week-05-hw-protein-design-part-ii
Assignment A
Part 1:
The human SOD1 sequence was retrieved from UniProt (P00441). The ALS-associated A4V mutation was introduced by substituting alanine with valine at position 5 of the full-length sequence (equivalent to position 4 after removal of the initiator methionine).
Original sequence obtained from uniprot:
MATKAVCVLKGDGPVQGIINFEQKESNGPVKVWGSIKGLTEGLHGFHVHEFGDNTAGCTSAGPHFNPLSRKHGGPKDEERHVGDLGNVTADKDGVADVSIEDSVISLSGDHCIIGRTLVVHEKADDLGKGGNEESTKTGNAGSRLACGVIGIAQ
Mutant A4V variant:
MATKVVCVLKGDGPVQGIINFEQKESNGPVKVWGSIKGLTEGLHGFHVHEFGDNTAGCTSAGPHFNPLSRKHGGPKDEERHVGDLGNVTADKDGVADVSIEDSVISLSGDHCIIGRTLVVHEKADDLGKGGNEESTKTGNAGSRLACGVIGIAQ
Generated Sequences with their corresponding perplexity scores next to them which pepMLMs confidence rating in them, 4th one is the known binder for reference:
| ID | Sequence | Score |
|---|---|---|
| 0 | WRYPAVALAHKX | 6.680728 |
| 1 | WLYYVVAAALGE | 17.121198 |
| 2 | WRYPAVAVRHKK | 15.373117 |
| 3 | WHYYAAALALKE | 15.080042 |
| 4 | FLYRWLPSRRGG | — |
Part 2:
I was given an error in alphafold because for my first generated peptide therte was an X given representing an unkown amino acid - im not quite sure why that was generated by pepMLM but i replaced it with a G for Glycine as it is the side chain-less amino acid and would cause least stearic clash and just act as a filler amino acid in place of the unknown X.
| Candidate | Peptide Sequence | ProteinMPNN Score | Boltz ipTM |
|---|---|---|---|
| 1 | WRYPAVALAHKG | 6.680728 | 0.37 |
| 2 | WLYYVVAAALGE | 17.121198 | 0.32 |
| 3 | WRYPAVAVRHKK | 15.373117 | 0.32 |
| 4 | WHYYAAALALKE | 15.080042 | 0.28 |
| 5 | FLYRWLPSRRGG | N/A | 0.38 |
So based off of these scores the peptide that actually performed the best comparative to the known binding peptide was the one with the unknown amino acid that I replaced with a Glycine residue. And it performed very closely with only a 0.01 difference.
Observations from the 3d molecular renderer: The designed peptide binder ran closely around the Beta Barrels strands and so its plausible to assume that these amino acids are conserved in this structure?
Part 3:
I ran the best and worst ipTM scored sequences through peptiverse and obtained the results below, it can be seen that the peptiverse ratings for each factor that represents if a peptide is therapeutically feasible corresponded well to the alphafold servers ipTM scores as the better scored sequence had better metrics on peptiverse.
