Welcome to my HTGAA Spring 2026 site. I’m interested in biotechnology, synthetic biology, protein engineering, and computational design. This page collects my coursework, lab work, and project development throughout the semester.
About me
I’m a Biotechnology student at Imperial College London with a strong interest in how engineering, biology, and computation can be combined to design useful systems. My current interests include:
Synthetic biology
Protein engineering
Biodesign
Computational tools for biology
Entrepreneurship in biotech
This site serves as a record of my progress, ideas, and outputs for HTGAA Spring 2026.
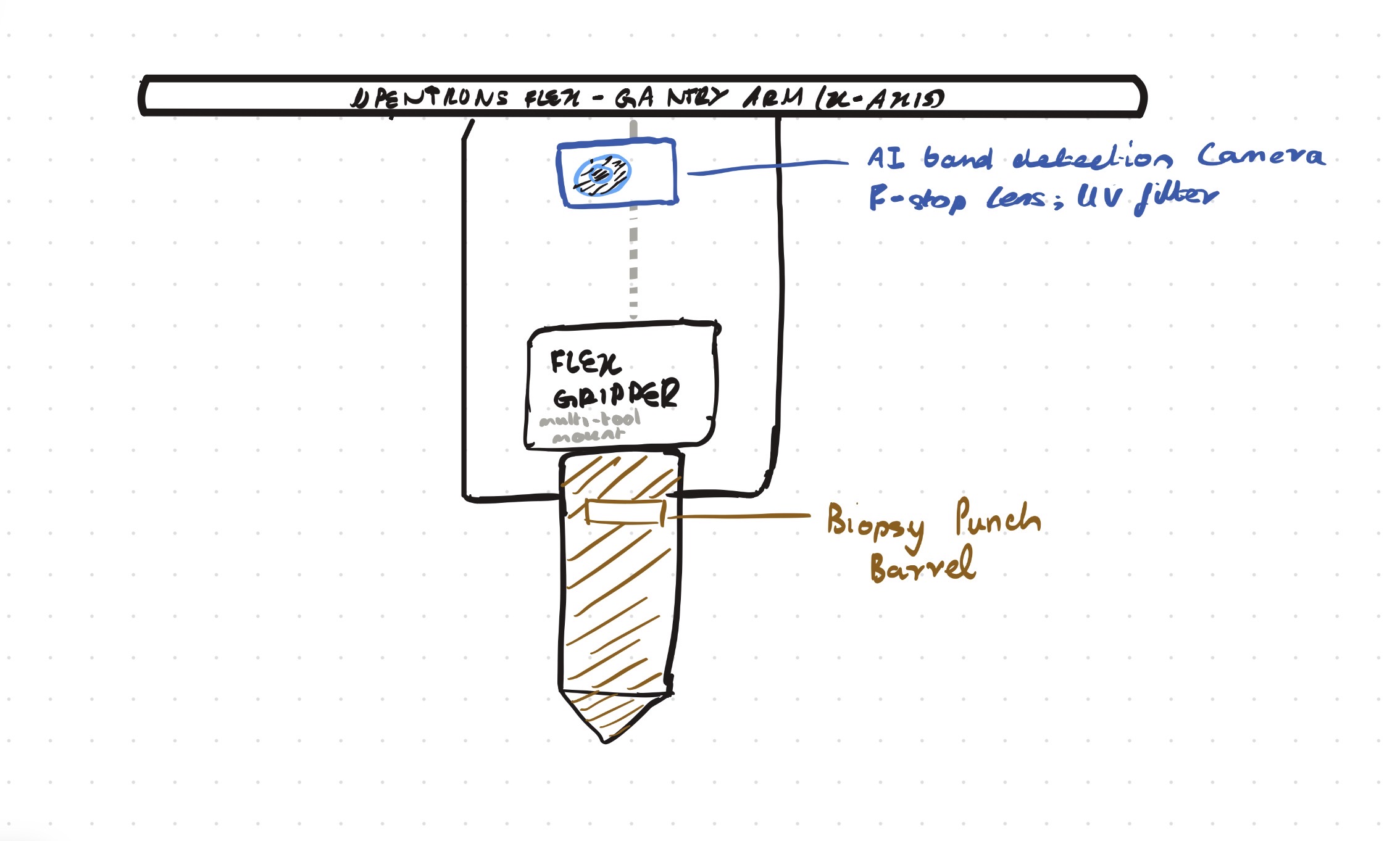
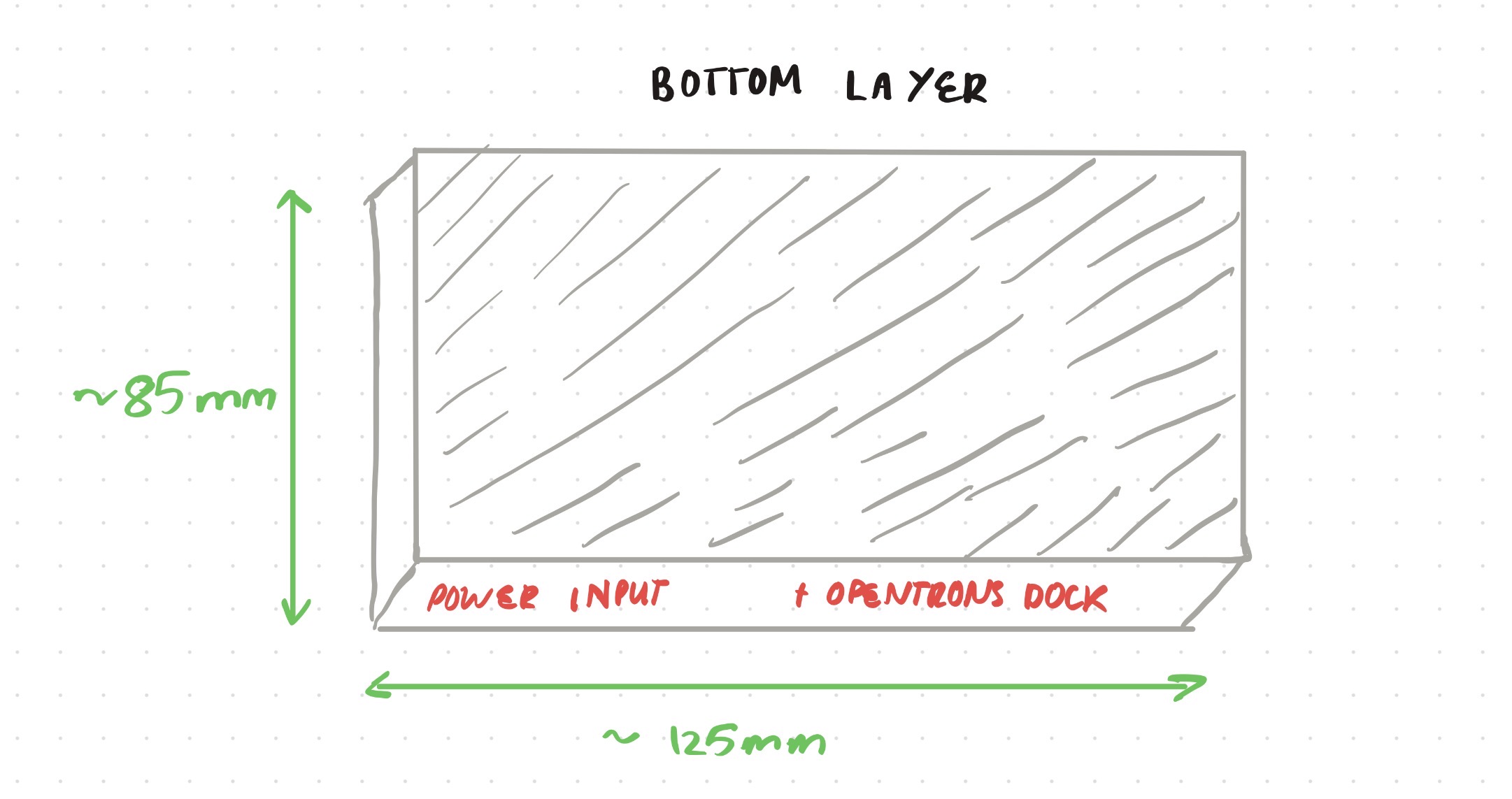
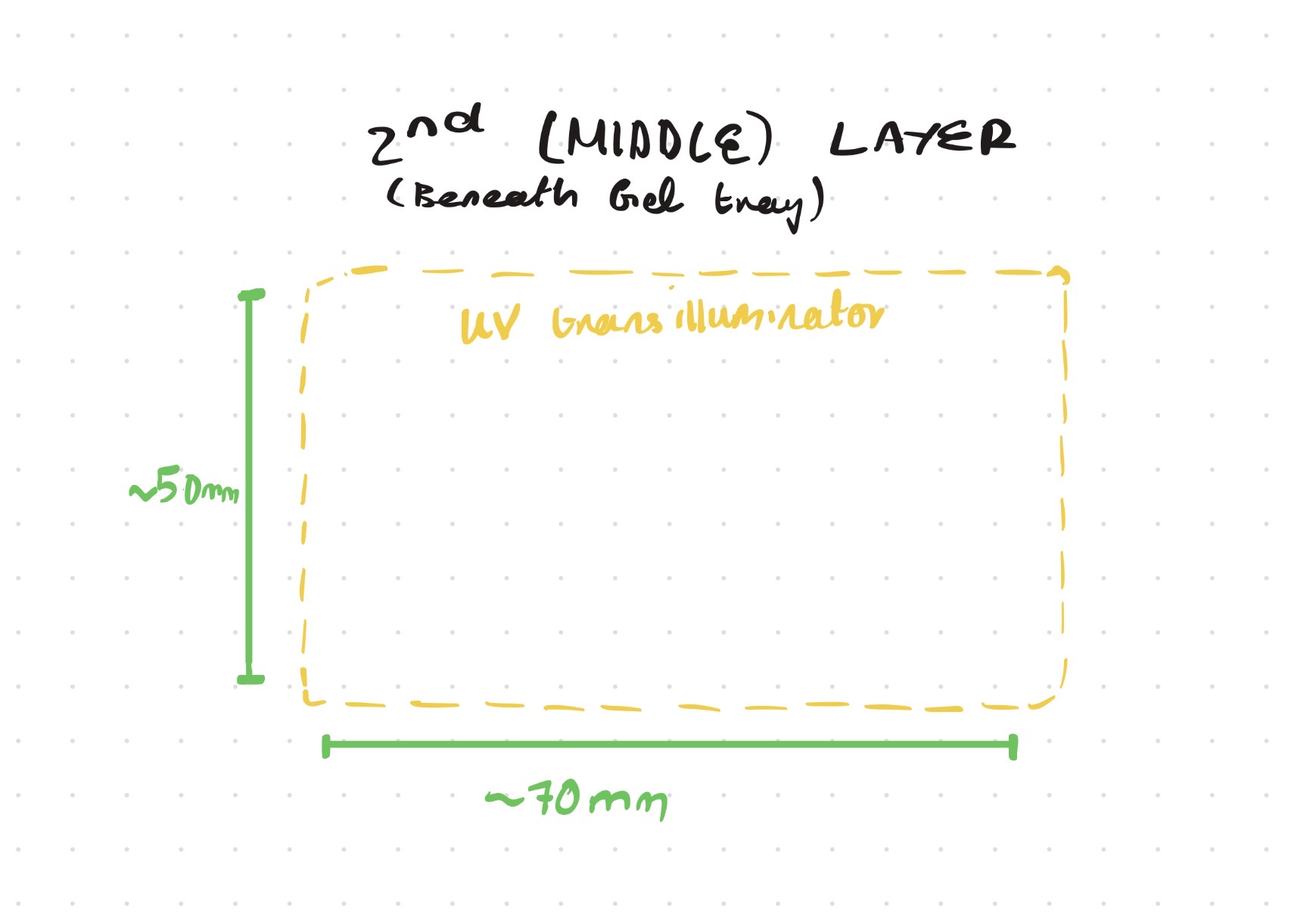
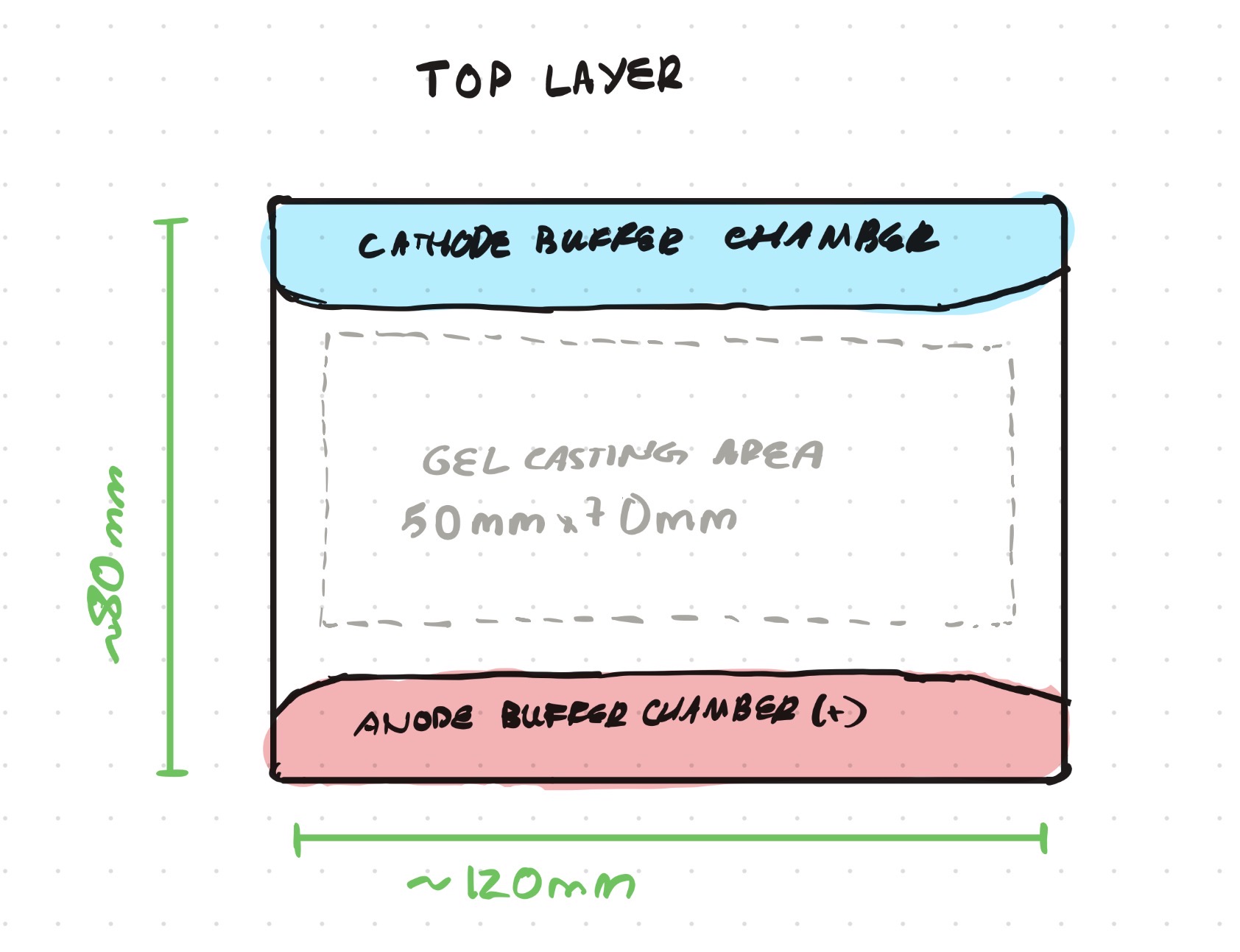
Opentrons-Integrated Modular Peptide Production Platform — full project proposal including experimental design, automation workflow, validation, and budget.
