Week 4 HW: Principles and Practices
Part A:
Question 1: How many molecules of amino acids do you take with a piece of 500 grams of meat? (on average an amino acid is ~100 Daltons)
To understand this its important to know that elements on the scale of amino acids masses are measured in units of Daltons, defined as the mass of 1/12th of a carbon atom or 1x10^-24 grams.
So in 500 grams of meat where following that there is 26 grams of protein per 100g of meat there is then 130 grams of protein.
This means there is 130 x 1.67 x 10^-24 daltons of protein (hence amino acids).
And since 1 amino acid is on average 100 Daltons we just have to divide by 100 to get the number of amino acid molecules present which ends up to be:
2.171 x 10^24 molecules of amino acids
Question 2: Why do humans eat beef but do not become a cow, eat fish but do not become fish?
This is because we have enzymes that cleave the proteins we eat into their individual amino acids, these amino acids are then used to build the human into its own unique structures and uses. The protein of each animal consumed isnt "directly transferred" into the human but its broken down into its amino acid constituents, carbohydrate sources etc.. which are then used as the building material. What decides how an organism looks or functions is to do with their genetic coding in their DNA, So the DNA acts as the building instructions and food/protein/amino acids consumed act as the raw mateerials used to carry out these nstructions.
Question 3: Why are there only 20 natural amino acids?
I Initially thought this was because a limitation in the triplet codon but then i remembered thats 4C3 which is 64 different combinations and they all only end up giving 20 different amino acids which is why they are degenerate. So I assume the reason theres only 20 amino acids in nature is because of a natural selection process that has occured throughout evolution as a combination of what amino acids provided useful and allowed for better survival.
Question 4 : Can you make other non-natural amino acids? Design some new amino acids
Yes this is possible - non-natural amino acids better known as noncanonical amino acids are those containing side chains beyond the side chains that DNA can code for.
By modifying the side chains of the basic glycine-derived α-amino acid structure, more complex functional groups can be introduced to design non-natural amino acids. These include D-amino acids, β-amino acids, and amino acids with modified side chains. Such modifications are crucial in biotechnology and pharmaceuticals for applications including the development of more stable therapeutic peptides, protein engineering, and the study of protein structure and function.
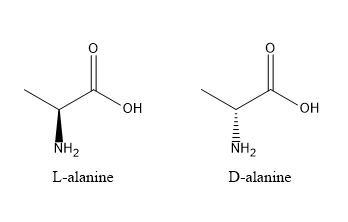
Noncanonical amino acids are commonly used to improve the stability of therapeutic peptides by reducing their susceptibility to proteolytic degradation. Proteases recognise specific sequence motifs and backbone geometries, so introducing a non-natural amino acid near a known cleavage site can disrupt this recognition. One effective strategy is the incorporation of D-amino acids, which have the same side chain but opposite stereochemistry to naturally occurring L-amino acids, making them poorly recognised by most proteases. For example, glucagon-like peptide-1 (GLP-1) is rapidly cleaved by DPP-4 at the Ala8–Glu9 bond. Replacing the alanine at position 8 with a D-amino acid such as D-alanine alters the local stereochemistry, reducing enzymatic cleavage and improving peptide half-life while maintaining biological function.
Question 5 : Where did amino acids come from before enzymes that make them, and before life started?
There are several proposed explanations for how amino acids formed before the origin of life. One key idea is that amino acids were produced through abiotic chemical reactions on early Earth. The early atmosphere is thought to have contained simple gases such as methane (CH₄), ammonia (NH₃), and water vapour (H₂O). With energy sources such as lightning and ultraviolet radiation, these gases could undergo chemical reactions to form organic molecules, including amino acids. This was demonstrated by the Miller-Urey experiment, which successfully produced amino acids under simulated early Earth conditions.
In addition, evidence from extraterrestrial sources supports this idea. The Murchison meteorite, which fell in 1969, was found to contain a wide range of organic molecules, including amino acids. This shows that amino acids can form in space and may have been delivered to early Earth via meteorites, contributing to the prebiotic chemical pool from which life eventually emerged.
Question 6: If you make an α-helix using D-amino acids, what handedness (right or left) would you expect?
Amino acids are chiral and exhibit stereospecificity, meaning their three-dimensional arrangement determines how they interact and fold. Natural proteins are composed of L-amino acids, which form right-handed α-helices due to the specific geometry of the peptide backbone and side chain orientation. If D-amino acids are used instead, the stereochemistry is inverted, resulting in a mirror-image structure. Consequently, D-amino acids form left-handed α-helices, as the backbone geometry and hydrogen bonding pattern are reversed.
Question 7: Can you discover additional helices in proteins?
