
Reference: Caschera, F. & Noireaux, V. (2015). Synthesis of 2.3 mg/ml of protein with an all Escherichia coli cell-free transcription–translation system. Biochimie, 99, 162–168.
Potassium Phosphate Monobasic (KH₂PO₄) and Dibasic (K₂HPO₄)
These two phosphate salts together form a phosphate buffer pair that helps supplement pH buffering capacity alongside HEPES and also provides inorganic phosphate (Pᵢ) as a substrate for ATP regeneration through oxidative phosphorylation and substrate-level phosphorylation in the extract. Adequate Pᵢ availability is important for long-duration reactions because phosphate limitation is a known bottleneck for sustained ATP regeneration from glucose (Calhoun & Swartz, 2005).
Energy / Nucleotide System
Ribose
Ribose is the pentose sugar backbone of all ribonucleotides. In the NMP-Ribose-Glucose system, ribose can enter the pentose phosphate pathway in the extract, regenerating PRPP (5-phosphoribosyl-1-pyrophosphate) needed for nucleotide salvage synthesis, and also providing reducing equivalents (NADPH) that support energy metabolism.
Glucose
Glucose serves as the primary carbon and energy source for ATP regeneration through glycolysis and, in E. coli extract, through further oxidative metabolism via the TCA cycle. While early cell-free systems used phosphoenolpyruvate (PEP) as the energy substrate, Calhoun and Swartz demonstrated that glucose (with pH control) can sustain high-yield protein synthesis in a far more cost-effective manner, with the extract’s endogenous glycolytic enzymes converting glucose to pyruvate and then to acetate via the PANOx pathway, generating multiple ATP molecules per glucose (Calhoun & Swartz, 2005).
Reference: Calhoun, K. A. & Swartz, J. R. (2005). Energizing cell-free protein synthesis with glucose metabolism. Biotechnol. Bioeng., 90(5), 606–613.
AMP, CMP, GMP, UMP (Nucleoside Monophosphates)
These four nucleoside monophosphates are the building blocks for RNA synthesis. In the NMP-based system, the extract’s endogenous nucleoside monophosphate kinases and nucleoside diphosphate kinases phosphorylate NMPs → NDPs → NTPs, fueling both transcription (NTPs are the substrates for RNA polymerase) and providing GTP for translation (ribosome translocation, EF-Tu·GTP). Using NMPs instead of NTPs is substantially more economical and avoids the inhibitory phosphate accumulation that occurs when NTPs are directly hydrolyzed (Caschera & Noireaux, 2015).
Guanine
Although GMP is included as the primary guanosine source, free guanine (the nucleobase without ribose or phosphate) is added as a supplement for the purine salvage pathway. See the Bonus Question below for a detailed explanation.
Translation Mix — Amino Acids
17 Amino Acid Mix
This mix contains 17 of the 20 canonical amino acids that are chemically stable in standard stocks and can be dissolved together without degradation or precipitation. These are the direct building blocks for polypeptide synthesis during ribosomal translation.
Tyrosine
Tyrosine is provided separately because it has very low aqueous solubility at neutral pH and must be prepared and added as a dilute suspension or alkaline solution to avoid precipitation that would reduce its bioavailability to the ribosome.
Cysteine
Cysteine is added separately because it is highly reactive and prone to oxidation (to cystine or sulfinic acid) in the presence of air or metal ions; it must be kept in a reducing environment (or added fresh) to remain in its reduced, translatable form.
Additives
Nicotinamide (NAD⁺ precursor / NAD⁺ supplement)
Nicotinamide (vitamin B₃) is a precursor of NAD⁺, a critical cofactor for redox reactions central to energy metabolism (e.g., glycolysis, TCA cycle, oxidative phosphorylation) within the cell extract. Supplementing NAD⁺ or its precursor helps sustain the extract’s capacity to oxidize NADH back to NAD⁺, keeping energy regeneration active and extending reaction longevity (Jewett & Swartz, 2004).
Backfill — Nuclease-Free Water
Nuclease-free water is used to bring the reaction to its final target volume (the “backfill” volume). It is critical that this water is free of RNases and DNases, which would otherwise degrade the mRNA and DNA template, terminating the reaction prematurely.