This semester I’m especially excited about exploring how biological systems can be designed, modelled, and engineered using both wet-lab and computational approaches.
Building at the intersection of biology, design, and engineering.
Subsections of Vithushan Varatharaj — HTGAA Spring 2026
Biological Engineering Application I propose developing an AI-assisted genomic analysis tool designed to predict the likelihood that a cell or tissue sample may become cancerous. The system would analyse DNA sequencing data to identify combinations of mutations, disrupted tumour suppressor genes, altered regulatory elements, and epigenetic markers associated with early stages of malignant transformation. By training machine-learning models on large cancer genomics datasets, the platform could detect patterns that indicate elevated cancer risk before visible symptoms or large tumours appear.
Lecture Overview – Lab Automation and Biosensors Introduction This week’s lecture focused on lab automation and biosensors, two technologies that are transforming modern life sciences research. Together, they enable scientists to run experiments faster, more accurately, and at a much larger scale than traditional manual laboratory techniques.
Part A: Question 1: How many molecules of amino acids do you take with a piece of 500 grams of meat? (on average an amino acid is ~100 Daltons) To understand this its important to know that elements on the scale of amino acids masses are measured in units of Daltons, defined as the mass of 1/12th of a carbon atom or 1x10-24 grams. So in 500 grams of meat where following that there is 26 grams of protein per 100g of meat there is then 130 grams of protein. This means there is 130 x 1.67 x 10-24 daltons of protein (hence amino acids). And since 1 amino acid is on average 100 Daltons we just have to divide by 100 to get the number of amino acid molecules present which ends up to be: 2.171 x 10^24 molecules of amino acids Question 2: Why do humans eat beef but do not become a cow, eat fish but do not become fish? This is because we have enzymes that cleave the proteins we eat into their individual amino acids, these amino acids are then used to build the human into its own unique structures and uses. The protein of each animal consumed isnt "directly transferred" into the human but its broken down into its amino acid constituents, carbohydrate sources etc.. which are then used as the building material. What decides how an organism looks or functions is to do with their genetic coding in their DNA, So the DNA acts as the building instructions and food/protein/amino acids consumed act as the raw mateerials used to carry out these nstructions. Question 3: Why are there only 20 natural amino acids? I Initially thought this was because a limitation in the triplet codon but then i remembered thats 4C3 which is 64 different combinations and they all only end up giving 20 different amino acids which is why they are degenerate. So I assume the reason theres only 20 amino acids in nature is because of a natural selection process that has occured throughout evolution as a combination of what amino acids provided useful and allowed for better survival. Question 4 : Can you make other non-natural amino acids? Design some new amino acids Yes this is possible - non-natural amino acids better known as noncanonical amino acids are those containing side chains beyond the side chains that DNA can code for. By modifying the side chains of the basic glycine-derived α-amino acid structure, more complex functional groups can be introduced to design non-natural amino acids. These include D-amino acids, β-amino acids, and amino acids with modified side chains. Such modifications are crucial in biotechnology and pharmaceuticals for applications including the development of more stable therapeutic peptides, protein engineering, and the study of protein structure and function. Noncanonical amino acids are commonly used to improve the stability of therapeutic peptides by reducing their susceptibility to proteolytic degradation. Proteases recognise specific sequence motifs and backbone geometries, so introducing a non-natural amino acid near a known cleavage site can disrupt this recognition. One effective strategy is the incorporation of D-amino acids, which have the same side chain but opposite stereochemistry to naturally occurring L-amino acids, making them poorly recognised by most proteases. For example, glucagon-like peptide-1 (GLP-1) is rapidly cleaved by DPP-4 at the Ala8–Glu9 bond. Replacing the alanine at position 8 with a D-amino acid such as D-alanine alters the local stereochemistry, reducing enzymatic cleavage and improving peptide half-life while maintaining biological function. Question 5 : Where did amino acids come from before enzymes that make them, and before life started? There are several proposed explanations for how amino acids formed before the origin of life. One key idea is that amino acids were produced through abiotic chemical reactions on early Earth. The early atmosphere is thought to have contained simple gases such as methane (CH₄), ammonia (NH₃), and water vapour (H₂O). With energy sources such as lightning and ultraviolet radiation, these gases could undergo chemical reactions to form organic molecules, including amino acids. This was demonstrated by the Miller-Urey experiment, which successfully produced amino acids under simulated early Earth conditions. In addition, evidence from extraterrestrial sources supports this idea. The Murchison meteorite, which fell in 1969, was found to contain a wide range of organic molecules, including amino acids. This shows that amino acids can form in space and may have been delivered to early Earth via meteorites, contributing to the prebiotic chemical pool from which life eventually emerged. Question 6: If you make an α-helix using D-amino acids, what handedness (right or left) would you expect? Amino acids are chiral and exhibit stereospecificity, meaning their three-dimensional arrangement determines how they interact and fold. Natural proteins are composed of L-amino acids, which form right-handed α-helices due to the specific geometry of the peptide backbone and side chain orientation. If D-amino acids are used instead, the stereochemistry is inverted, resulting in a mirror-image structure. Consequently, D-amino acids form left-handed α-helices, as the backbone geometry and hydrogen bonding pattern are reversed. Question 7: Can you discover additional helices in proteins? The α-helix is one of the most common protein secondary structures, characterised by a right-handed coil with approximately 3.6 amino acid residues per turn. It has a pitch of 5.4 Å and a rise of 1.5 Å per residue, and is stabilised by hydrogen bonds between the carbonyl oxygen of residue i and the amide hydrogen of residue i+4. In principle, other helical structures can exist by altering the number of residues per turn or the hydrogen bonding pattern, such as the 3₁₀ helix (i → i+3) or π-helix (i → i+5), and even left-handed helices under certain conditions. However, the formation of helices in nature is constrained by backbone geometry and energetic stability. Only specific hydrogen bonding patterns and torsional angles are energetically favourable, meaning that while alternative helices are theoretically possible, only a limited number form stable and commonly observed structures in proteins. Question 9: Why do β-sheets tend to aggregate? What is the driving force for β-sheet aggregation? The main idea is that the forces are more stabilised when these sheets aggregate, stacking and slotting into one another. The β-sheets tend to aggregate because their extended backbone structure allows for extensive intermolecular hydrogen bonding between neighbouring strands or sheets. In addition, hydrophobic side chains can align and interact between sheets, further stabilising aggregation. This combination of backbone hydrogen bonding and hydrophobic interactions leads to the formation of highly stable, stacked β-sheet structures, such as those found in amyloid fibrils. Question 10: Why do amyloid diseases form B-sheets? Amyloid diseases arise when normally soluble proteins misfold into structures rich in β-sheets. These β-sheets form highly stable aggregates due to extensive intermolecular hydrogen bonding and hydrophobic interactions. The sheets stack to form insoluble fibrils with a characteristic cross-β structure. These fibrils accumulate in tissues, where they disrupt cellular function and become toxic, ultimately leading to tissue damage and disease. Part B: Protein Analysis and Visualization Hemoglobin is a well-studied and highly important protein found in red blood cells, where it functions as the primary carrier of oxygen throughout the body. It has a quaternary structure composed of four subunits (two α and two β chains), each containing a heme group capable of binding oxygen. Its structure is particularly interesting because it exhibits allosteric, cooperative binding, meaning that the binding of one oxygen molecule increases the affinity for subsequent oxygen molecules, enabling efficient oxygen uptake in the lungs and release in tissues. Since its a quarternary structure of 4 subunits, the alpha and beta chains have two different respective amnino acid sequences. α: VLSPADKTNVKAAWGKVGAHAGEYGAEALERMFLSFPTTKTYFPHF DLSHGSAQVKGHGKKVADALTNAVAHVDDMPNALSALSDLHAHKLR VDPVNFKLLSHCLLVTLAAHLPAEFTPAVHASLDKFLASVSTVLTS KYR β: MVHLTPEEKSAVTALWGKVNVDEVGGEALGRLLVVYPWTQRFFESF GDLSTPDAVMGNPKVKAHGKKVLGAFSDGLAHLDNLKGTFATLSEL HCDKLHVDPENFRLLGNVLVCVLAHHFGKEFTPPVQAAYQKVVAGV ANALAHKYH The structure of Hemoglobin can be found on the RCSB Protein Data Bank (e.g. PDB ID: 1A3N). It was first solved in the 1960s–1970s, with modern structures having a resolution of around 2.7 Å, indicating good quality as lower resolution values correspond to more precise structural detail. The solved structure contains not only the protein itself but also additional molecules such as heme groups with Fe²⁺ ions, which are essential for oxygen binding, as well as ligands like oxygen or water molecules. Hemoglobin belongs to the globin protein family, which is characterised by predominantly α-helical structures and a conserved heme-binding pocket. I then uploaded Haemoglobins PDB file to pymol and rendered the following structure visualising it as a cartoon: And then the following two renderings are the same structure but visualised as a ribbon and Ball-and-stick representation:
Assignment A Part 1: The human SOD1 sequence was retrieved from UniProt (P00441). The ALS-associated A4V mutation was introduced by substituting alanine with valine at position 5 of the full-length sequence (equivalent to position 4 after removal of the initiator methionine).
Original sequence obtained from uniprot:
What are some components in the Phusion High-Fidelity PCR Master Mix and what is their purpose? The Phusion High-Fidelity PCR Master Mix contains several key components that enable accurate DNA amplification. It includes Phusion DNA polymerase, a high-fidelity enzyme with proofreading activity that reduces errors during DNA synthesis. The mix also contains dNTPs, which serve as the building blocks for new DNA strand formation. An optimized reaction buffer provides the correct pH and ionic conditions, including magnesium ions (Mg²⁺), which are essential cofactors for polymerase activity. Additionally, stabilizers and enhancers are included to improve enzyme performance and allow efficient amplification of difficult templates such as GC-rich regions. Some versions also contain a tracking dye, enabling direct loading of PCR products onto a gel for analysis.