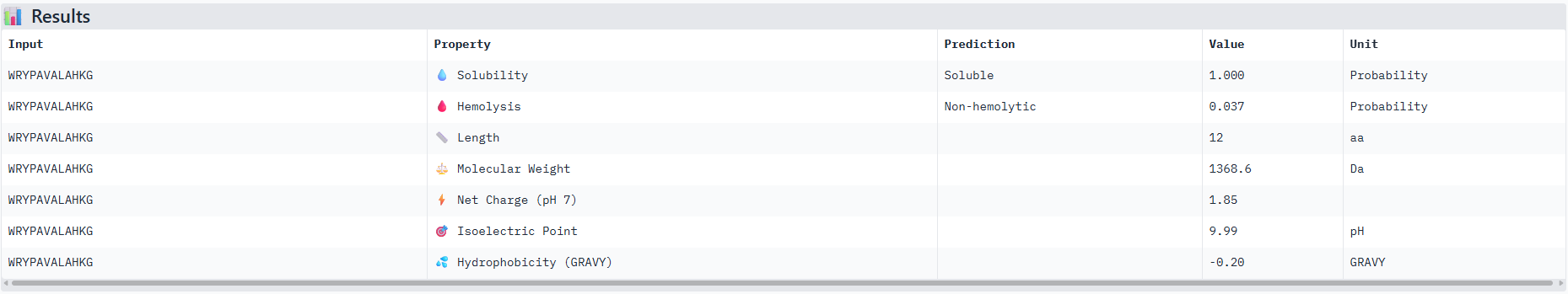
Results for peptide 0:
Results for peptide 2:
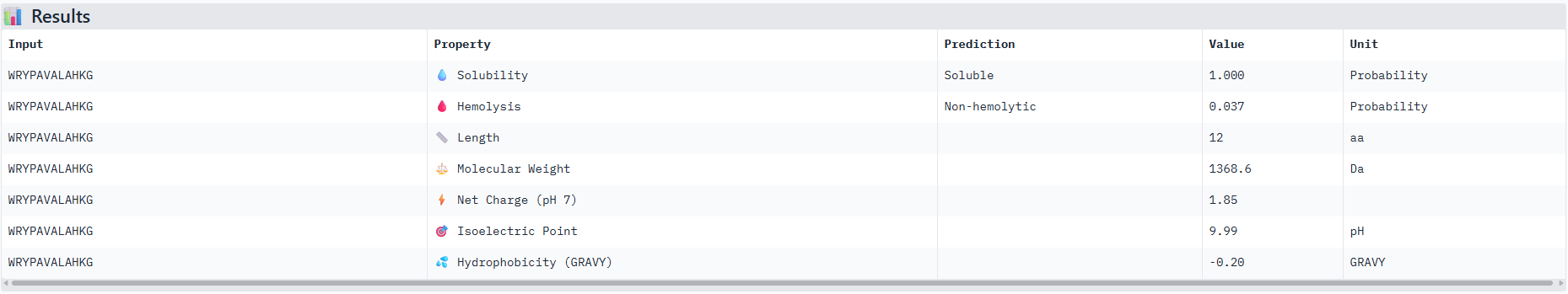
I would advance with peptide 0 as it has th best ipTM score from the alphafold server and also has good therapeutic scoring metrics as seen from the peptiverse.
Part 4
This took much longer than any other as the code was running for a long time but ended up producing results with much better and more improved scores than the previous methods. In terms of testing its validity I assume we could pass through the amino acid sequences its given us through peptiverse too to check again.
| Peptide | Hemolysis | Solubility | Affinity | Motif |
|---|---|---|---|---|
| EKCYGCHYGYQL | 0.951 | 0.917 | 6.38 | 0.818 |
| KEVVRELCCGRP | 0.953 | 0.667 | 7.74 | 0.811 |
| RKYGYQNDCCYA | 0.937 | 0.917 | 6.85 | 0.815 |
Assignment C
The notebook code initially had some issues when running but I managed to fix them with the help of Gemini.