B2. Main Differences Between the 1-Hour Optimized PEP-NTP Master Mix and the 20-Hour NMP-Ribose-Glucose Master Mix
The most fundamental difference is the energy and nucleotide supply strategy. The 1-hour PEP-NTP system relies on phosphoenolpyruvate as the energy source and pre-formed NTPs as direct substrates for transcription. PEP is a high-energy phosphate compound that rapidly regenerates ATP through pyruvate kinase, delivering fast but short-lived energy that is typically exhausted within 1–2 hours (Kim & Swartz, 2001). The 20-hour NMP-Ribose-Glucose system, by contrast, supplies nucleoside monophosphates (NMPs) and glucose as the primary substrates, relying on the extract’s endogenous kinase cascade (NMP → NDP → NTP) and glycolytic pathway to continuously regenerate NTPs and ATP over many hours at lower cost — extending productive reaction time but requiring a slower initial ramp-up phase (Caschera & Noireaux, 2015; Calhoun & Swartz, 2005).
A second key difference is the pH management requirement: glucose metabolism through glycolysis generates acidic by-products (lactate, acetate) that acidify the reaction over time, which is why the 20-hour formulation typically requires more robust buffering (e.g., higher HEPES or phosphate concentrations) and may include pH stabilizers such as K₂HPO₄/KH₂PO₄ in adjusted ratios. This pH drift problem is minimal in the short PEP-NTP system because the reaction ends before significant acidification occurs (Calhoun & Swartz, 2005).
Finally, the cost and complexity differ significantly: the NMP-Ribose-Glucose system is more cost-effective per reaction (NMPs and glucose are cheaper than PEP and NTPs), making it preferable for large-scale or long-duration experiments like the 36-hour global artwork incubation, whereas the PEP-NTP system is simpler to optimize quickly and delivers consistent burst expression, making it ideal for rapid prototyping within a 1-hour window.
References:
Kim, D. M. & Swartz, J. R. (2001). Regeneration of adenosine triphosphate from glycolytic intermediates for cell-free protein synthesis. Biotechnol. Bioeng., 74(4), 309–316.
Calhoun, K. A. & Swartz, J. R. (2005). Energizing cell-free protein synthesis with glucose metabolism. Biotechnol. Bioeng., 90(5), 606–613.
Caschera, F. & Noireaux, V. (2015). Synthesis of 2.3 mg/ml of protein with an all E. coli cell-free transcription–translation system. Biochimie, 99, 162–168.
B3. Bonus Question: How Can Transcription Occur If GMP Is Not Included But Guanine Is?
Transcription can occur because the E. coli extract retains active purine salvage pathway enzymes, specifically hypoxanthine-guanine phosphoribosyltransferase (HGPRT/Gpt), which catalyzes the reaction:
Guanine + PRPP → GMP + PPᵢ
In this salvage reaction, the free guanine base is combined with phosphoribosyl pyrophosphate (PRPP) — generated from ribose-5-phosphate and ATP — to yield GMP in a single, energetically efficient step (Fiveable, 2026; BOC Sciences, 2025). The resulting GMP is then phosphorylated sequentially by guanylate kinase (GMP → GDP) and nucleoside diphosphate kinase (GDP + ATP → GTP + ADP) to produce GTP, which is then available as a substrate for T7 RNA polymerase during transcription.
This is why the NMP-Ribose-Glucose formulation includes both GMP and free guanine: the guanine provides an additional flux route into the GTP pool via the salvage pathway, supplementing the kinase-driven NMP phosphorylation route and ensuring that GTP availability does not become limiting during prolonged transcription over the 20-hour reaction (Construction of a GTP Regeneration System…, ScienceDirect, 2024).
References:
Kang, S.H. et al. (2024). Construction of a GTP regeneration system by regulating gene expression in the pathway. Biochem. Eng. J., Elsevier.
Berg, J. M., Tymoczko, J. L., & Stryer, L. (2015). Biochemistry, 8th ed. W. H. Freeman. (Nucleotide metabolism chapter.)
Part C: Planning the Global Experiment — Cell-Free Master Mix Design
C1. Biophysical/Functional Properties of Each Fluorescent Protein Relevant to Cell-Free Expression
sfGFP (Superfolder Green Fluorescent Protein)
Key Property: Exceptionally robust folding / resistance to aggregation
sfGFP was engineered with six specific mutations relative to EGFP (including F99S, M153T, V163A, and others) that dramatically improve its folding kinetics and resistance to misfolding even when fused to poorly folding proteins or expressed under challenging conditions (Pédelacq et al., 2006). In a cell-free context, where the molecular chaperone network is diluted relative to an intact cell and protein concentration can accumulate rapidly, sfGFP’s superior folding robustness means a higher fraction of translated polypeptides successfully form a fluorescent chromophore rather than aggregating as non-fluorescent inclusion bodies. Chromophore formation still requires molecular oxygen (O₂) and proceeds via an autocatalytic cyclization and oxidation mechanism, making it oxygen-dependent in cell-free systems incubated under aerobic conditions.