Assignment Part 1: Intracellular Artificial Neural Networks (IANNs) What advantages do IANNs have over traditional genetic circuits, whose input/output behaviors are Boolean functions? Advantages include the fact that since IANNs connections between nodes can have varying continuous weightages this is very important when it comes to modelling the effect a gene has on a biological outcome as its very likely that they hold a non-linear relationship and so being able to model for this discrepancy is important as boolean functions assume two discrete states which isnt the case.
General Homework Questions Explain the main advantages of cell-free protein synthesis over traditional in vivo methods, specifically in terms of flexibility and control over experimental variables. Name at least two cases where cell-free expression is more beneficial than cell production. One advantage includes the fact that cell-free protein synthesis gives direct control over reaction composition which means we can precisely control the exact concenctrations of factors like DNA, amino acids and modified nucleotides. No cell membrane barrier means everything is immediately accessible and modifiable The speed at which you can cycle designs and tests is much faster thant that if you’d have to do the usual process of cloning then transform then grow You can precisely and exactly control the specific environments its in, setting the exact pH, temperature and also be able to eliminate toxic constraints. The bread and butter for cell free systems is the cell extract which includes the ribosomes that are the machinery for protein synthesis, tRNAs to deliver amino acids, translation factors and also the enzymes required for and involved in metabolism.
Final project Homework Waters Part 1 - Molecular Weight Using the website https://web.expasy.org/compute_pi/ it can be calculated that the molecular weight of the given eGFP Sequence: MVSKGEELFTG VVPILVELDG DVNGHKFSVS GEGEGDATYG KLTLKFICTT GKLPVPWPTL VTTLTYGVQC FSRYPDHMKQ HDFFKSAMPE GYVQERTIFF KDDGNYKTRA EVKFEGDTLV NRIELKGIDF KEDGNILGHK LEYNYNSHNV YIMADKQKNG IKVNFKIRHN IEDGSVQLAD HYQQNTPIGD GPVLLPDNHY LSTQSALSKD PNEKRDHMVL LEFVTAAGIT LGMDELYKLE HHHHHH
I propose developing an AI-assisted genomic analysis tool designed to predict the likelihood that a cell or tissue sample may become cancerous. The system would analyse DNA sequencing data to identify combinations of mutations, disrupted tumour suppressor genes, altered regulatory elements, and epigenetic markers associated with early stages of malignant transformation. By training machine-learning models on large cancer genomics datasets, the platform could detect patterns that indicate elevated cancer risk before visible symptoms or large tumours appear.
The goal of this tool would be to enable earlier detection and preventative intervention in oncology. Current cancer diagnostics often identify disease only after significant cellular transformation has occurred. A predictive genomic system could instead flag high-risk cellular states earlier, allowing clinicians to monitor patients more closely or apply targeted preventative treatments.
In the long term, such technology could improve cancer outcomes by shifting medicine from reactive treatment toward proactive risk prediction and prevention.
2. Governance and Policy Goals
Goal 1: Genetic Privacy and Discrimination Prevention
All patients and individuals involved must be guaranteed that their genomic data is kept private in order to prevent potential discrimination in external contexts such as employment or insurance. Genetic information is highly sensitive, and misuse could lead to unfair treatment if individuals are identified as having a higher risk of developing certain diseases.
In addition, individuals’ privacy and autonomy must be respected. Genomic data should not be used, shared, or analysed without the explicit consent and permission of the individual from whom the data was obtained. Clear policies for data ownership, consent, and access must therefore be established to ensure ethical use of the technology.
Goal 2: Democratise the Use of the Technology and Promote Equitable Access
Another important policy goal is to ensure that this technology is accessible globally rather than restricted to wealthy institutions or countries. Because predictive cancer diagnostics could significantly improve health outcomes and longevity, limiting access would risk widening existing healthcare inequalities.
Sub-goals therefore include building systems and distribution frameworks that allow countries across different economic levels to access the technology. This could involve international collaborations, subsidised programs, or integration with public healthcare systems to ensure that individuals from diverse socioeconomic backgrounds are able to benefit from the tool.
3. Governance actions in response
Governance Action 1
Purpose: Protecting the Privacy and Security of Genomic Data
The purpose of this governance action is to ensure that individuals’ genomic data is protected from misuse. As predictive genomic tools become more common, large amounts of highly sensitive biological data will be collected. Without appropriate safeguards, this information could potentially be accessed or exploited by governments, corporations, or other actors, particularly during periods of political instability. Establishing strong governance mechanisms is therefore necessary to maintain trust and prevent the misuse of biodata.
Design
One potential approach would be the creation of an independent international organisation responsible for managing and securing genomic health data. This organisation would operate independently from national governments and establish global standards for data storage, access control, and ethical use. Its role would be to ensure that genomic information collected for medical purposes cannot be repurposed for surveillance, discrimination, or political misuse.
Assumptions
This proposal assumes that the creation of a truly independent global organisation is feasible. In practice, international cooperation in healthcare governance can be difficult to achieve. It is also uncertain whether governments would be willing to allow an external body to manage sensitive health data generated within their jurisdictions.
Risks of Failure and “Success”
This system could fail if the organisation itself becomes corrupt or compromised, as it would ultimately rely on trust and strong oversight. Even if successful, ethical questions may arise regarding who governs the organisation, how decisions are made, and whether all countries would accept its authority.
Governance Action 2
Purpose: Promoting Equitable Access to Predictive Genomic Healthcare
The purpose of this governance action is to ensure that emerging genomic diagnostic technologies do not worsen existing healthcare inequalities. Currently, advanced medical technologies are often disproportionately accessible to wealthier populations, while less affluent communities face financial barriers to care. If predictive genomic tools are introduced without equitable distribution mechanisms, they could primarily benefit those with greater economic resources.
Design
To address this issue, governments could support the subsidisation and large-scale deployment of genomic diagnostic infrastructure. Public funding could be used to minimise the cost of implementing the technology and to distribute diagnostic systems according to population needs. By integrating such tools into public healthcare systems, access could be expanded beyond private healthcare markets.
Assumptions
This proposal assumes that governments would be willing to allocate sufficient funding to support large-scale implementation. It also assumes that the technology itself can be developed and deployed at a cost that makes widespread access feasible.
Risks of Failure and “Success”
This strategy could fail if funding is insufficient or if political priorities shift away from healthcare investment. Additionally, even if the technology becomes widely available, wealthier individuals may still gain preferential access through private healthcare systems, potentially limiting the intended benefits of equitable distribution.
Does the option:
Option 1: International Genomic Data Governance Organisation
Option 2: Government Funding for Equitable Access
Option 3: Regulation and Clinical Validation of AI Diagnostic Tools
Enhance Biosecurity
By preventing incidents
1
3
2
By helping respond
2
3
2
Foster Lab Safety
By preventing incidents
2
3
1
By helping respond
2
3
1
Protect the environment
By preventing incidents
2
3
2
By helping respond
2
3
2
Other considerations
Minimizing costs and burdens to stakeholders
3
2
2
Feasibility
3
2
2
Not impede research
2
2
2
Promote constructive applications
2
1
2
Policy Of Urgence
I consider ensuring the privacy and security of genomic data to be the highest priority among the governance policies considered. Genomic information is highly sensitive and uniquely identifiable, and large-scale genomic databases could potentially be misused if appropriate safeguards are not implemented. In extreme cases, access to detailed genetic information could be exploited not only against individuals, for example through discrimination or surveillance, but potentially at a broader scale in contexts such as targeted biological threats.
For this reason, it is essential that large volumes of genomic data are stored securely and governed by strict access controls. Robust governance frameworks should ensure that such data are used exclusively for legitimate medical research and healthcare purposes. Protecting genomic privacy is therefore critical for maintaining public trust and ensuring that advances in predictive genomic technologies contribute positively to society.
Week 2 HW: DNA Read,Write and Edit
Part 1: Benchling and In-silico Gel Art
I followed the instructions and made a free account using benchling.com - a website used to design experiments, manage biological data, and collaborate on research where in our case for the purpose of this homework we will be using it edit DNA plasmid structures with different restriction enzyme digests.
The next step was to import Lambda DNA into the website as a new project, this DNA would act as the template for which we would perform restriction site digests with different enzymes.
I was a little confused as to why the link for Lambda DNA redirected me to an ordering page and I tried searching through it to see if it had a download link but couldnt find anything so I then directed my search to google and found it on the following page:
Next I uploaded the .fasta file I was able to downloaded as a linear topology, in metadata there was an error to identify what schema was being used which was confusing as I uploaded itas a DNA sequence already.
Ignoring that error, I moved ontto simulation of restriction Enzyme Digestion for the following enzymes:
EcoRI
HindIII
BamHI
KpnI
EcoRV
SacI
SalI
After performing the digest with all the above enzymes I recieved the following results:
And then to add a bit of an artistic approach to the gel lanes i tried to make a mirror image stair case which ended up looking like this:
PART 3: DNA Design Challenge
Protein chosen: Retatrutide. An emerging game-changer in obesity pharmacotherapy, retatrutide represents the forefront of a broader shift toward the normalised use of peptide-based therapeutics, not only for treating metabolic diseases but also for expanding applications in cosmetic and lifestyle medicine.
Biological assembly of Retratrutide Structure:
As retatrutide is a synthetic peptide drug rather than a naturally occurring protein, it is not listed as a canonical entry in UniProt. Therefore, we instead reference one of the endogenous hormones it is derived from, GLP-1.
GLP-1 amino acid sequence:
HAEGTFTSDVSSYLEGQAAKEFIAWLVKGRG
Using a reverse translation the following possible DNA sequence is obtained:
Codon optimisation is necessary because the genetic code is degenerate, meaning that most amino acids can be encoded by multiple different codons. Although these codons specify the same amino acid, they are not used equally across all organisms. Different species show codon bias, where certain synonymous codons are preferred over others. This preference is largely linked to the availability of corresponding tRNAs in the host organism, which can strongly influence the efficiency and accuracy of translation.
In this project, codon optimisation was used to adapt the DNA sequence so that it matches the codon usage preferences of the chosen expression host. This helps to improve protein expression by reducing the likelihood of slow or inefficient translation.
In our case, E. coli was selected as the host organism because of its rapid growth rate, ease of transformation, low cost, and widespread use in recombinant protein expression. These features make it a practical and efficient system for producing the target peptide.
And so following codon optimization suited for E. coli the following is obtained:
part IV Preparing my first trial twist DNA Synthesis Order
week-03 HW: Lab-automation
Lecture Overview – Lab Automation and Biosensors
Introduction
This week’s lecture focused on lab automation and biosensors, two technologies that are transforming modern life sciences research. Together, they enable scientists to run experiments faster, more accurately, and at a much larger scale than traditional manual laboratory techniques.
Automation reduces human error and increases throughput, while biosensors allow researchers to observe biological changes in real time. These tools are particularly valuable in areas such as drug discovery, synthetic biology, and molecular diagnostics.
Lab Automation
Why Automation Matters
Traditional laboratory experiments often involve manual pipetting, where researchers transfer very small volumes of liquid between wells or tubes. While precise, manual work is:
Time-consuming
Physically repetitive
Prone to human error
Difficult to scale for large experiments
Lab automation addresses these limitations by using robotic systems to handle liquid transfers and experimental workflows.
Key Advantages
Speed – Automated systems can run experiments continuously and much faster than manual methods.
Accuracy and Precision – Robotic pipettes can consistently transfer extremely small volumes with high reproducibility.
High Throughput – Multiple experimental conditions can be tested simultaneously.
Reproducibility – Automated protocols reduce variability between experiments and researchers.
These capabilities are especially important in drug discovery and biological screening, where thousands of chemical compounds or genetic variants may need to be tested against biological targets.
Robotic Pipetting and Parallel Testing
One major application of automation is robotic pipetting platforms. These systems can:
Dispense microlitre or nanolitre volumes of liquids
Follow programmable experimental protocols
Operate on multi-well plates (such as 96-well or 384-well plates)
This enables parallel experimentation, where many experimental conditions are tested simultaneously in a compact layout.
For example, researchers can:
Test multiple drug candidates against a biological target
Compare different genetic modifications
Measure biological responses across a range of concentrations
Because each well contains a different experimental condition, researchers can gather large datasets from a single automated run.
One platform discussed in the lecture is Opentrons, an open-source robotic pipetting system. It allows researchers and students to:
Program laboratory protocols using code
Automate repetitive liquid-handling tasks
Prototype experimental workflows quickly
This system will also be used in the Art & Design homework, where we will get to explore creative and experimental uses of automated pipetting.
Biosensors
What Are Biosensors?
Biosensors are biological or bioengineered systems that detect and report changes inside living cells or biological environments.
They provide real-time feedback about biological processes such as:
Gene expression (RNA production)
DNA activity
Protein interactions
Metabolic changes
Cellular stress or environmental signals
By translating biological activity into a detectable signal, biosensors allow scientists to monitor what is happening inside cells during experiments.