The α-helix is one of the most common protein secondary structures, characterised by a right-handed coil with approximately 3.6 amino acid residues per turn. It has a pitch of 5.4 Å and a rise of 1.5 Å per residue, and is stabilised by hydrogen bonds between the carbonyl oxygen of residue i and the amide hydrogen of residue i+4.
In principle, other helical structures can exist by altering the number of residues per turn or the hydrogen bonding pattern, such as the 3₁₀ helix (i → i+3) or π-helix (i → i+5), and even left-handed helices under certain conditions. However, the formation of helices in nature is constrained by backbone geometry and energetic stability. Only specific hydrogen bonding patterns and torsional angles are energetically favourable, meaning that while alternative helices are theoretically possible, only a limited number form stable and commonly observed structures in proteins.
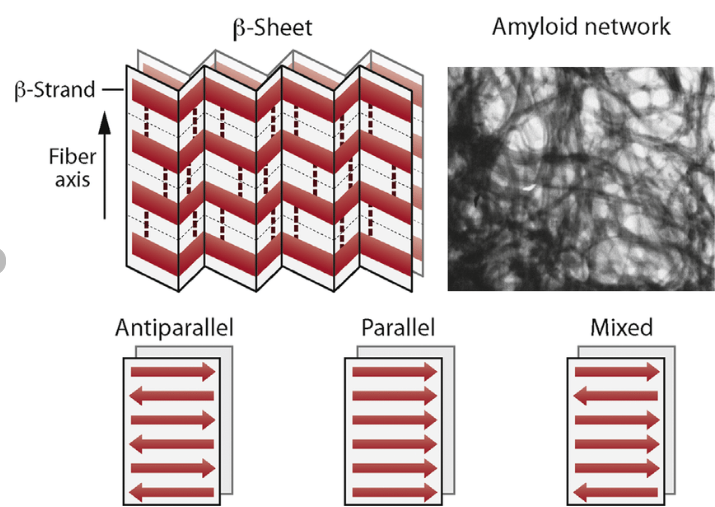
Question 9: Why do β-sheets tend to aggregate? What is the driving force for β-sheet aggregation?
The main idea is that the forces are more stabilised when these sheets aggregate, stacking and slotting into one another. The β-sheets tend to aggregate because their extended backbone structure allows for extensive intermolecular hydrogen bonding between neighbouring strands or sheets. In addition, hydrophobic side chains can align and interact between sheets, further stabilising aggregation. This combination of backbone hydrogen bonding and hydrophobic interactions leads to the formation of highly stable, stacked β-sheet structures, such as those found in amyloid fibrils.
Question 10: Why do amyloid diseases form B-sheets?
Amyloid diseases arise when normally soluble proteins misfold into structures rich in β-sheets. These β-sheets form highly stable aggregates due to extensive intermolecular hydrogen bonding and hydrophobic interactions. The sheets stack to form insoluble fibrils with a characteristic cross-β structure. These fibrils accumulate in tissues, where they disrupt cellular function and become toxic, ultimately leading to tissue damage and disease.
Part B: Protein Analysis and Visualization
- Hemoglobin is a well-studied and highly important protein found in red blood cells, where it functions as the primary carrier of oxygen throughout the body. It has a quaternary structure composed of four subunits (two α and two β chains), each containing a heme group capable of binding oxygen. Its structure is particularly interesting because it exhibits allosteric, cooperative binding, meaning that the binding of one oxygen molecule increases the affinity for subsequent oxygen molecules, enabling efficient oxygen uptake in the lungs and release in tissues.
- Since its a quarternary structure of 4 subunits, the alpha and beta chains have two different respective amnino acid sequences. α: VLSPADKTNVKAAWGKVGAHAGEYGAEALERMFLSFPTTKTYFPHF DLSHGSAQVKGHGKKVADALTNAVAHVDDMPNALSALSDLHAHKLR VDPVNFKLLSHCLLVTLAAHLPAEFTPAVHASLDKFLASVSTVLTS KYR β: MVHLTPEEKSAVTALWGKVNVDEVGGEALGRLLVVYPWTQRFFESF GDLSTPDAVMGNPKVKAHGKKVLGAFSDGLAHLDNLKGTFATLSEL HCDKLHVDPENFRLLGNVLVCVLAHHFGKEFTPPVQAAYQKVVAGV ANALAHKYH
- The structure of Hemoglobin can be found on the RCSB Protein Data Bank (e.g. PDB ID: 1A3N). It was first solved in the 1960s–1970s, with modern structures having a resolution of around 2.7 Å, indicating good quality as lower resolution values correspond to more precise structural detail. The solved structure contains not only the protein itself but also additional molecules such as heme groups with Fe²⁺ ions, which are essential for oxygen binding, as well as ligands like oxygen or water molecules. Hemoglobin belongs to the globin protein family, which is characterised by predominantly α-helical structures and a conserved heme-binding pocket.