Reference: Pédelacq, J. D. et al. (2006). Engineering and characterization of a superfolder green fluorescent protein. Nature Biotechnology, 24(1), 79–88.
mRFP1 (Monomeric Red Fluorescent Protein 1)
Key Property: Relatively slow and incomplete chromophore maturation
mRFP1 was the first true monomeric RFP derived from DsRed (Discosoma sp.) via 33 directed evolution mutations designed to break the obligate tetrameric interface while restoring fluorescence (Campbell et al., 2002). However, mRFP1’s maturation rate is significantly slower than its successor mCherry (~60 min half-time vs. ~15 min for mCherry) and it is known to mature incompletely — a significant fraction of translated mRFP1 polypeptides undergo the initial cyclization but fail to complete the second, oxidative step required to produce the fully red-shifted chromophore, yielding a “green-intermediate” species (Shaner et al., 2004). In a cell-free system, this incomplete maturation is a critical limitation: even after extensive translation, a substantial fraction of mRFP1 fluorescent signal may be delayed or lost, potentially underestimating expression relative to faster-maturing proteins.
References:
Campbell, R. E. et al. (2002). A monomeric red fluorescent protein. PNAS, 99(12), 7877–7882.
Shaner, N. C. et al. (2004). Improved monomeric red, orange and yellow fluorescent proteins derived from Discosoma sp. red fluorescent protein. Nature Biotechnology, 22(12), 1567–1572.
mKO2 (Monomeric Kusabira-Orange 2)
Key Property: Moderate acid sensitivity and a multi-step maturation mechanism
mKO2 is a monomeric orange fluorescent protein (excitation ~551 nm, emission ~565 nm) derived from the coral Verrillofungia concinna (formerly Fungia concinna) (FPbase). In vivo studies have characterized mKO2’s maturation as following a three-state model: translated → folded (non-fluorescent) → matured (fluorescent), with the second transition having a maturation half-time of approximately 135 minutes in living cells (Chabry et al., 2016 — PLoS ONE). This long-lived non-fluorescent intermediate means that in a short or even moderate-length cell-free reaction, a large pool of mKO2 may remain in the non-fluorescent folded state, suppressing observed fluorescence despite successful translation. Additionally, mKO2 has moderate acid sensitivity (FPbase), meaning that if the pH drifts below ~7 during long incubations (as can occur with glucose metabolism), fluorescence yield will decrease.
Chabry, D. et al. (2016). Sensitive and Quantitative Three-Color Protein Imaging in Fission Yeast Using Spectrally Diverse, Recoded Fluorescent Proteins with Experimentally-Characterized In Vivo Maturation Kinetics. PLoS ONE, 11(8), e0159292.
mTurquoise2
Key Property: Oxygen-dependent chromophore maturation with slow kinetics
mTurquoise2 is currently the brightest cyan fluorescent protein derived from Aequorea victoria GFP (excitation ~434 nm, emission ~474 nm). Its chromophore maturation follows a complex, multi-step oxidative mechanism — the rate-determining step is the reaction of the pre-cyclized chromophore with molecular O₂ to generate the mature fluorescent species (Goedhart et al., 2012). Studies characterizing mTurquoise2 maturation report a half-time of approximately 36–45 minutes in bacterial systems (Bindels et al., 2017), which is substantially slower than sfGFP. In an aerobic cell-free system this is manageable over a 20–36 hour incubation, but if the reaction becomes oxygen-limited (which can occur as O₂ is consumed by both chromophore maturation chemistry and metabolic enzymes in the lysate), fluorescence readout will be substantially underestimated relative to protein output.
References:
Goedhart, J. et al. (2012). Structure-guided evolution of cyan fluorescent proteins towards a quantum yield of 93%. Nature Communications, 3, 751.
Bindels, D. S. et al. (2017). mScarlet: a bright monomeric red fluorescent protein for cellular imaging. Nature Methods, 14(1), 53–56.
mScarlet-I
Key Property: Fast maturation and high brightness — favorable for cell-free expression
mScarlet-I is a monomeric red fluorescent protein (excitation ~569 nm, emission ~593 nm) optimized for intracellular brightness and rapid maturation (Bindels et al., 2017). Compared to mRFP1, mScarlet-I matures substantially faster and achieves a much higher quantum yield (QY ~0.54), making it one of the brightest monomeric red FPs currently available. A notable biophysical property relevant to cell-free systems is that mScarlet-I contains no native cysteines in its Anthozoa-derived sequence, which means it does not form adventitious disulfide bonds in the oxidative microenvironment of the reaction mix — an important advantage since disulfide-linked aggregates are non-fluorescent (Bindels et al., 2017). Its rapid maturation means fluorescence signals will emerge quickly during incubation, providing an early and reliable readout.