Fluorescent Proteins and Visual Signals
One of the most widely used biosensing methods relies on fluorescent proteins.
Scientists have discovered and engineered proteins that emit visible light when exposed to specific wavelengths of ultraviolet (UV) or blue light. These proteins glow in different colours, including:
Green
Red
Yellow
Cyan
By linking fluorescent proteins to specific biological mechanisms, researchers can create systems where a biological event produces a visible signal.
Example
A genetic circuit might be designed so that:
When a particular gene is activated → a fluorescent protein is produced
The cell begins to glow under UV light
The colour or brightness indicates the level of activity
This allows researchers to visually track biological changes during experiments without destroying the sample.
Fluorescent biosensors are widely used in:
Cell biology
Synthetic biology
Drug testing
Biomedical research
Automation Systems Covered in the Lecture
1. Microfluidics (Electrowetting Systems)
Microfluidic systems manipulate very small volumes of liquid (often nanolitres or picolitres) within tiny channels or droplets.
An electrowetting system moves droplets across a surface using controlled electric fields. By changing electrical signals, the system can:
Move droplets
Merge droplets
Split droplets
Mix reagents
Advantages
Extremely small reagent volumes
Rapid reaction times
Highly parallel experimentation
Compact experimental platforms
Microfluidics is often used in high-throughput screening and diagnostic devices.
2. Opentrons
Opentrons is a robotic liquid-handling platform designed for laboratory automation.
Key features include:
Programmable pipetting protocols
Compatibility with standard labware (multi-well plates, tubes, etc.)
Open-source software and hardware design
Accessible pricing compared with industrial lab robots
Because it is programmable, experiments can be designed as automated workflows, allowing researchers to run complex procedures with minimal manual intervention.
3. Nebula
Nebula is another automation platform designed for distributed and cloud-connected laboratory experimentation.
These types of systems enable:
Remote experiment control
Automated experimental pipelines
Integration with digital lab notebooks and data analysis systems
By combining automation with networked infrastructure, platforms like Nebula support scalable experimentation and collaborative research environments.
Key Takeaways
Lab automation dramatically increases experimental speed, precision, and scalability.
Robotic pipetting systems allow researchers to run many experiments simultaneously in multi-well plates.
Biosensors, particularly fluorescent protein systems, provide real-time insight into biological processes.
Technologies such as microfluidics, Opentrons, and Nebula represent different approaches to automating laboratory workflows.
Together, these technologies are reshaping how modern biological research is conducted, enabling larger experiments, faster discoveries, and more reproducible science.
Homework: Opentrons Design Challenge
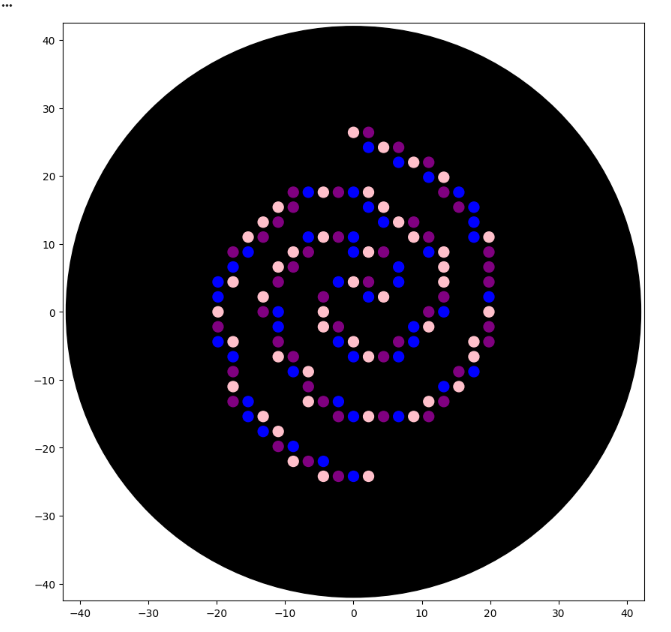
The main task for this homework is to design an art using the opentrons automation art website and then implement this into a python program that the opentrons pipetting machine can then use to pipette it onto an agar plate - the design will then glow different colours (Blue,pink and purple for LifeFabs assigned colours) under UV light to show case a living E. Coli artwork of our design.
Step 1: Making a design using the automartion art designer website by Ronan
Step 2: Next step was to implement these coordinates into the python program, I struggled a bit at the start but with the help of claude code I found my way around issues I ran into
Heres my code for reference:
fromopentronsimporttypesmetadata={'Vithushan':'','OpentronArtDesign':'','description':'','source':'HTGAA 2026 Opentrons Lab','apiLevel':'2.20'}################################################################################# Robot deck setup constants - don't change these##############################################################################TIP_RACK_DECK_SLOT=9COLORS_DECK_SLOT=6AGAR_DECK_SLOT=5PIPETTE_STARTING_TIP_WELL='A1'well_colors={'A1':'Pink','B1':'Purple','C1':'Blue'}# mKate2 TF design coordinates (x, y) in mmmkate2_tf_points=[(0,26.4),(2.2,26.4),(2.2,24.2),(4.4,24.2),(6.6,24.2),(6.6,22),(8.8,22),(11,22),(11,19.8),(13.2,19.8),(-8.8,17.6),(-6.6,17.6),(-4.4,17.6),(-2.2,17.6),(0,17.6),(2.2,17.6),(13.2,17.6),(15.4,17.6),(-11,15.4),(-8.8,15.4),(2.2,15.4),(4.4,15.4),(15.4,15.4),(17.6,15.4),(-13.2,13.2),(-11,13.2),(4.4,13.2),(6.6,13.2),(8.8,13.2),(17.6,13.2),(-15.4,11),(-13.2,11),(-6.6,11),(-4.4,11),(-2.2,11),(0,11),(8.8,11),(11,11),(17.6,11),(19.8,11),(-17.6,8.8),(-15.4,8.8),(-8.8,8.8),(-6.6,8.8),(0,8.8),(2.2,8.8),(4.4,8.8),(11,8.8),(13.2,8.8),(19.8,8.8),(-17.6,6.6),(-11,6.6),(-8.8,6.6),(6.6,6.6),(13.2,6.6),(19.8,6.6),(-19.8,4.4),(-17.6,4.4),(-11,4.4),(-2.2,4.4),(0,4.4),(2.2,4.4),(6.6,4.4),(13.2,4.4),(19.8,4.4),(-19.8,2.2),(-13.2,2.2),(-4.4,2.2),(2.2,2.2),(4.4,2.2),(13.2,2.2),(19.8,2.2),(-19.8,0),(-13.2,0),(-11,0),(-4.4,0),(11,0),(13.2,0),(19.8,0),(-19.8,-2.2),(-11,-2.2),(-4.4,-2.2),(-2.2,-2.2),(8.8,-2.2),(11,-2.2),(19.8,-2.2),(-19.8,-4.4),(-17.6,-4.4),(-11,-4.4),(-2.2,-4.4),(0,-4.4),(6.6,-4.4),(8.8,-4.4),(17.6,-4.4),(19.8,-4.4),(-17.6,-6.6),(-11,-6.6),(-8.8,-6.6),(0,-6.6),(2.2,-6.6),(4.4,-6.6),(6.6,-6.6),(17.6,-6.6),(-17.6,-8.8),(-8.8,-8.8),(-6.6,-8.8),(15.4,-8.8),(17.6,-8.8),(-17.6,-11),(-6.6,-11),(13.2,-11),(15.4,-11),(-17.6,-13.2),(-15.4,-13.2),(-6.6,-13.2),(-4.4,-13.2),(-2.2,-13.2),(11,-13.2),(13.2,-13.2),(-15.4,-15.4),(-13.2,-15.4),(-2.2,-15.4),(0,-15.4),(2.2,-15.4),(4.4,-15.4),(6.6,-15.4),(8.8,-15.4),(11,-15.4),(-13.2,-17.6),(-11,-17.6),(-11,-19.8),(-8.8,-19.8),(-8.8,-22),(-6.6,-22),(-4.4,-22),(-4.4,-24.2),(-2.2,-24.2),(0,-24.2),(2.2,-24.2)]# Assign each point a colour based on its index (cycling through A1=Pink, B1=Purple, C1=Blue)defassign_color(index):colors=['Pink','Purple','Blue']returncolors[index%3]defrun(protocol):################################################################################# Load labware, modules and pipettes############################################################################### Tipstips_20ul=protocol.load_labware('opentrons_96_tiprack_20ul',TIP_RACK_DECK_SLOT,'Opentrons 20uL Tips')# Pipettespipette_20ul=protocol.load_instrument("p20_single_gen2","right",[tips_20ul])# Modulestemperature_module=protocol.load_module('temperature module gen2',COLORS_DECK_SLOT)# Temperature Module Platetemperature_plate=temperature_module.load_labware('opentrons_96_aluminumblock_generic_pcr_strip_200ul','Cold Plate')color_plate=temperature_plate# Agar Plateagar_plate=protocol.load_labware('htgaa_agar_plate',AGAR_DECK_SLOT,'Agar Plate')center_location=agar_plate['A1'].top()pipette_20ul.starting_tip=tips_20ul.well(PIPETTE_STARTING_TIP_WELL)################################################################################# Patterning##############################################################################deflocation_of_color(color_string):forwell,colorinwell_colors.items():ifcolor.lower()==color_string.lower():returncolor_plate[well]raiseValueError(f"No well found with color {color_string}")defdispense_and_detach(pipette,volume,location):assertisinstance(volume,(int,float))above_location=location.move(types.Point(z=location.point.z+5))pipette.move_to(above_location)pipette.dispense(volume,location)pipette.move_to(above_location)###### mKate2 TF design — pink, purple, blue cycling pattern###current_color=Nonefori,(x,y)inenumerate(mkate2_tf_points):point_color=assign_color(i)# Pick up a new tip when the colour changes (or at the very first point)ifpoint_color!=current_color:ifpipette_20ul.has_tip:pipette_20ul.drop_tip()pipette_20ul.pick_up_tip()pipette_20ul.aspirate(2,location_of_color(point_color))current_color=point_color# Calculate the absolute position on the agar platedispense_location=center_location.move(types.Point(x=x,y=y,z=0))dispense_and_detach(pipette_20ul,1,dispense_location)# Drop the final tipifpipette_20ul.has_tip:pipette_20ul.drop_tip()
Week 4 HW: Principles and Practices
Part A:
Question 1: How many molecules of amino acids do you take with a piece of 500 grams of meat? (on average an amino acid is ~100 Daltons)
To understand this its important to know that elements on the scale of amino acids masses are measured in units of Daltons, defined as the mass of 1/12th of a carbon atom or 1x10^-24 grams.
So in 500 grams of meat where following that there is 26 grams of protein per 100g of meat there is then 130 grams of protein.
This means there is 130 x 1.67 x 10^-24 daltons of protein (hence amino acids).
And since 1 amino acid is on average 100 Daltons we just have to divide by 100 to get the number of amino acid molecules present which ends up to be:
2.171 x 10^24 molecules of amino acids
Question 2: Why do humans eat beef but do not become a cow, eat fish but do not become fish?
This is because we have enzymes that cleave the proteins we eat into their individual amino acids, these amino acids are then used to build the human into its own unique structures and uses. The protein of each animal consumed isnt "directly transferred" into the human but its broken down into its amino acid constituents, carbohydrate sources etc.. which are then used as the building material. What decides how an organism looks or functions is to do with their genetic coding in their DNA, So the DNA acts as the building instructions and food/protein/amino acids consumed act as the raw mateerials used to carry out these nstructions.
Question 3: Why are there only 20 natural amino acids?
I Initially thought this was because a limitation in the triplet codon but then i remembered thats 4C3 which is 64 different combinations and they all only end up giving 20 different amino acids which is why they are degenerate. So I assume the reason theres only 20 amino acids in nature is because of a natural selection process that has occured throughout evolution as a combination of what amino acids provided useful and allowed for better survival.
Question 4 : Can you make other non-natural amino acids? Design some new amino acids
Yes this is possible - non-natural amino acids better known as noncanonical amino acids are those containing side chains beyond the side chains that DNA can code for.
By modifying the side chains of the basic glycine-derived α-amino acid structure, more complex functional groups can be introduced to design non-natural amino acids. These include D-amino acids, β-amino acids, and amino acids with modified side chains. Such modifications are crucial in biotechnology and pharmaceuticals for applications including the development of more stable therapeutic peptides, protein engineering, and the study of protein structure and function.
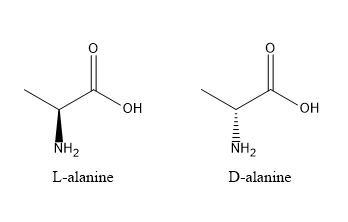
Noncanonical amino acids are commonly used to improve the stability of therapeutic peptides by reducing their susceptibility to proteolytic degradation. Proteases recognise specific sequence motifs and backbone geometries, so introducing a non-natural amino acid near a known cleavage site can disrupt this recognition. One effective strategy is the incorporation of D-amino acids, which have the same side chain but opposite stereochemistry to naturally occurring L-amino acids, making them poorly recognised by most proteases. For example, glucagon-like peptide-1 (GLP-1) is rapidly cleaved by DPP-4 at the Ala8–Glu9 bond. Replacing the alanine at position 8 with a D-amino acid such as D-alanine alters the local stereochemistry, reducing enzymatic cleavage and improving peptide half-life while maintaining biological function.