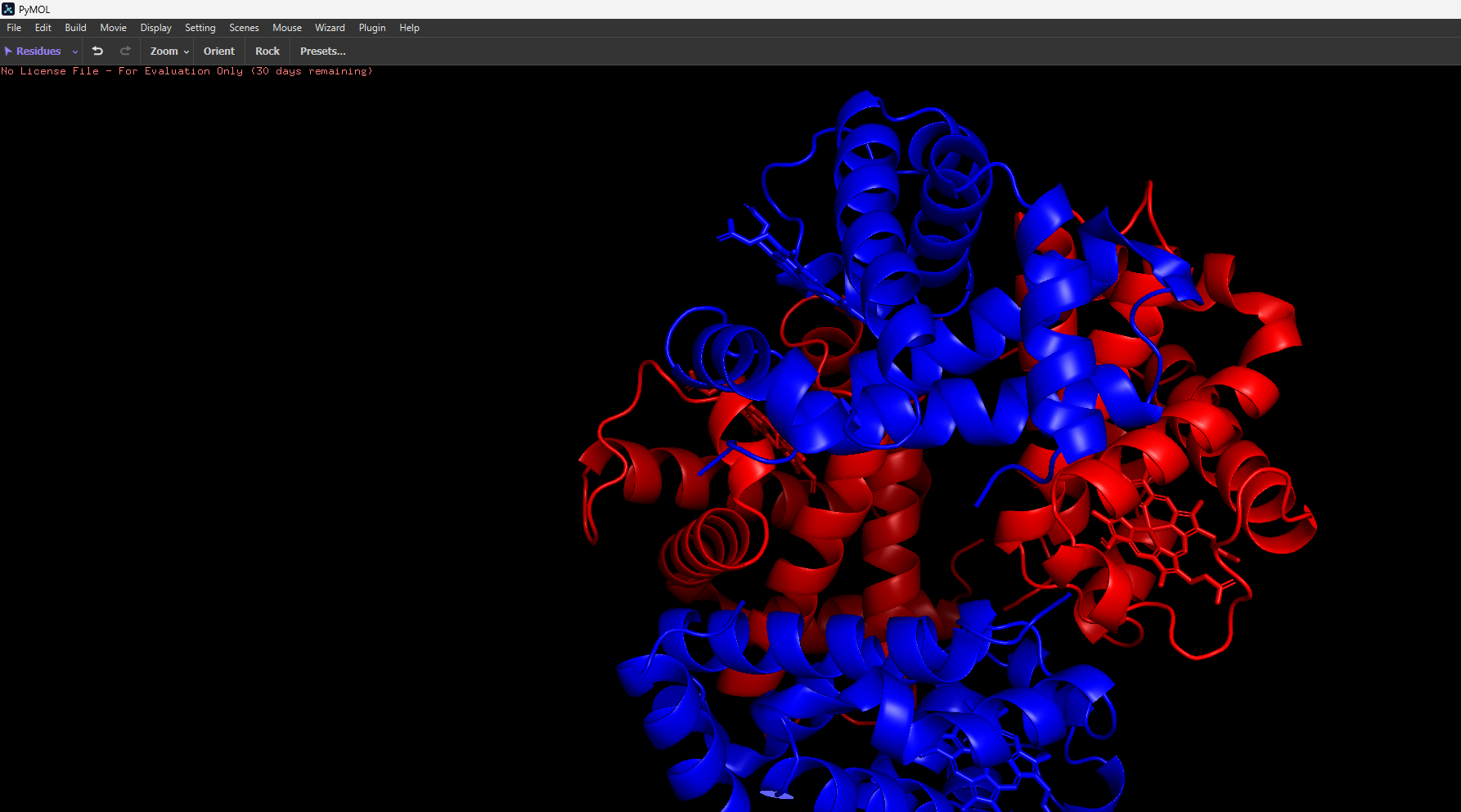
- I then uploaded Haemoglobins PDB file to pymol and rendered the following structure visualising it as a cartoon:
And then the following two renderings are the same structure but visualised as a ribbon and Ball-and-stick representation:

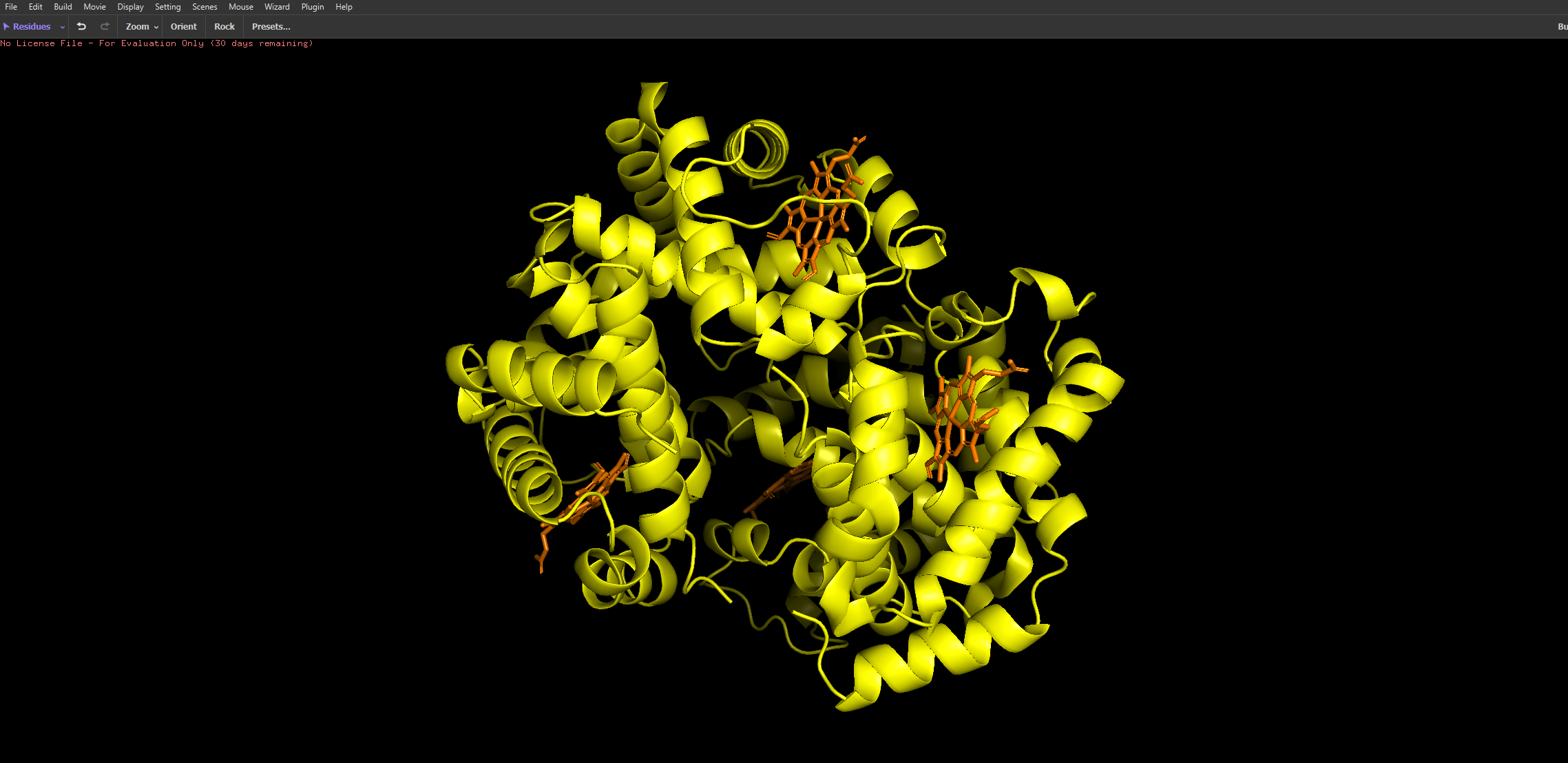
The ball and stick representation is particularly interesting as it allows us to to see the porphyrin rings that hold the iron group where the oxygen binds to one by one and changes the conformation of the entire structure hence the affinity due to its allosteric nature
After colouring the protein by secondary structure, it is clear that haemoglobin is composed almost entirely of α-helices, with very little to no β-sheet content. These helices are connected by short loop (coil) regions, forming a compact globular structure known as the globin fold. This predominance of α-helical structure is characteristic of oxygen-binding proteins and supports the formation of a stable hydrophobic pocket for the heme group.