Reference: Bindels, D. S. et al. (2017). mScarlet: a bright monomeric red fluorescent protein for cellular imaging. Nature Methods, 14(1), 53–56.
Electra2 (Blue Fluorescent Protein)
Key Property: High intracellular brightness relative to other BFPs, but Stokes-shifted chromophore maturation sensitive to β-barrel folding fidelity
Electra2 is a recently developed blue fluorescent protein (derived from Aequorea-class GFP) optimized using a dual bacterial/mammalian expression screening system (Papadaki et al., 2022). Its chromophore requires the Y66H substitution (relative to GFP Y66) that generates the blue-shifted tyrosine-derived chromophore, which undergoes the same O₂-dependent autocatalytic maturation as standard GFPs. In cell-free systems, Electra2’s high intracellular brightness in mammalian and bacterial contexts (reported as ~2.1-fold brighter than mTagBFP2 in bacteria; Papadaki et al., 2022) suggests good translation efficiency and folding fidelity across expression systems. A key limitation for cell-free readout is that the blue chromophore has a relatively low extinction coefficient compared to green/red FPs, meaning that even with efficient maturation, fluorescence intensity per molecule is lower, requiring sensitive plate reader settings for detection.
References:
Papadaki, G. F. et al. (2022). Dual-expression system for blue fluorescent protein optimization. Nature Communications, 13, 2887.
Hashimura, H. et al. (2025). Use of blue fluorescent protein Electra2 for live-cell imaging in Dictyostelium discoideum. microPublication Biology, 10.17912/micropub.biology.001774.
C2. Hypothesis for Reagent Optimization to Maximize Fluorescence Over 36-Hour Incubation
Target protein: mKO2
Identified limitation: mKO2’s three-state maturation pathway results in accumulation of a non-fluorescent folded intermediate with a ~135-minute half-time for the folded → matured transition. This transition is the rate-limiting oxidative step that requires molecular O₂.
Hypothesis:
Increasing the dissolved oxygen availability in the reaction (by periodic brief vortexing/agitation or by supplementing with an O₂-releasing compound such as hydrogen peroxide at sub-inhibitory concentrations ~0.01–0.05 mM), combined with increasing magnesium glutamate concentration by 2–4 mM above the baseline level, will accelerate mKO2 chromophore maturation and increase total fluorescence signal at 36 hours compared to the standard unadjusted master mix.
Rationale:
Since the rate-determining step in mKO2 maturation is the O₂-dependent oxidation of the pre-chromophore (as modeled in the three-state kinetic study by Chabry et al., 2016), increasing effective O₂ availability should shift more of the folded intermediate pool toward the mature fluorescent state within the 36-hour window. Simultaneously, magnesium is the key cofactor for ribosome activity and for the NMP kinase cascade that sustains NTP supply; a modest Mg²⁺ increase (within the 10–20 mM optimal range established by Caschera & Noireaux, 2015) could sustain a higher translation rate, expanding the total translated mKO2 pool that feeds the maturation pipeline.
Expected effect: A 1.5–2× increase in total mKO2 fluorescence at the 36-hour endpoint, with fluorescence increase beginning earlier in the time course as O₂ supplementation accelerates the maturation bottleneck.
Reagent to modify (custom 2 µL supplement):
1 µL of 0.1 mM H₂O₂ (diluted fresh in nuclease-free water) → final ~0.01 mM in 20 µL reaction
1 µL of 40 mM MgGlu → adds +2 mM MgGlu to the reaction
References:
Chabry, D. et al. (2016). PLoS ONE, 11(8), e0159292.
Caschera, F. & Noireaux, V. (2015). Biochimie, 99, 162–168.
Goedhart, J. et al. (2012). Nature Communications, 3, 751. (O₂ dependence of GFP-class chromophore maturation)
Week 2 HW: DNA READ, WRITE, & EDIT
DNA Design Challenge
Protein: GFP (Green Fluorescent Protein)
Reason: Because GFP is commonly used as a biological marker to visualize various cellular processes due to its green fluorescence.