Question 5 : Where did amino acids come from before enzymes that make them, and before life started?
There are several proposed explanations for how amino acids formed before the origin of life. One key idea is that amino acids were produced through abiotic chemical reactions on early Earth. The early atmosphere is thought to have contained simple gases such as methane (CH₄), ammonia (NH₃), and water vapour (H₂O). With energy sources such as lightning and ultraviolet radiation, these gases could undergo chemical reactions to form organic molecules, including amino acids. This was demonstrated by the Miller-Urey experiment, which successfully produced amino acids under simulated early Earth conditions.
In addition, evidence from extraterrestrial sources supports this idea. The Murchison meteorite, which fell in 1969, was found to contain a wide range of organic molecules, including amino acids. This shows that amino acids can form in space and may have been delivered to early Earth via meteorites, contributing to the prebiotic chemical pool from which life eventually emerged.
Question 6: If you make an α-helix using D-amino acids, what handedness (right or left) would you expect?
Amino acids are chiral and exhibit stereospecificity, meaning their three-dimensional arrangement determines how they interact and fold. Natural proteins are composed of L-amino acids, which form right-handed α-helices due to the specific geometry of the peptide backbone and side chain orientation. If D-amino acids are used instead, the stereochemistry is inverted, resulting in a mirror-image structure. Consequently, D-amino acids form left-handed α-helices, as the backbone geometry and hydrogen bonding pattern are reversed.
Question 7: Can you discover additional helices in proteins?
The α-helix is one of the most common protein secondary structures, characterised by a right-handed coil with approximately 3.6 amino acid residues per turn. It has a pitch of 5.4 Å and a rise of 1.5 Å per residue, and is stabilised by hydrogen bonds between the carbonyl oxygen of residue i and the amide hydrogen of residue i+4.
In principle, other helical structures can exist by altering the number of residues per turn or the hydrogen bonding pattern, such as the 3₁₀ helix (i → i+3) or π-helix (i → i+5), and even left-handed helices under certain conditions. However, the formation of helices in nature is constrained by backbone geometry and energetic stability. Only specific hydrogen bonding patterns and torsional angles are energetically favourable, meaning that while alternative helices are theoretically possible, only a limited number form stable and commonly observed structures in proteins.
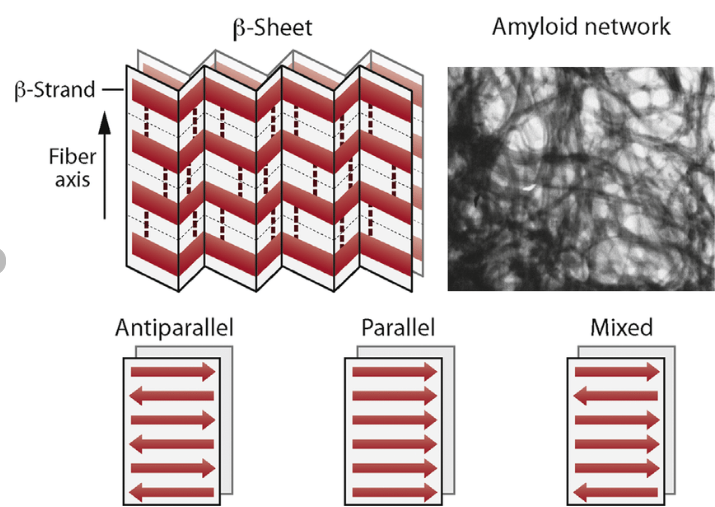
Question 9: Why do β-sheets tend to aggregate? What is the driving force for β-sheet aggregation?
The main idea is that the forces are more stabilised when these sheets aggregate, stacking and slotting into one another. The β-sheets tend to aggregate because their extended backbone structure allows for extensive intermolecular hydrogen bonding between neighbouring strands or sheets. In addition, hydrophobic side chains can align and interact between sheets, further stabilising aggregation. This combination of backbone hydrogen bonding and hydrophobic interactions leads to the formation of highly stable, stacked β-sheet structures, such as those found in amyloid fibrils.
Question 10: Why do amyloid diseases form B-sheets?
Amyloid diseases arise when normally soluble proteins misfold into structures rich in β-sheets. These β-sheets form highly stable aggregates due to extensive intermolecular hydrogen bonding and hydrophobic interactions. The sheets stack to form insoluble fibrils with a characteristic cross-β structure. These fibrils accumulate in tissues, where they disrupt cellular function and become toxic, ultimately leading to tissue damage and disease.
Part B: Protein Analysis and Visualization
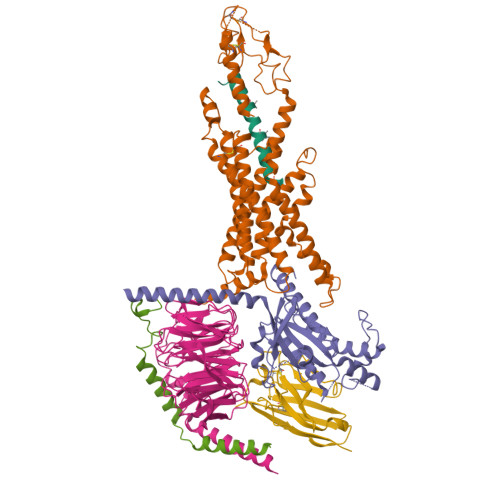
Hemoglobin is a well-studied and highly important protein found in red blood cells, where it functions as the primary carrier of oxygen throughout the body. It has a quaternary structure composed of four subunits (two α and two β chains), each containing a heme group capable of binding oxygen. Its structure is particularly interesting because it exhibits allosteric, cooperative binding, meaning that the binding of one oxygen molecule increases the affinity for subsequent oxygen molecules, enabling efficient oxygen uptake in the lungs and release in tissues.