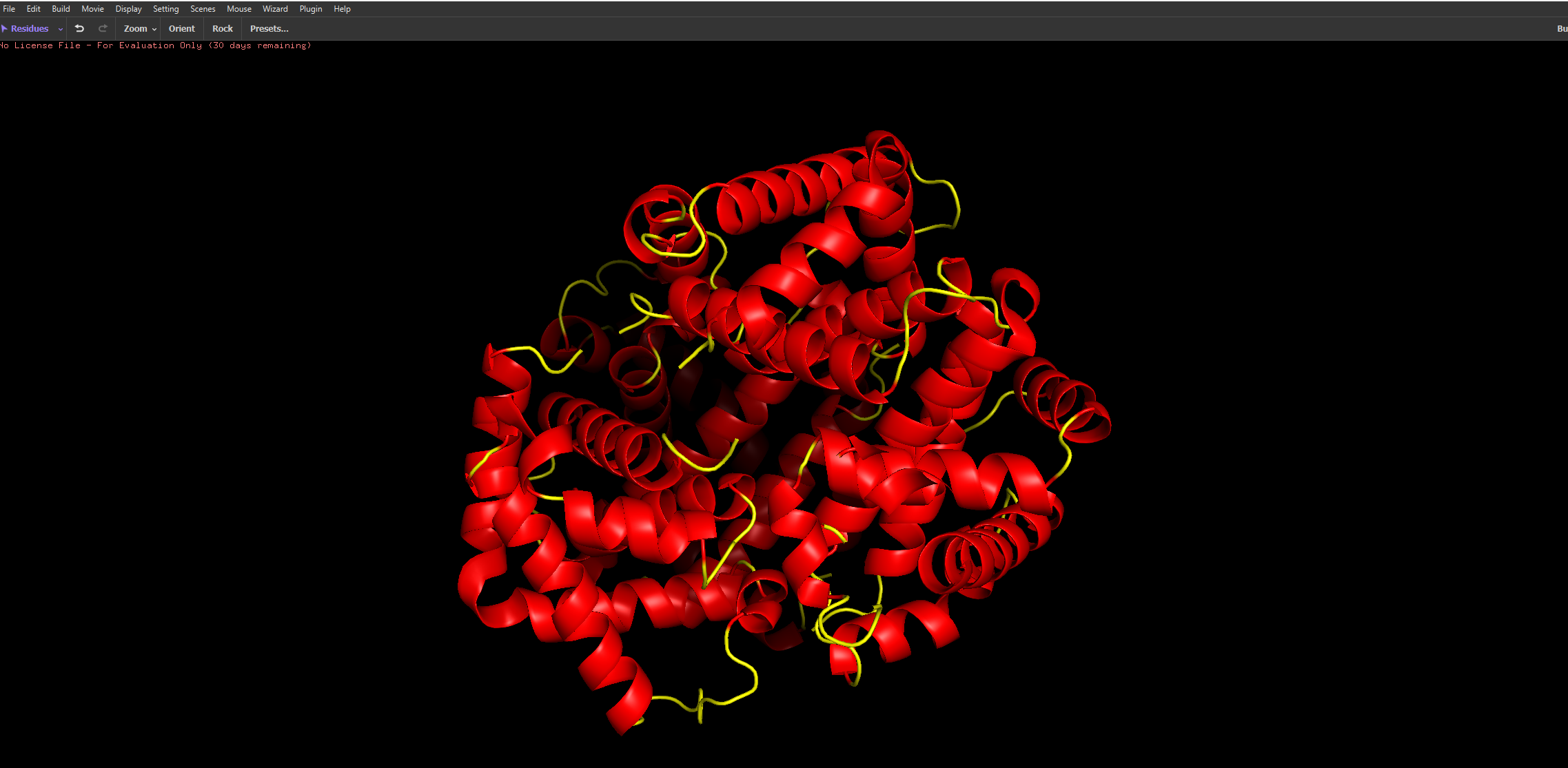
When coloured by residue type, with cyan representing hydrophilic residues and orange/red representing hydrophobic residues, it is evident that hydrophilic residues are predominantly located on the exterior of the protein, where they interact with the aqueous environment of the blood. This enhances the protein’s solubility, which is essential for its role in oxygen transport. In contrast, hydrophobic residues are largely buried within the core of the protein, where they pack closely together between α-helices, forming stabilising hydrophobic interactions. This arrangement contributes to the overall structural stability of haemoglobin and helps maintain the integrity of the heme-binding pocket.

Part C: Using ML-Based Protein Design Tools
I Chose to go with the GFP protein amino acid sequence to run the following computational analysises.
When seeing its mutation scan the following heatmap was produced
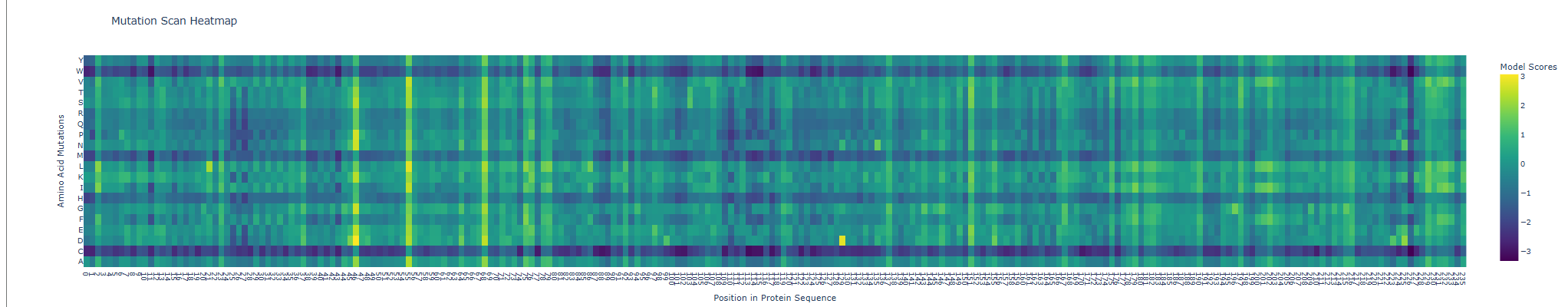
For the next comparison with experimental values I first exported my results from the deep scan heatmap produced and extracted the scoring values onto a .csv spreadsheet.