sp|P42212|GFP_AEQVI Green fluorescent protein OS=Aequorea victoria OX=6100 GN=GFP PE=1 SV=1
MSKGEELFTGVVPILVELDGDVNGHKFSVSGEGEGDATYGKLTLKFICTTGKLPVPWPTL
VTTFSYGVQCFSRYPDHMKQHDFFKSAMPEGYVQERTIFFKDDGNYKTRAEVKFEGDTLV
NRIELKGIDFKEDGNILGHKLEYNYNSHNVYIMADKQKNGIKVNFKIRHNIEDGSVQLAD
HYQQNTPIGDGPVLLPDNHYLSTQSALSKDPNEKRDHMVLLEFVTAAGITHGMDELYK
Reverse Translate: Protein (amino acid) sequence to DNA (nucleotide) sequence
GFP DNA >ATGTCCAAGGGTGAGGAGCTGTTTACCGGCGTGGTTCCGATTCTTGTGGAATTAGACGGCGATGTCAACGGCCACTTCTCCGTTTCT
GGCGAGGGCGAGGGAGGCGACGCCACGTATGGCAAATTGACCCTGAAGTTTATTTGCACGACCGGAAAATTGCCTGTACCGTGGCCCACACTTTGGT
CACTACCGTTATCAATGTTTCTCGCTATCCGGACCACATGAAGCAGCATGACTTCTTTAAAAGTGCAATGCCCGAGGGTTATGTTCAAGAGCGGACCA
TCTTTTTTAAAAGACGACGGCAACTACAAGACGCGCGAGGTGAAGTTCGAGGGCGACACGCTGGTGAATCGGATTGAGTTAAAAGGAATTGACTTTAA
AAGATGACGGCAACATCCTTGGACATAAGTTAGAGTACAATTATAATTCAAACCACGTGTACATCATGGCCGACAAACAAAAAAACGGCATCAAGGTA
AACTTTAAAATTAGACATAATATCGAGGATGGCAGTGTTCAATTAGCCGACCATTACCAACAGAACACCGATAGGCGGACGGTCCTGTATTACCTGAC
AACCATTACCTTAGCACGCAGTCTGCACTGTCCAAGGACCCAAATGAGAAACGAGGACCATATGGTGTTGCTAGAGTTCGTTACCGCAGCAGGAATAAC
Codon optimization
The selected organism: Escherichia coli (E. coli)
Reason: This is because E. coli is a common model organism for the production of recombinant proteins
due to its speed, low cost and ease of manipulation.
Cell-dependent method in this case, focused on E. coli:
Cloning: An insertion of the optimized sequence is carried out in a plasmid.
Transformation: In this step, what is done is that the plasmid is introduced into E. coli.
Expression: With this, an induction is carried out with IPTG to produce GFP.
Purification: This step seeks to use affinity chromatography.
Cell free method: For this purpose, E. coli extracts are used to produce GFP directly.
DNA Read/Write/Edit
DNA Read
(i) What DNA Would You Edit?
The DNA to be sequenced would be the F9 gene locus extracted from the patient’s liver cells following CRISPR-Cas9 gene editing. The core purpose is verification — confirming that the correction
worked exactly as intended and that no unintended damage was introduced elsewhere in the genome.
There are three specific sequencing goals. First, confirming the edit — reading across the exact mutation site to verify that the correct nucleotide was restored by Homology-Directed Repair,
and that no indels were introduced by the messier NHEJ pathway instead. Second, off-target analysis — scanning the broader genome for any locations where Cas9 may have made unintended cuts,
which is a critical safety check before any edited cells are returned to the patient. Third, monitoring gene expression — by sequencing RNA transcripts (via RNA-seq), it is possible to confirm
that the corrected F9 gene is actually being transcribed and translated into functional Factor IX protein, not just sitting silently in the genome.
(ii) In lecture, a variety of sequencing technologies were mentioned. What technology or technologies would you use to perform sequencing on your DNA and why?
Nanopore Sequencing (Oxford Nanopore Technologies)
For verifying CRISPR edits in the F9 gene, Nanopore sequencing is the strongest choice. The F9 gene involves complex structural mutations, and Nanopore’s ability
to read very long stretches of DNA in a single pass makes it ideal for confirming whether the edit succeeded and detecting any off-target cuts elsewhere in the genome.
Generation
Nanopore is a third-generation sequencing technology. Unlike first-generation Sanger sequencing (slow, single fragment) or second-generation Illumina (short reads, requires amplification),
Nanopore reads single DNA molecules in real time without amplification, and uniquely detects bases electrically rather than optically.
Input & Preparation
The input is high-molecular-weight genomic DNA extracted from the patient’s edited liver cells. After extraction, DNA quality is checked using NanoDrop and gel electrophoresis.
The ends are then repaired and dephosphorylated to create clean blunt ends, followed by ligation of Oxford Nanopore’s proprietary motor protein adapters. Importantly, no PCR amplification
is needed, which avoids bias and preserves the DNA’s native state.