Since its a quarternary structure of 4 subunits, the alpha and beta chains have two different respective amnino acid sequences. α: VLSPADKTNVKAAWGKVGAHAGEYGAEALERMFLSFPTTKTYFPHF
DLSHGSAQVKGHGKKVADALTNAVAHVDDMPNALSALSDLHAHKLR
VDPVNFKLLSHCLLVTLAAHLPAEFTPAVHASLDKFLASVSTVLTS
KYR
β: MVHLTPEEKSAVTALWGKVNVDEVGGEALGRLLVVYPWTQRFFESF
GDLSTPDAVMGNPKVKAHGKKVLGAFSDGLAHLDNLKGTFATLSEL
HCDKLHVDPENFRLLGNVLVCVLAHHFGKEFTPPVQAAYQKVVAGV
ANALAHKYH
The structure of Hemoglobin can be found on the RCSB Protein Data Bank (e.g. PDB ID: 1A3N). It was first solved in the 1960s–1970s, with modern structures having a resolution of around 2.7 Å, indicating good quality as lower resolution values correspond to more precise structural detail. The solved structure contains not only the protein itself but also additional molecules such as heme groups with Fe²⁺ ions, which are essential for oxygen binding, as well as ligands like oxygen or water molecules. Hemoglobin belongs to the globin protein family, which is characterised by predominantly α-helical structures and a conserved heme-binding pocket.
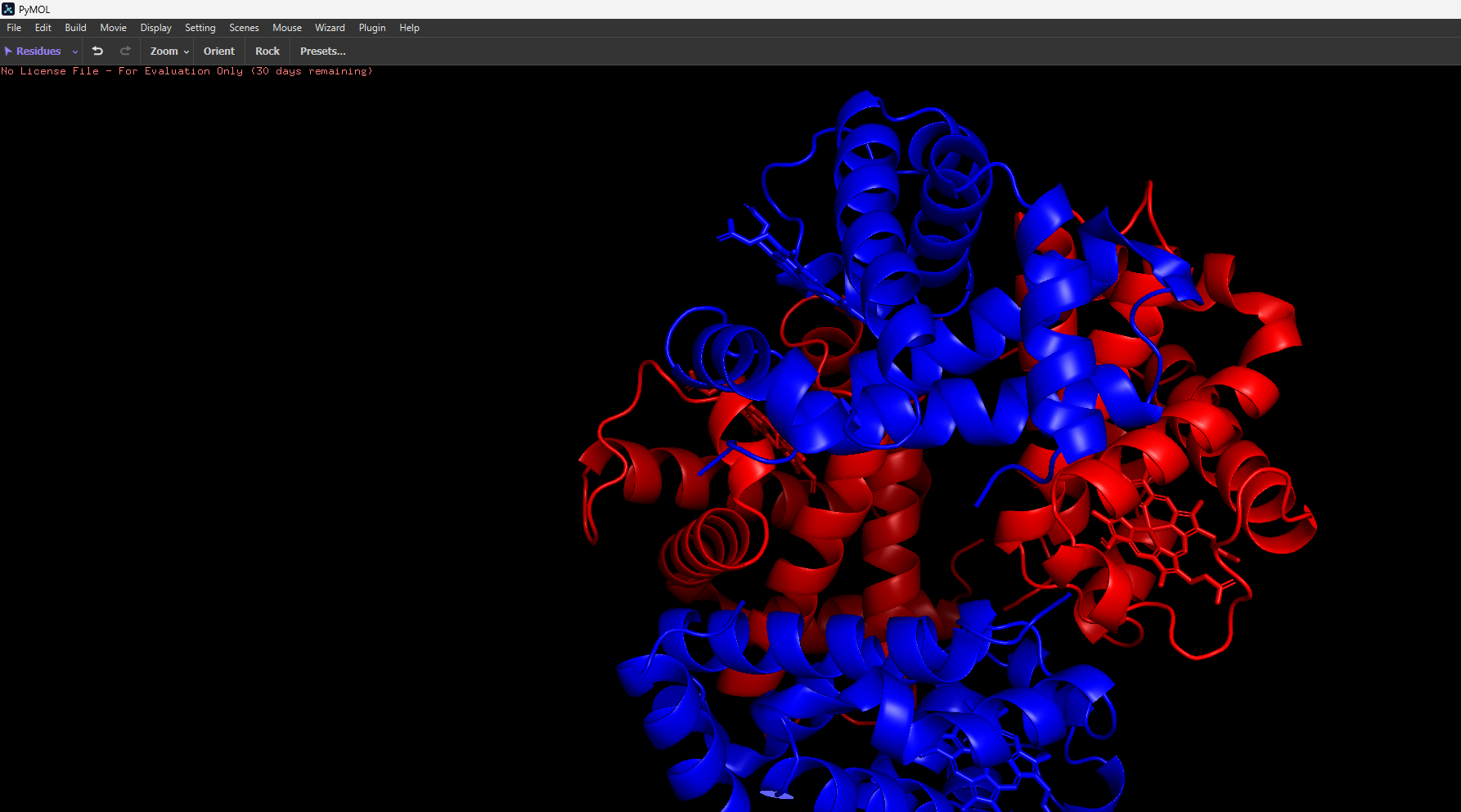
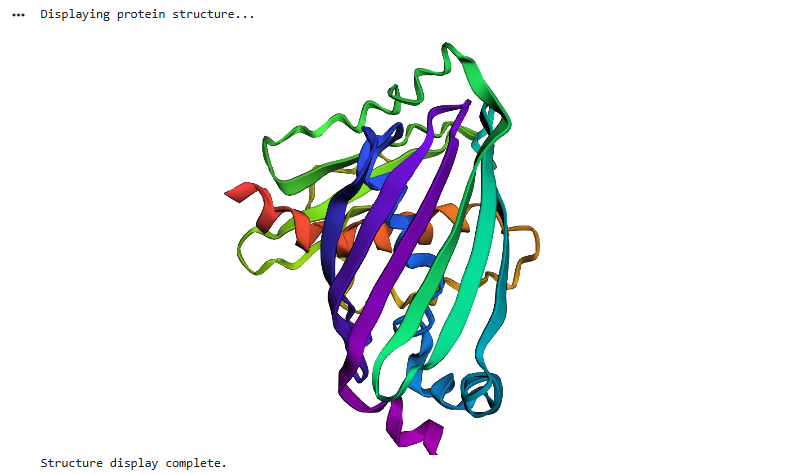
I then uploaded Haemoglobins PDB file to pymol and rendered the following structure visualising it as a cartoon:
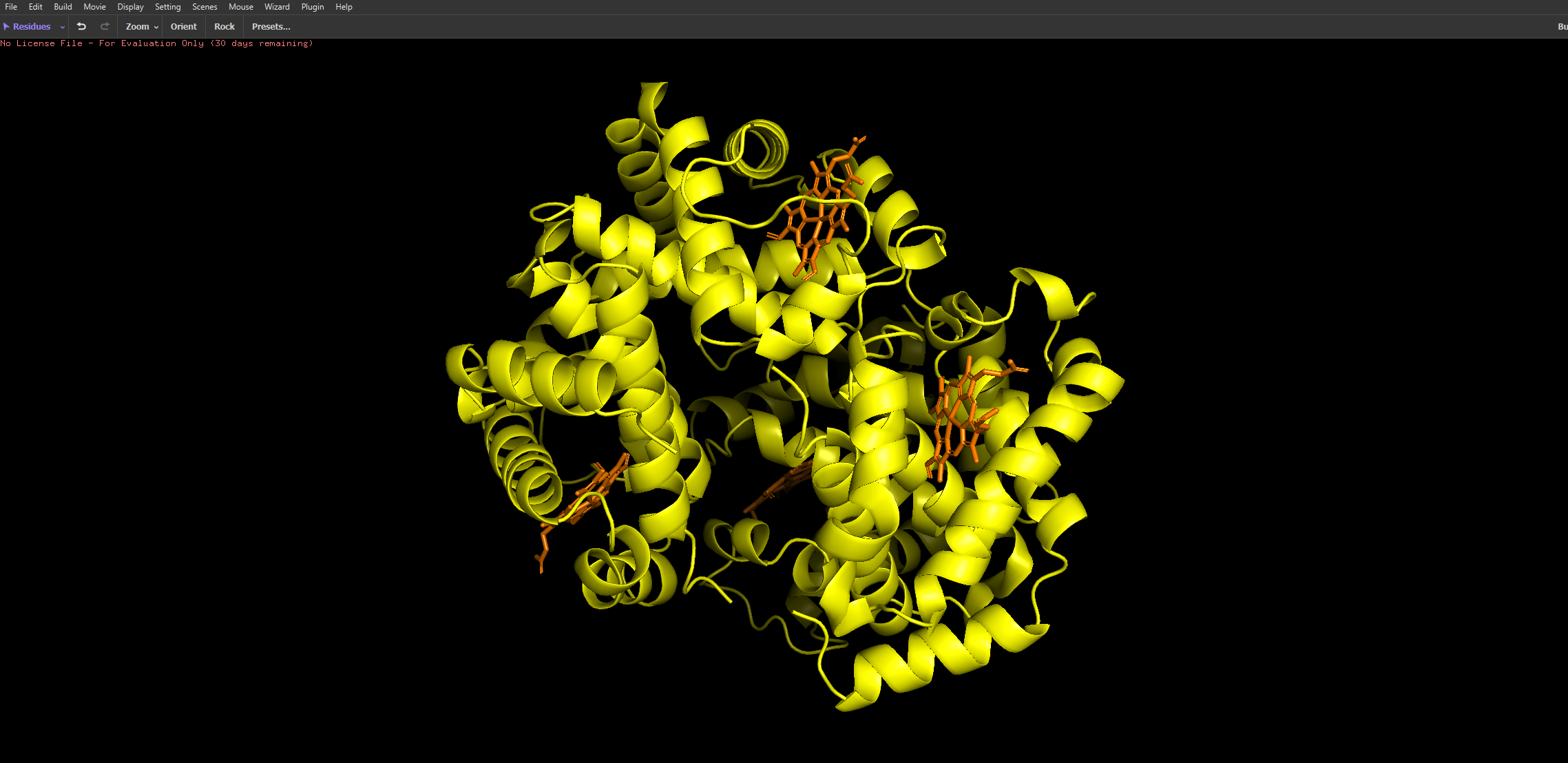
And then the following two renderings are the same structure but visualised as a ribbon and Ball-and-stick representation:
The ball and stick representation is particularly interesting as it allows us to to see the porphyrin rings that hold the iron group where the oxygen binds to one by one and changes the conformation of the entire structure hence the affinity due to its allosteric nature
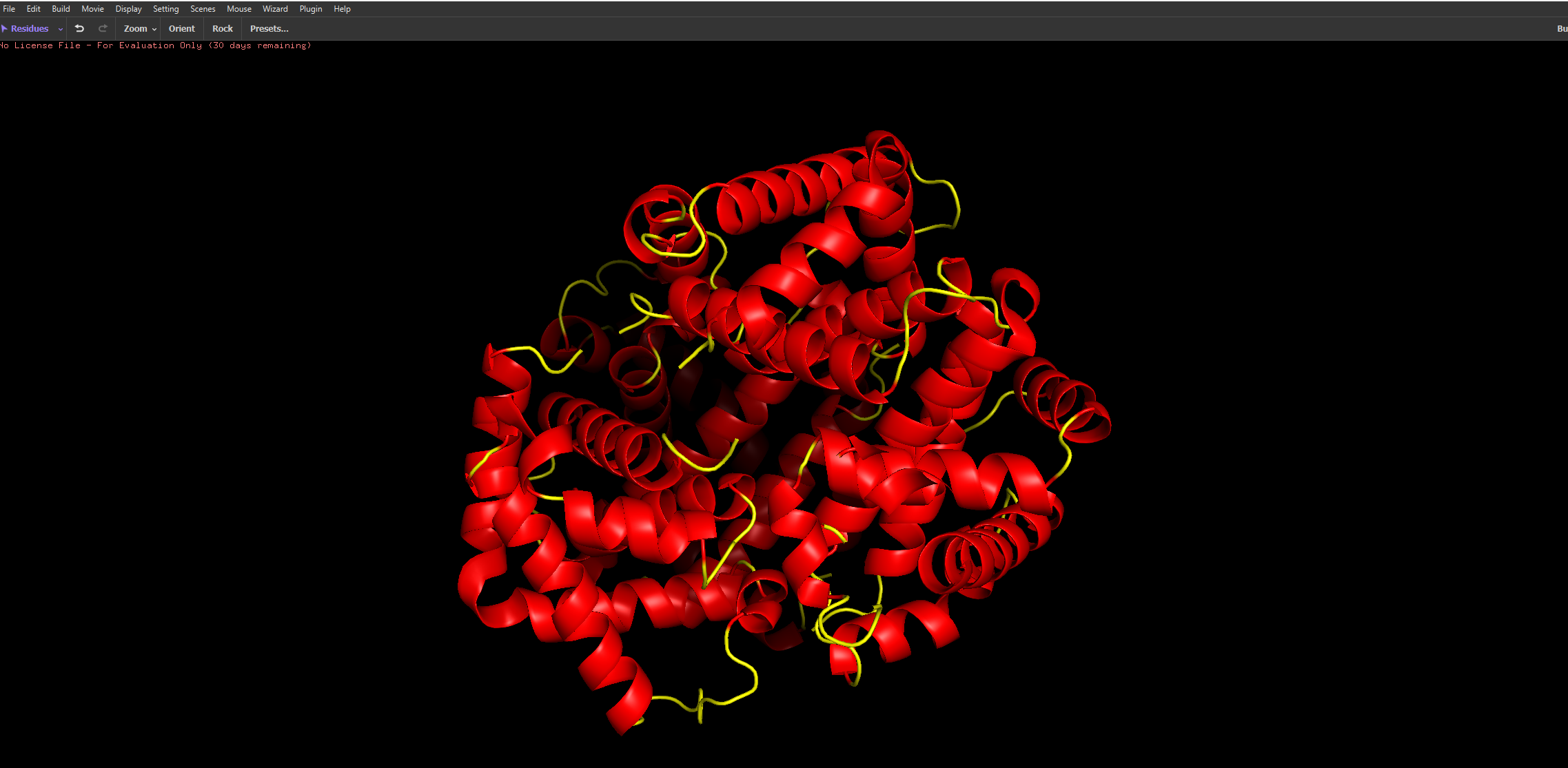
After colouring the protein by secondary structure, it is clear that haemoglobin is composed almost entirely of α-helices, with very little to no β-sheet content. These helices are connected by short loop (coil) regions, forming a compact globular structure known as the globin fold. This predominance of α-helical structure is characteristic of oxygen-binding proteins and supports the formation of a stable hydrophobic pocket for the heme group.
When coloured by residue type, with cyan representing hydrophilic residues and orange/red representing hydrophobic residues, it is evident that hydrophilic residues are predominantly located on the exterior of the protein, where they interact with the aqueous environment of the blood. This enhances the protein’s solubility, which is essential for its role in oxygen transport. In contrast, hydrophobic residues are largely buried within the core of the protein, where they pack closely together between α-helices, forming stabilising hydrophobic interactions. This arrangement contributes to the overall structural stability of haemoglobin and helps maintain the integrity of the heme-binding pocket.
Part C: Using ML-Based Protein Design Tools
I Chose to go with the GFP protein amino acid sequence to run the following computational analysises.
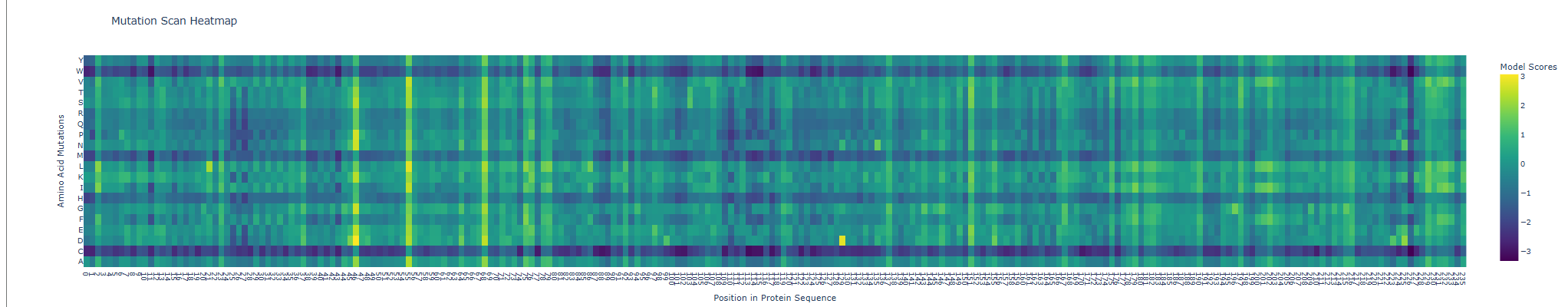
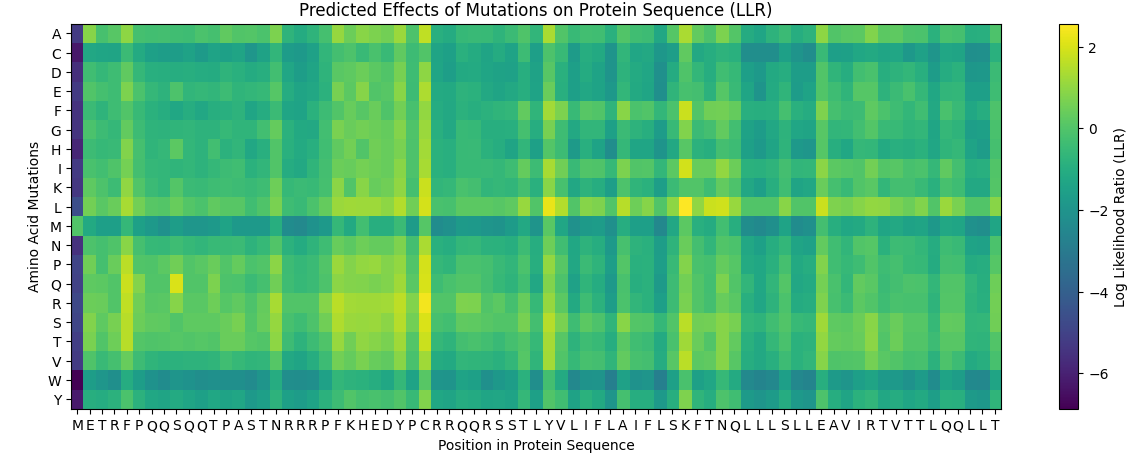
When seeing its mutation scan the following heatmap was produced
For the next comparison with experimental values I first exported my results from the deep scan heatmap produced and extracted the scoring values onto a .csv spreadsheet.
Then, the heatmap output was converted into a more interpretable format by constructing mutation labels in the form wild-type residue + position + mutated residue (e.g. Y66A). These mutations were paired with their corresponding ESM-2 likelihood scores.
To evaluate the model’s predictions, these scores were compared against experimental fluorescence measurements obtained from deep mutational scanning studies of GFP, where fluorescence acts as a proxy for protein function. In theory, a positive correlation is expected, as mutations predicted to be more compatible (higher ESM score) should better preserve protein structure and therefore maintain fluorescence.
This trend was observed for several mutations in the dataset. For example, Y66A showed both a low ESM score and very low fluorescence, which is biologically consistent because Tyr66 is part of the chromophore responsible for GFP fluorescence. However, not all mutations followed this trend, and some discrepancies were observed.