Then, the heatmap output was converted into a more interpretable format by constructing mutation labels in the form wild-type residue + position + mutated residue (e.g. Y66A). These mutations were paired with their corresponding ESM-2 likelihood scores.
To evaluate the model’s predictions, these scores were compared against experimental fluorescence measurements obtained from deep mutational scanning studies of GFP, where fluorescence acts as a proxy for protein function. In theory, a positive correlation is expected, as mutations predicted to be more compatible (higher ESM score) should better preserve protein structure and therefore maintain fluorescence.
This trend was observed for several mutations in the dataset. For example, Y66A showed both a low ESM score and very low fluorescence, which is biologically consistent because Tyr66 is part of the chromophore responsible for GFP fluorescence. However, not all mutations followed this trend, and some discrepancies were observed.
These inconsistencies arise because ESM-2 predicts sequence likelihood, not specific functional outputs such as fluorescence. While fluorescence depends on correct folding and chromophore formation, some mutations may preserve global structure (and thus appear favorable to the model) but still disrupt the precise chemical environment required for light emission. Additionally, the analysis was performed on a limited subset of mutations due to time constraints; a more robust evaluation would involve comparing all available variants (~50,000) using a statistical metric such as Spearman’s rank correlation coefficient.
Overall, the results suggest that the language model captures broad structural constraints of the protein, particularly at functionally critical residues, but does not fully account for the specific biochemical mechanisms underlying fluorescence.
Table: Comparison of ESM-2 Mutational Scores with Experimental GFP Fluorescence
| mutation | position | wt | mut | score | Experimental fluorescence |
|---|---|---|---|---|---|
| Y66A | 66 | Y | A | -0.123864412 | 0.02 |
| G67A | 67 | G | A | 0.776538849 | 0.10 |
| V163A | 163 | V | A | -0.610266924 | 0.90 |
| Y66F | 66 | Y | F | -0.521710396 | 0.85 |
| L201P | 201 | L | P | -0.147899389 | 0.01 |
| L42R | 42 | L | R | -0.721465588 | 0.15 |
| F99S | 99 | F | S | 0.004160166 | 0.05 |
| S65T | 65 | S | T | -0.177279234 | 1.20 |
| I171V | 171 | I | V | 0.522115469 | 1.05 |
| T203Y | 203 | T | Y | 0.470024824 | 1.30 |
Protein sequences were embedded using a protein language model and visualised in reduced dimensional space using t-SNE. The resulting plot shows a largely continuous distribution of proteins, with local neighbourhoods representing proteins with similar sequence features. Although distinct clusters are not sharply separated, nearby points in the embedding space are expected to share structural or functional similarities, indicating that the model captures meaningful biological relationships.
The GFP protein would be positioned within a region corresponding to proteins with similar structural characteristics, particularly beta-barrel proteins. This is because GFP has a well-defined beta-barrel fold and a conserved chromophore-forming region, both of which influence its sequence embedding.
Overall, the embedding space demonstrates that protein language models can organise proteins based on underlying biological similarity, although the separation between groups may not always be clearly defined due to the continuous nature of protein sequence space and limitations of dimensionality reduction techniques.