Sequencing & Base Calling
A protein nanopore is embedded in a membrane with a constant ionic current flowing through it. The motor protein feeds the DNA strand through the pore one base at a time, and each base
disrupts the current by a characteristic amount. These electrical signals are recorded in real time and translated into a DNA sequence by a neural network-based base calling algorithm such as Dorado.
Output
The output is a set of long reads — sometimes hundreds of thousands of base pairs — stored in FASTQ format, containing both the sequence and per-base quality scores. These are aligned to the
reference genome to confirm the F9 correction, identify any NHEJ-caused indels, and flag potential off-target edits.
DNA Write(i) What DNA Would You Synthesize and Why?
The target for synthesis would be a corrected copy of the F9 gene, which encodes Clotting Factor IX — the protein absent or dysfunctional in Hemophilia B patients. Rather than relying solely on
CRISPR to repair the mutation in place, synthesizing a complete, healthy F9 sequence allows it to be used as the donor template in Homology-Directed Repair, or delivered independently as a gene
therapy construct. The F9 gene spans approximately 34,000 base pairs, and the synthesized version would carry the correct nucleotide at the mutation site, restoring the gene’s ability to produce
functional Factor IX. This approach is compelling because it offers a one-time, permanent fix rather than the lifelong Factor IX infusions patients currently depend on.
(ii) What technology or technologies would you use to perform this DNA synthesis and why?
Because the F9 gene is too long to synthesize as a single error-free strand using standard chemical methods, the same two-stage approach applies. Silicon-based oligonucleotide synthesis
(as used by platforms like Twist Bioscience) prints thousands of short, overlapping DNA fragments in parallel using phosphoramidite chemistry, building the sequence one nucleotide at a
time directly onto a chip.
Once detached, these fragments are fed into Gibson Assembly, where an exonuclease exposes overlapping single-stranded ends, a polymerase fills any gaps, and a ligase seals the joins —
stitching all the pieces into the complete, corrected F9 sequence in a single reaction.
The synthesized F9 construct would then be packaged into an AAV vector (Adeno-Associated Virus), specifically AAV5 or AAV8, which has a strong affinity for liver cells where Factor
IX is naturally produced, and delivered intravenously to the patient. The primary limitations are error rates — longer constructs accumulate more synthesis errors, requiring thorough
verification with Nanopore sequencing — and the AAV’s limited cargo capacity, which at roughly 4.7kb is smaller than the full F9 gene, necessitating the use of a compact promoter and
careful construct design to fit within that constraint.
DNA Edit(i) What DNA would you want to edit and why?
The DNA to be edited is the F9 gene located on the X chromosome, which encodes Clotting Factor IX — a protein essential to the blood clotting cascade. Hemophilia B occurs when mutations
in this gene prevent the body from producing functional Factor IX, meaning even minor injuries or internal bleeds can become life-threatening without immediate medical intervention.
The most common severe mutations include point mutations and small deletions that either truncate the protein or render it completely nonfunctional.
The technology of choice is CRISPR-Cas9, delivered to liver cells via an AAV8 viral vector. CRISPR-Cas9 is the most practical option because it is programmable, relatively affordable,
and precise enough to target a single mutation within the F9 gene. AAV8 is paired with it specifically because this serotype has a strong natural affinity for hepatocytes — the liver
cells where Factor IX is produced — making intravenous delivery efficient without requiring cells to be removed from the patient.
How It Works
A custom Guide RNA (gRNA) is designed to match the exact sequence surrounding the F9 mutation. The Cas9 protein escorts this gRNA through the cell, scanning the genome until it finds
its complementary sequence and binds. It then executes a Double-Strand Break, cutting through both strands of the DNA helix at the target site. At this point, a donor DNA template
carrying the correct F9 sequence is provided alongside the CRISPR machinery. The cell’s repair system uses this template as a blueprint to fix the break through Homology-Directed
Repair (HDR), precisely overwriting the mutation with the healthy sequence.
Preparation & Input
The editing package consists of four components: the Cas9 nuclease, the custom gRNA, the corrective donor DNA template, and the AAV8 vector to carry everything into liver cells.
The gRNA must be carefully designed to be unique to the F9 locus to minimize the risk of cutting elsewhere. The entire construct is packaged into AAV8 and delivered intravenously,
where it naturally homes to the liver.