These inconsistencies arise because ESM-2 predicts sequence likelihood, not specific functional outputs such as fluorescence. While fluorescence depends on correct folding and chromophore formation, some mutations may preserve global structure (and thus appear favorable to the model) but still disrupt the precise chemical environment required for light emission. Additionally, the analysis was performed on a limited subset of mutations due to time constraints; a more robust evaluation would involve comparing all available variants (~50,000) using a statistical metric such as Spearman’s rank correlation coefficient.
Overall, the results suggest that the language model captures broad structural constraints of the protein, particularly at functionally critical residues, but does not fully account for the specific biochemical mechanisms underlying fluorescence.
Table: Comparison of ESM-2 Mutational Scores with Experimental GFP Fluorescence
mutation
position
wt
mut
score
Experimental fluorescence
Y66A
66
Y
A
-0.123864412
0.02
G67A
67
G
A
0.776538849
0.10
V163A
163
V
A
-0.610266924
0.90
Y66F
66
Y
F
-0.521710396
0.85
L201P
201
L
P
-0.147899389
0.01
L42R
42
L
R
-0.721465588
0.15
F99S
99
F
S
0.004160166
0.05
S65T
65
S
T
-0.177279234
1.20
I171V
171
I
V
0.522115469
1.05
T203Y
203
T
Y
0.470024824
1.30
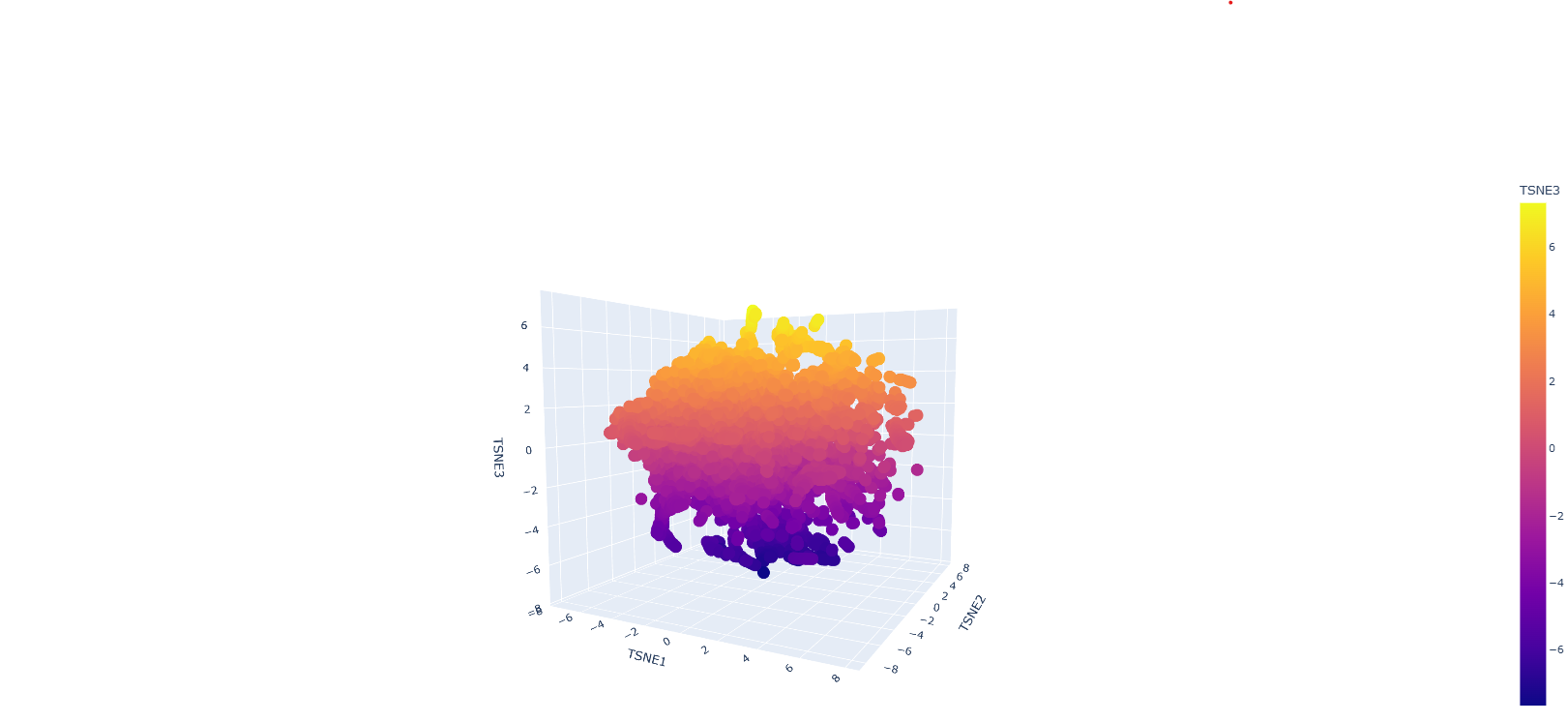
Protein sequences were embedded using a protein language model and visualised in reduced dimensional space using t-SNE. The resulting plot shows a largely continuous distribution of proteins, with local neighbourhoods representing proteins with similar sequence features. Although distinct clusters are not sharply separated, nearby points in the embedding space are expected to share structural or functional similarities, indicating that the model captures meaningful biological relationships.
The GFP protein would be positioned within a region corresponding to proteins with similar structural characteristics, particularly beta-barrel proteins. This is because GFP has a well-defined beta-barrel fold and a conserved chromophore-forming region, both of which influence its sequence embedding.
Overall, the embedding space demonstrates that protein language models can organise proteins based on underlying biological similarity, although the separation between groups may not always be clearly defined due to the continuous nature of protein sequence space and limitations of dimensionality reduction techniques.
Protein sequences were embedded using a protein language model and visualised in reduced dimensional space using t-SNE. The resulting plot shows a largely continuous distribution of proteins, with local neighbourhoods representing proteins with similar sequence features. Although distinct clusters are not sharply separated, nearby points in the embedding space are expected to share structural or functional similarities, indicating that the model captures meaningful biological relationships.
The GFP protein would be positioned within a region corresponding to proteins with similar structural characteristics, particularly beta-barrel proteins. This is because GFP has a well-defined beta-barrel fold and a conserved chromophore-forming region, both of which influence its sequence embedding.
Overall, the embedding space demonstrates that protein language models can organise proteins based on underlying biological similarity, although the separation between groups may not always be clearly defined due to the continuous nature of protein sequence space and limitations of dimensionality reduction techniques.
e GFP structure predicted using ESMFold closely matches the experimentally known structure, displaying the characteristic β-barrel fold composed of multiple β-strands forming a cylindrical shape. This indicates that the model accurately captures the global topology and structural organisation of the protein.
When small mutations were introduced, the overall structure remained largely unchanged, demonstrating that the protein fold is resilient to minor sequence perturbations. However, mutations at key residues, such as those involved in chromophore formation, can significantly affect function without drastically altering the structure.
In contrast, larger sequence modifications led to noticeable structural disruption, with loss of the β-barrel integrity and reduced folding stability. This suggests that while protein structures are robust to small mutations, they are sensitive to larger changes that interfere with critical folding interactions.
Overall, these results show that protein structure is more conserved than sequence, and that ESMFold can reliably predict structural consequences of mutations.
For the final part, inverse folding was performed using the GFP PDB backbone as input to ProteinMPNN in order to generate a sequence predicted to be compatible with the original structure. The resulting sequence was substantially different from the native GFP amino acid sequence, showing that multiple sequences may be compatible with a similar backbone geometry. However, the designed sequence did not retain the characteristic Ser65–Tyr66–Gly67 chromophore-forming motif required for GFP fluorescence, suggesting that structural compatibility does not necessarily preserve the original function. This highlights an important distinction between designing a sequence that can adopt a fold and preserving the precise residues needed for biochemical activity. The designed sequence was then intended to be passed through ESMFold to test whether it would refold into a structure resembling the original GFP beta-barrel.
Part D. Group Brainstorm on Bacteriophage Engineering
No group, just myself as I hadn’t realised our node was required this until marking day:((
I propose to computationally engineer the MS2 L lysis protein with a primary focus on increased stability and a secondary focus on improving lysis effectiveness through altered host interaction. This is a realistic starting point because the L protein is central to host-cell lysis, and to my current knowledge and expertise I would specifically frame stability as the easiest engineering target while toxicity/function is a bit more ambitious on my own. HTGAA’s Week 4 materials also explicitly position this assignment around applying ML-based protein design tools to engineer a better bacteriophage.
Proposed computational tools and approaches
Start from the wild-type L protein sequence and gather any available prior knowledge on residues involved in membrane insertion, oligomerization, or host interactions.
Use a protein language model for in silico mutagenesisto generate and score single-point or small combinatorial mutants, a software such as ESM from earlier could be used to generate those values we saw in the heat map and downloaded into a .csv file#
Filter candidates to remove obviously disruptive mutations, especially in regions likely required for membrane localization or core function.
Use a computational tool ssuch as AlphaFold-Multimer on selected variants to test whether mutations may alter interactions with relevant host factors such as E. coli DnaJ, since MS2 L-mediated lysis has been reported to depend on the host chaperone DnaJ.
Rank variants based on a combination of sequence plausibility, predicted structural confidence, and predicted interaction changes.
Protein language models such as ESM are useful because they can estimate which amino acid substitutions are more likely to be tolerated while preserving overall protein fitness. That makes them a practical first-pass screen before more expensive structural modeling. Recent work shows that protein language models can be effective for predicting mutational effects and other sequence-function relationships.
AlphaFold-based tools may help because our problem is partly structural: if we want a more stable L protein, then mutations that severely disrupt folding or membrane-associated assembly are poor candidates. For the secondary goal, AlphaFold-Multimer could help us test whether specific mutations might weaken or strengthen predicted contacts with host proteins. This is especially relevant because published work on MS2-L indicates that DnaJ is involved in its lytic activity, and more recent in vitro characterization suggests that MS2-L forms high-order oligomeric states after membrane insertion.
One limitation is that prediction is not proof. A mutation that looks favorable in silico may still reduce true phage fitness in vivo. Another issue is that transmembrane and lysis proteins are harder to model accurately than soluble globular proteins, so structure confidence may not perfectly reflect real function.
Proposed Pipeline Schematic:
Wild-type L protein sequence
→ Protein language model mutational scan
→ Filter top candidate mutations
→ AlphaFold structure prediction of mutants
→ AlphaFold-Multimer with host target(s), e.g. DnaJ
→ Rank variants by predicted stability + interaction effects
→ Nominate best variants for experimental testing
week-05-hw-protein-design-part-ii
Assignment A
Part 1:
The human SOD1 sequence was retrieved from UniProt (P00441). The ALS-associated A4V mutation was introduced by substituting alanine with valine at position 5 of the full-length sequence (equivalent to position 4 after removal of the initiator methionine).
Generated Sequences with their corresponding perplexity scores next to them which pepMLMs confidence rating in them, 4th one is the known binder for reference:
ID
Sequence
Score
0
WRYPAVALAHKX
6.680728
1
WLYYVVAAALGE
17.121198
2
WRYPAVAVRHKK
15.373117
3
WHYYAAALALKE
15.080042
4
FLYRWLPSRRGG
—
Part 2:
I was given an error in alphafold because for my first generated peptide therte was an X given representing an unkown amino acid - im not quite sure why that was generated by pepMLM but i replaced it with a G for Glycine as it is the side chain-less amino acid and would cause least stearic clash and just act as a filler amino acid in place of the unknown X.