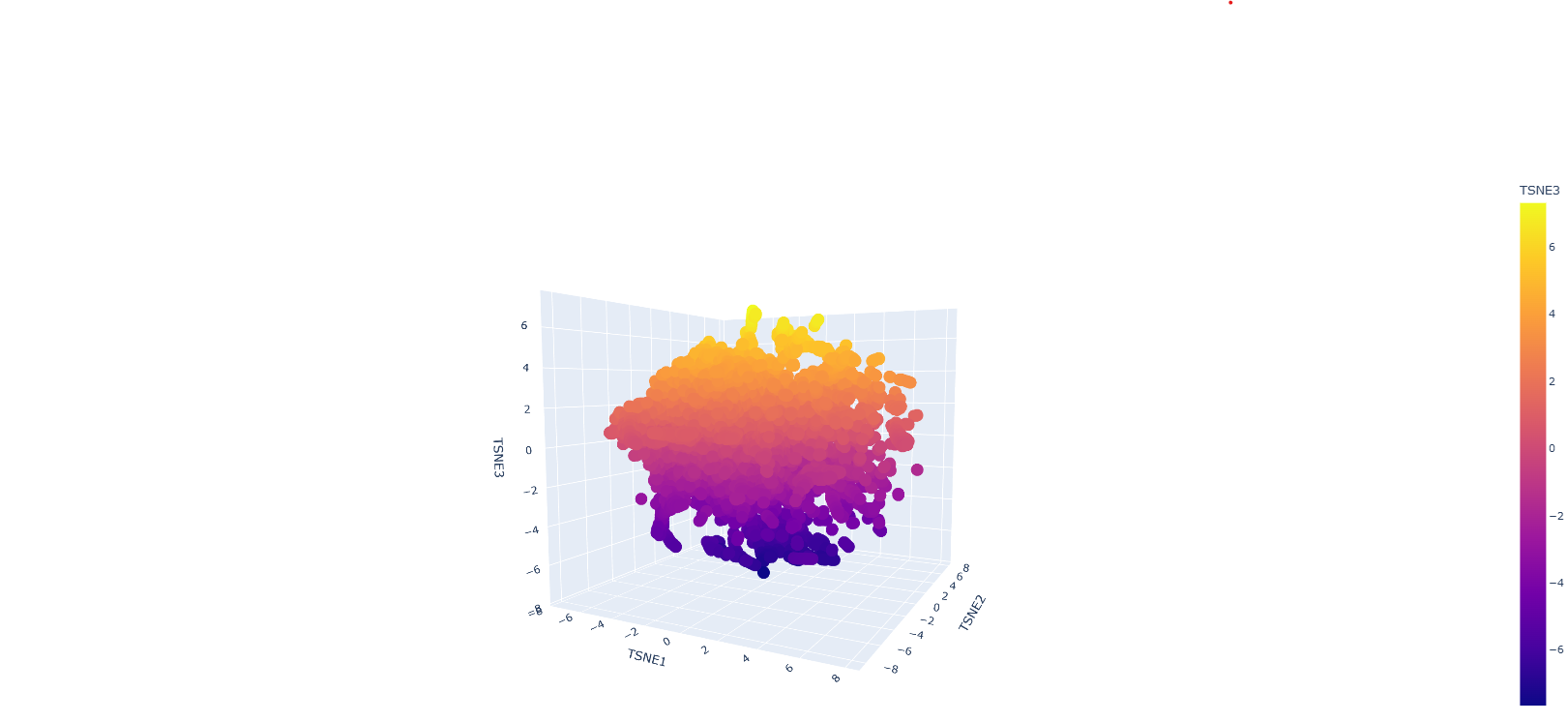
Protein sequences were embedded using a protein language model and visualised in reduced dimensional space using t-SNE. The resulting plot shows a largely continuous distribution of proteins, with local neighbourhoods representing proteins with similar sequence features. Although distinct clusters are not sharply separated, nearby points in the embedding space are expected to share structural or functional similarities, indicating that the model captures meaningful biological relationships.
The GFP protein would be positioned within a region corresponding to proteins with similar structural characteristics, particularly beta-barrel proteins. This is because GFP has a well-defined beta-barrel fold and a conserved chromophore-forming region, both of which influence its sequence embedding.
Overall, the embedding space demonstrates that protein language models can organise proteins based on underlying biological similarity, although the separation between groups may not always be clearly defined due to the continuous nature of protein sequence space and limitations of dimensionality reduction techniques.
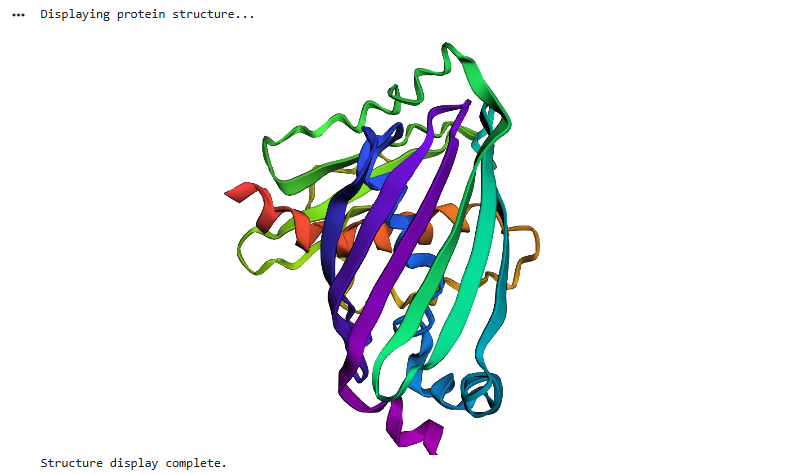
e GFP structure predicted using ESMFold closely matches the experimentally known structure, displaying the characteristic β-barrel fold composed of multiple β-strands forming a cylindrical shape. This indicates that the model accurately captures the global topology and structural organisation of the protein.