Limitations
The three main limitations are efficiency, off-target effects, and immune response. HDR is inherently inefficient in adult liver cells, as these are largely non-dividing — many
cells will default to the imprecise NHEJ repair pathway, potentially worsening the mutation rather than correcting it. Off-target cuts remain a safety concern; if Cas9 mistakes
a similar genomic sequence for its intended target, it could disrupt a critical gene. Finally, some patients carry pre-existing immune responses to AAV vectors, which can neutralize
the delivery system before it reaches the liver, limiting the therapy’s effectiveness.
Generated Peptides:
We obtained approximately 4 peptides plus one control peptide, each showing a perplexity score. The sequence modification made was Alanine to Valine substitution at position 4.
Pép-1: WVRFLPYRKGGL (Perplexity: 12.4)
Pép-2: KLVFAVGPSTRR (Perplexity: 14.8)
Pép-3: RRLYWAIPRGGF (Perplexity: 11.2)
Pép-4: VVFQRLSSTGRR (Perplexity: 15.1)
Control: FLYRWLPSRRGG (Known peptide)
The low perplexity observed in Pép-3 suggests that the model has high confidence that this sequence “fits” in the evolutionary and structural context of the mutated SOD1. Lower perplexity indicates greater model confidence in the sequence’s contextual appropriateness.
Part 2: Evaluation with AlphaFold3 (AF3)
When these sequences were loaded into AF3 along with SOD1 A4V, the following results were obtained:
Pép-3 (RRLYWAIPRGGF):
iPTM (interaction Predicted TM-score): 0.78
Location: Near the N-terminal region
Function: Appears to stabilize the beta sheet destabilized by the A4V mutation
Strategic positioning: Close to the mutation site (position 4)
Both iPTM values exceed 0.7, indicating high confidence in the interactions. iPTM scores above 0.7 are generally considered excellent predictors of binding reliability. In the context of our final project, the MeR peptide binds near the metal-binding site (such as the Cys-Cys site in MerA), which is fundamental for preventing mercury-induced protein unfolding before reduction occurs.
Part 3: Property Assessment in PeptiVerse
Peptide
Affinity (pKd)
Solubility
Hemolysis
Charge (pH 7)
Pép-3
8.2
High
Low
+3
Pép-1
7.5
Medium
Medium
+2
Control
8.5
Low
High
+3
Although the control peptide exhibits the highest affinity (8.5), its low solubility and high hemolytic potential make it a poor therapeutic candidate. Therefore, Pép-3 is selected as the optimal candidate, achieving an excellent balance between affinity and favorable physicochemical properties. Its positive charge (+3) and high solubility facilitate interaction with acidic regions of SOD1 (or the MerA enzyme used in our final project), promoting proper protein folding in the bacterial cytoplasm during bioremediation processes.
Part 4: Optimization with moPPIt
In moPPIt, we do not simply generate random sequences; instead, we guide the design process toward specific objectives which are:
Design a peptide that specifically binds to residues 4-10 (A4V mutation site)
Target zinc-binding residues (positions 80, 71, 63)
However it is important to value these things:
PepMLM: Proposes “natural” sequences based on language model training
moPPIt: Generates mathematically optimized sequences specifically designed for enhanced affinity
This two-stage approach combines evolutionary relevance (PepMLM) with computational optimization (moPPIt) to create peptides with superior binding characteristics.
Part 5: Application in MerR for Bioremediation (Final Project)
My final project is a -> mercury biosensor design:
Using the MerR protein as a heavy metal sensor (particularly for mercury), we employ moPPIt to design peptides that mimic the Hg(II) binding site. This approach allows us to:
Create more sensitive biosensors by engineering peptide sequences that specifically recognize mercury ions
Improve binding specificity through optimized interactions at the metal-binding interface
Enhance bioremediation efficiency by tailoring peptide-protein interactions to target specific mercury-binding mechanisms
🔬 Project Documentation SECTION 1: ABSTRACT Significance: Heavy metal contamination, specifically mercury ($Hg^{2+}$), poses a critical threat due to its persistence and bioaccumulation in living organisms. In industrial and mining regions like Lima, Peru, traditional detection relies on expensive analytical equipment (ICP-MS), which is inaccessible to local communities. This project addresses the urgent need for a low-cost “sense-and-respond” biological system.
Heavy Metal Detection and Removal Using Biosensor Microbes
🔬 Project Documentation
SECTION 1: ABSTRACT
Significance:
Heavy metal contamination, specifically mercury ($Hg^{2+}$), poses a critical threat due to its persistence and bioaccumulation in living organisms. In industrial and mining regions like Lima, Peru, traditional detection relies on expensive analytical equipment (ICP-MS), which is inaccessible to local communities. This project addresses the urgent need for a low-cost “sense-and-respond” biological system.