And the following results were obtained where yellow represents a likely positive change to the folding and a more dark blue colour represents a negative change for the proteins folding ability due to the mutation:
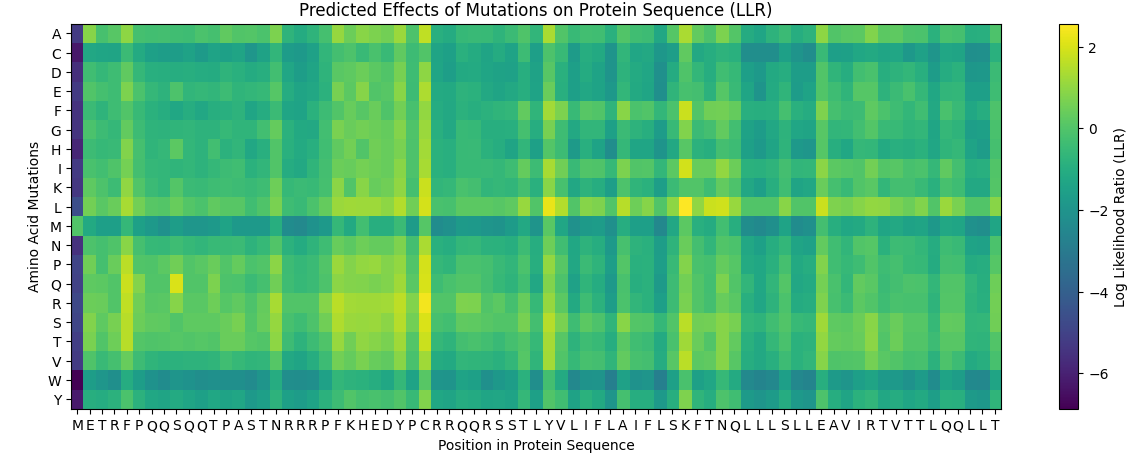
And this was the table computationally obtained for the top 10 Scores:
| Mutation | Position | Wild-Type Residue | Mutated Residue | LLR Score |
|---|---|---|---|---|
| K50L | 50 | K | L | 2.561 |
| C29R | 29 | C | R | 2.395 |
| Y39L | 39 | Y | L | 2.242 |
| C29S | 29 | C | S | 2.043 |
| S9Q | 9 | S | Q | 2.014 |
| C29Q | 29 | C | Q | 1.997 |
| C29P | 29 | C | P | 1.971 |
| C29L | 29 | C | L | 1.961 |
| K50I | 50 | K | I | 1.929 |
| N53L | 53 | N | L | 1.865 |
The results seen experimentally do contrast in comparison to the computationally obtained results in a few, to display this below is a table showing the top 10 list with the experimental results as of whether or not they could still perform lysis and also if the protein was still expressed or not
| Mutation | Position | Wild-Type Residue | Mutated Residue | LLR Score | Lysis | Protein Expression |
|---|---|---|---|---|---|---|
| K50L | 50 | K | L | 2.561 | Y | N |
| C29R | 29 | C | R | 2.395 | N | N |
| Y39L | 39 | Y | L | 2.242 | Y | Y |
| C29S | 29 | C | S | 2.043 | Y | Y |
| S9Q | 9 | S | Q | 2.014 | Y | Y |
| C29Q | 29 | C | Q | 1.997 | N | N |
| C29P | 29 | C | P | 1.971 | N | N |
| C29L | 29 | C | L | 1.961 | N | N |
| K50I | 50 | K | I | 1.929 | Y | N |
| N53L | 53 | N | L | 1.865 | Y | Y |
Comparison of computational LLR scores with experimental data shows that mutations with higher predicted scores often retain lysis activity and detectable protein expression. However, several high-scoring mutations (e.g., C29R) fail to produce functional protein, indicating that the computational model captures some but not all structural constraints governing L protein stability and function. Hence for the following selection of the 5 mutations chosen I used a combination of both data, first checking if experimentally they protein was still succesfully expressed and also still had the ability to perform lysis, and then checking if they produced a significant LLR score.
Soluble Region
Mutations in the soluble region must occur between amino acids 1–40 of the L protein.
Two mutations were selected from this region based on their high LLR scores and positive experimental outcomes for both protein expression and lysis activity, indicating that these variants are likely to produce functional proteins.
The selected soluble-region mutations are:
- S9Q
- C29S
Both mutations show detectable protein expression and successful lysis, making them strong candidates for functional L-protein variants.
Transmembrane Region
Mutations in the transmembrane region must occur between amino acids 41–75, which correspond to the membrane-spanning helix responsible for pore formation and bacterial lysis.
Two mutations were selected from this region:
N53L — This mutation shows positive protein expression and successful lysis activity, suggesting that the substitution does not disrupt membrane insertion or oligomerization of the lysis protein.
K50L — This mutation has the highest computational LLR score, indicating strong predicted sequence compatibility. However, experimental data shows no detectable protein expression, suggesting that despite the favorable computational prediction, the mutation may destabilize the protein or prevent proper folding.
Additional Mutation
The final mutation selected is Y39L, located near the boundary of the soluble and transmembrane regions.
This mutation was chosen because it shows:
- a high LLR score
- successful lysis activity
- detectable protein expression
These properties suggest that the mutation is well tolerated and likely preserves the functional structure of the L protein.