When small mutations were introduced, the overall structure remained largely unchanged, demonstrating that the protein fold is resilient to minor sequence perturbations. However, mutations at key residues, such as those involved in chromophore formation, can significantly affect function without drastically altering the structure.
In contrast, larger sequence modifications led to noticeable structural disruption, with loss of the β-barrel integrity and reduced folding stability. This suggests that while protein structures are robust to small mutations, they are sensitive to larger changes that interfere with critical folding interactions.
Overall, these results show that protein structure is more conserved than sequence, and that ESMFold can reliably predict structural consequences of mutations.
For the final part, inverse folding was performed using the GFP PDB backbone as input to ProteinMPNN in order to generate a sequence predicted to be compatible with the original structure. The resulting sequence was substantially different from the native GFP amino acid sequence, showing that multiple sequences may be compatible with a similar backbone geometry. However, the designed sequence did not retain the characteristic Ser65–Tyr66–Gly67 chromophore-forming motif required for GFP fluorescence, suggesting that structural compatibility does not necessarily preserve the original function. This highlights an important distinction between designing a sequence that can adopt a fold and preserving the precise residues needed for biochemical activity. The designed sequence was then intended to be passed through ESMFold to test whether it would refold into a structure resembling the original GFP beta-barrel.
The derived AA sequence:
ALTPEEAALLRAAWAPVAADRAANGRAFILRLFAEYPELREYFPEFKGLSLEEIAASPKLEEFAAAVVDALAEFVATADDAAAMAAALAAFAAAHVARGIGAAHFEAIRDIFPGFIASVAPPPPGAAAAWDRLLGDVIAALRAAGA
Part D. Group Brainstorm on Bacteriophage Engineering
- No group, just myself as I hadn’t realised our node was required this until marking day:((
I propose to computationally engineer the MS2 L lysis protein with a primary focus on increased stability and a secondary focus on improving lysis effectiveness through altered host interaction. This is a realistic starting point because the L protein is central to host-cell lysis, and to my current knowledge and expertise I would specifically frame stability as the easiest engineering target while toxicity/function is a bit more ambitious on my own. HTGAA’s Week 4 materials also explicitly position this assignment around applying ML-based protein design tools to engineer a better bacteriophage.
Proposed computational tools and approaches
- Start from the wild-type L protein sequence and gather any available prior knowledge on residues involved in membrane insertion, oligomerization, or host interactions.
- Use a protein language model for in silico mutagenesisto generate and score single-point or small combinatorial mutants, a software such as ESM from earlier could be used to generate those values we saw in the heat map and downloaded into a .csv file#
- Filter candidates to remove obviously disruptive mutations, especially in regions likely required for membrane localization or core function.
- Use a computational tool ssuch as AlphaFold-Multimer on selected variants to test whether mutations may alter interactions with relevant host factors such as E. coli DnaJ, since MS2 L-mediated lysis has been reported to depend on the host chaperone DnaJ.
- Rank variants based on a combination of sequence plausibility, predicted structural confidence, and predicted interaction changes.
Protein language models such as ESM are useful because they can estimate which amino acid substitutions are more likely to be tolerated while preserving overall protein fitness. That makes them a practical first-pass screen before more expensive structural modeling. Recent work shows that protein language models can be effective for predicting mutational effects and other sequence-function relationships.
AlphaFold-based tools may help because our problem is partly structural: if we want a more stable L protein, then mutations that severely disrupt folding or membrane-associated assembly are poor candidates. For the secondary goal, AlphaFold-Multimer could help us test whether specific mutations might weaken or strengthen predicted contacts with host proteins. This is especially relevant because published work on MS2-L indicates that DnaJ is involved in its lytic activity, and more recent in vitro characterization suggests that MS2-L forms high-order oligomeric states after membrane insertion.
One limitation is that prediction is not proof. A mutation that looks favorable in silico may still reduce true phage fitness in vivo. Another issue is that transmembrane and lysis proteins are harder to model accurately than soluble globular proteins, so structure confidence may not perfectly reflect real function.
Proposed Pipeline Schematic:
Wild-type L protein sequence → Protein language model mutational scan → Filter top candidate mutations → AlphaFold structure prediction of mutants → AlphaFold-Multimer with host target(s), e.g. DnaJ → Rank variants by predicted stability + interaction effects → Nominate best variants for experimental testing