Broad Objective:
The overall goal of this project is to engineer Pseudomonas putida to serve as a dual-action biosensor and bioremediator that can simultaneously detect mercury concentrations and initiate physical sequestration.
Hypothesis:
This project tests the principle that a MerR-regulated genetic circuit can exhibit high sensitivity to $Hg^{2+}$ ions, triggering a proportional expression of Green Fluorescent Protein (GFP) for quantification and SmtA (a bacterial metallothionein) for sequestration.
Specific Aims:
Characterize the sensitivity of the MerR-based sensing module.
Engineer a surface-display module for metal binding.
Develop a strategy for community-led deployment in contaminated sites.
Methods:
The technical approach utilizes Golden Gate Assembly to create a bicistronic operon under the control of the mercury-responsive $P_{mer}$ promoter. Detection is measured via fluorometry (488nm/509nm). Remediation is achieved through the expression of cysteine-rich Metallothioneins (MTs). These proteins use thiol-group coordination to bind $Hg^{2+}$ ions with high affinity. To prevent cellular toxicity, these proteins are fused to the Lpp-OmpA system for display on the outer membrane of the cell.
SECTION 2: PROJECT AIMS
Aim 1: Experimental Aim (The Sensing Circuit)
The first aim of my final project is to construct and validate a mercury-responsive genetic circuit in Pseudomonas putida by utilizing the MerR regulatory protein and the $P_{mer}$ promoter. MerR acts as a specialized transcription factor; when $Hg^{2+}$ binds to its conserved cysteine residues, it induces a conformational twist in the DNA, allowing RNA polymerase to initiate transcription of the GFP reporter. This aim will establish the operational detection limit of the biosensor.
Aim 2: Development Aim (The Binding Module)
The next step following the successful creation of the sensor is to engineer a remediation module by expressing SmtA Metallothioneins. These proteins are highly enriched in Cysteine (Cys) residues, which provide sulfur atoms for stable coordination of heavy metals. To maximize capture efficiency, I will utilize a surface-display strategy (fusing SmtA to the OmpA protein) so the bacteria can sequester mercury directly from the external environment without requiring intracellular transport.
Aim 3: Visionary Aim (Autonomous Remediation)
The long-term vision is to democratize environmental monitoring by deploying these microbes in a “Living Filter” system. This involves immobilizing the engineered P. putida in a porous matrix to create a self-regenerating water treatment device. This challenges the current paradigm of centralized waste management by providing local communities with a cost-effective, autonomous method for both monitoring and cleaning their water sources.
SECTION 3: BACKGROUND
Background and Literature Context
The MerR protein is a highly specific metalloregulatory protein. Research by Brown et al. (2003) demonstrates that MerR binds to the $P_{mer}$ operator as a homodimer; upon mercury binding, it rotates the DNA to activate transcription. For sequestration, Metallothioneins (MTs) like SmtA are utilized. As discussed by Romero-Isart and Vasak (2002), these proteins form metal-thiolate clusters, allowing a single protein to bind multiple mercury ions with high stability. Currently, most technologies offer “sensing only” or “remediation only”; an integrated, field-robust system in a resilient chassis like P. putida is a notable gap in current research.
Innovation and Novelty
This project is innovative because it integrates detection and remediation into a single, automated “sense-and-respond” circuit.
Integrated Logic: Using a bicistronic design ($P_{mer} \to GFP + MT$) ensures that remediation effort is strictly proportional to the contamination detected.
Surface Display: Instead of internalizing toxins, the project uses Lpp-OmpA fusions to keep mercury on the cell exterior, protecting the host’s metabolism and making metal recovery easier.
Host Selection: By utilizing P. putida instead of E. coli, the project applies synthetic biology to a “non-model” organism that is naturally optimized for survival in harsh industrial waste.
Impact and Significance
The Problem: Mercury is a “forever toxin” that accumulates in the food chain, causing neurological damage in local populations.
Societal Contribution: This project provides a “Green Chemistry” alternative to chemical precipitation, creating a low-cost tool for environmental justice in marginalized mining communities.
Knowledge Advancement: Achieving these aims will provide critical data on the metabolic cost of maintaining dual-action circuits in environmentally-robust bacteria.
Field-level Change: If successful, this changes the field from passive monitoring to active surveillance, where cleanup and detection occur simultaneously and autonomously at the source of contamination.
🧬 System Architecture
[ merR Regulator ] --(Constitutive)--> [ MerR Protein ]
|
v
[ Pmer Promoter ] --(Induced by Hg2+)--> [ RBS ] --> [ GFP Reporter ] --> [ RBS ] --> [ SmtA-OmpA Fusion ]