Candidate
Peptide Sequence
ProteinMPNN Score
Boltz ipTM
1
WRYPAVALAHKG
6.680728
0.37
2
WLYYVVAAALGE
17.121198
0.32
3
WRYPAVAVRHKK
15.373117
0.32
4
WHYYAAALALKE
15.080042
0.28
5
FLYRWLPSRRGG
N/A
0.38
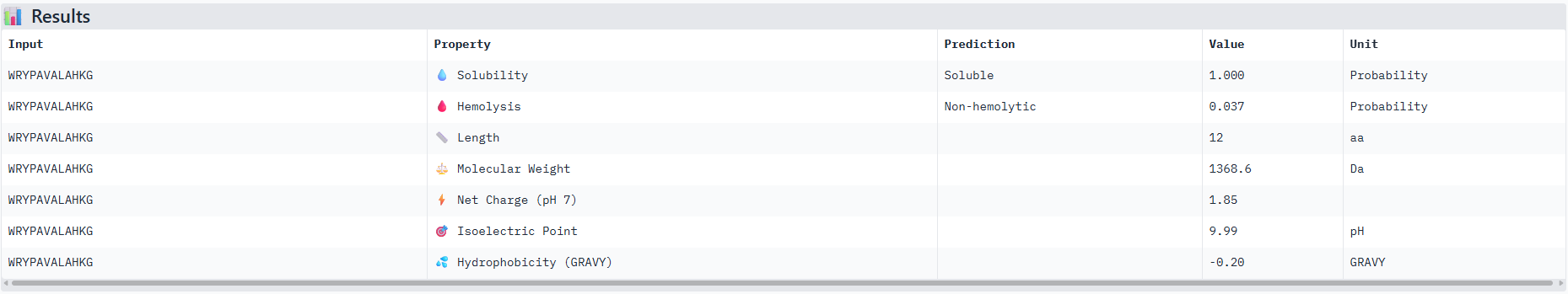
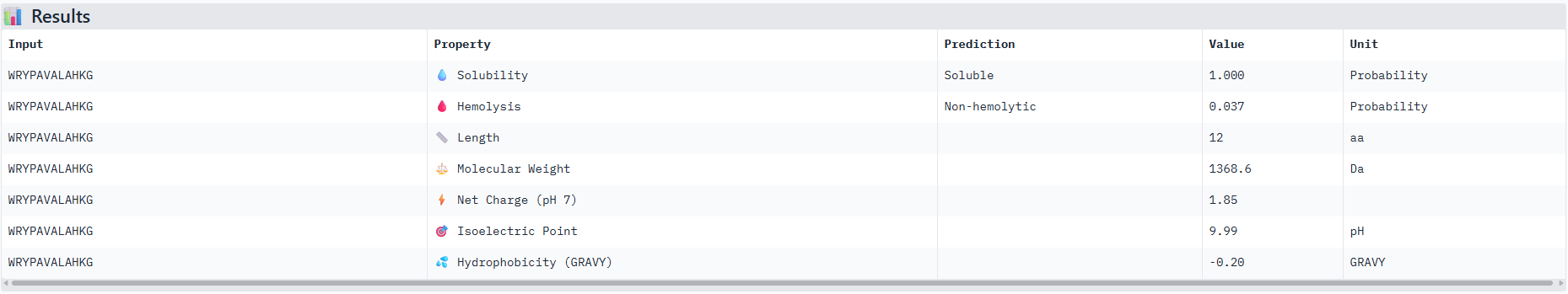
So based off of these scores the peptide that actually performed the best comparative to the known binding peptide was the one with the unknown amino acid that I replaced with a Glycine residue. And it performed very closely with only a 0.01 difference.
Observations from the 3d molecular renderer: The designed peptide binder ran closely around the Beta Barrels strands and so its plausible to assume that these amino acids are conserved in this structure?
Part 3:
I ran the best and worst ipTM scored sequences through peptiverse and obtained the results below, it can be seen that the peptiverse ratings for each factor that represents if a peptide is therapeutically feasible corresponded well to the alphafold servers ipTM scores as the better scored sequence had better metrics on peptiverse.
Results for peptide 0:
Results for peptide 2:
I would advance with peptide 0 as it has th best ipTM score from the alphafold server and also has good therapeutic scoring metrics as seen from the peptiverse.
Part 4
This took much longer than any other as the code was running for a long time but ended up producing results with much better and more improved scores than the previous methods. In terms of testing its validity I assume we could pass through the amino acid sequences its given us through peptiverse too to check again.
Peptide
Hemolysis
Solubility
Affinity
Motif
EKCYGCHYGYQL
0.951
0.917
6.38
0.818
KEVVRELCCGRP
0.953
0.667
7.74
0.811
RKYGYQNDCCYA
0.937
0.917
6.85
0.815
Assignment C
The notebook code initially had some issues when running but I managed to fix them with the help of Gemini.
And the following results were obtained where yellow represents a likely positive change to the folding and a more dark blue colour represents a negative change for the proteins folding ability due to the mutation:
And this was the table computationally obtained for the top 10 Scores:
Mutation
Position
Wild-Type Residue
Mutated Residue
LLR Score
K50L
50
K
L
2.561
C29R
29
C
R
2.395
Y39L
39
Y
L
2.242
C29S
29
C
S
2.043
S9Q
9
S
Q
2.014
C29Q
29
C
Q
1.997
C29P
29
C
P
1.971
C29L
29
C
L
1.961
K50I
50
K
I
1.929
N53L
53
N
L
1.865
The results seen experimentally do contrast in comparison to the computationally obtained results in a few, to display this below is a table showing the top 10 list with the experimental results as of whether or not they could still perform lysis and also if the protein was still expressed or not
Mutation
Position
Wild-Type Residue
Mutated Residue
LLR Score
Lysis
Protein Expression
K50L
50
K
L
2.561
Y
N
C29R
29
C
R
2.395
N
N
Y39L
39
Y
L
2.242
Y
Y
C29S
29
C
S
2.043
Y
Y
S9Q
9
S
Q
2.014
Y
Y
C29Q
29
C
Q
1.997
N
N
C29P
29
C
P
1.971
N
N
C29L
29
C
L
1.961
N
N
K50I
50
K
I
1.929
Y
N
N53L
53
N
L
1.865
Y
Y
Comparison of computational LLR scores with experimental data shows that mutations with higher predicted scores often retain lysis activity and detectable protein expression. However, several high-scoring mutations (e.g., C29R) fail to produce functional protein, indicating that the computational model captures some but not all structural constraints governing L protein stability and function.
Hence for the following selection of the 5 mutations chosen I used a combination of both data, first checking if experimentally they protein was still succesfully expressed and also still had the ability to perform lysis, and then checking if they produced a significant LLR score.
Soluble Region
Mutations in the soluble region must occur between amino acids 1–40 of the L protein. Two mutations were selected from this region based on their high LLR scores and positive experimental outcomes for both protein expression and lysis activity, indicating that these variants are likely to produce functional proteins.
The selected soluble-region mutations are:
S9Q
C29S
Both mutations show detectable protein expression and successful lysis, making them strong candidates for functional L-protein variants.
Transmembrane Region
Mutations in the transmembrane region must occur between amino acids 41–75, which correspond to the membrane-spanning helix responsible for pore formation and bacterial lysis.
Two mutations were selected from this region:
N53L — This mutation shows positive protein expression and successful lysis activity, suggesting that the substitution does not disrupt membrane insertion or oligomerization of the lysis protein.
K50L — This mutation has the highest computational LLR score, indicating strong predicted sequence compatibility. However, experimental data shows no detectable protein expression, suggesting that despite the favorable computational prediction, the mutation may destabilize the protein or prevent proper folding.
Additional Mutation
The final mutation selected is Y39L, located near the boundary of the soluble and transmembrane regions.
This mutation was chosen because it shows:
a high LLR score
successful lysis activity
detectable protein expression
These properties suggest that the mutation is well tolerated and likely preserves the functional structure of the L protein.
week-06-hw-genetic-circuits-part-i
1. What are some components in the Phusion High-Fidelity PCR Master Mix and what is their purpose?
The Phusion High-Fidelity PCR Master Mix contains several key components that enable accurate DNA amplification. It includes Phusion DNA polymerase, a high-fidelity enzyme with proofreading activity that reduces errors during DNA synthesis. The mix also contains dNTPs, which serve as the building blocks for new DNA strand formation. An optimized reaction buffer provides the correct pH and ionic conditions, including magnesium ions (Mg²⁺), which are essential cofactors for polymerase activity. Additionally, stabilizers and enhancers are included to improve enzyme performance and allow efficient amplification of difficult templates such as GC-rich regions. Some versions also contain a tracking dye, enabling direct loading of PCR products onto a gel for analysis.
2. What are some factors that determine primer annealing temperature during PCR?
Several factors determine the optimal primer annealing temperature in PCR, as it must allow specific binding without nonspecific interactions. The most important factor is the melting temperature of the primers, which depends on their length and nucleotide composition. Primers with a higher GC content have higher Tm values because G–C base pairs form three hydrogen bonds, making them more stable than A–T pairs. Primer length also influences Tm, with longer primers generally requiring higher annealing temperatures. The sequence composition matters as well, since secondary structures like hairpins or primer dimers can affect binding efficiency. Additionally, the salt concentration and buffer conditions in the reaction can influence primer-template interactions and stability. Typically, the annealing temperature is set a few degrees (about 3–5°C) below the primer Tm to ensure specific and efficient binding.
3. There are two methods from this class that create linear fragments of DNA: PCR, and restriction enzyme digests. Compare and contrast these two methods, both in terms of protocol as well as when one may be preferable to use over the other.
Though they both fragment of DNA, PCR creates a ver large scale volume of repliucants of the exact same linear fragment and when it does this its joining fresh nucleotides together forming new DNA, whereas with restriction enzyme digests they are acting as molecular scissors and are break a pre existing DNA molecule into seperate fragments at specific restriction sites.
4. How can you ensure that the DNA sequences that you have digested and PCR-ed will be appropriate for Gibson cloning?
Gibson Assembly is a cloning method that joins DNA fragments with overlapping ends in a single isothermal reaction. It uses an exonuclease to create single-stranded overhangs, allowing complementary fragments to anneal. And so the important part is to ensure that the adjacent fragments contain matcvhing overlapping end sequences, usually around 20-40 base pairs long - these overlaps must be designed so that each fragment can anneal specifically to the next fragment or to the linearized vector.
5. How does the plasmid DNA enter the E. coli cells during transformation?
Plasmid DNA enters E. coli cells during transformation by first making the cells competent, meaning their membranes are temporarily permeable to DNA. In chemical transformation, cells are treated with calcium chloride, which helps neutralize the negative charges on both the DNA and the cell membrane. A brief heat shock (e.g., 42 °C) then creates a thermal imbalance that allows the plasmid DNA to pass through the membrane and into the cell.
6. Describe another assembly method in detail (such as Golden Gate Assembly)
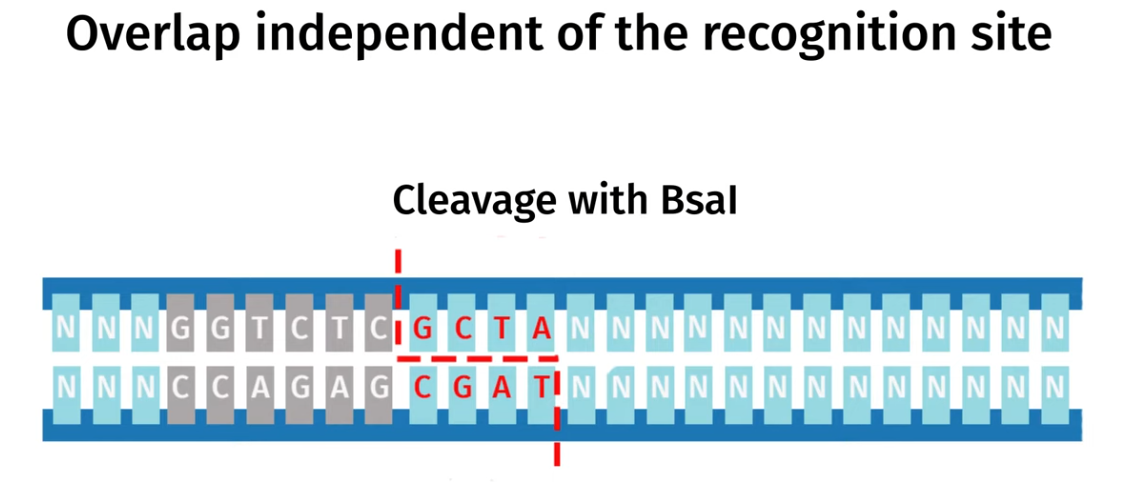
Golden Gate Assembly is another molecular cloning method that allows multiple DNA fragments to be joined together in single reaciton using special Type IIS estriction enzymes (such as BsaI) and DNA ligase,. The Type IIS restriction eznymes cut DNA outside their recognition sequence and leave a specific custome designed sticky ends that are unique for each fragment and so this allows the DNA fragments to be assembled in a specific order and orientation.
DNA ligase simultaeneuosuly joins these fragments together as they are being cut and once the fragments are correctly assembled the recognition sites are removed so the final construct doesnt get cut again.
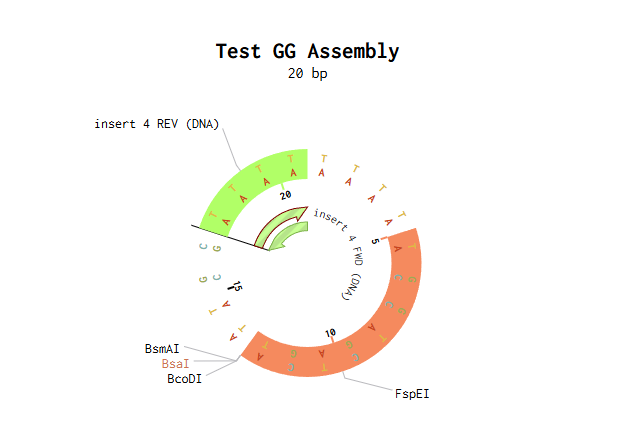
I modelled Golden Gate Assembly in Benchling by creating a test backbone and insert sequences and adding restriction sites for a Type IIS enzyme, BsaI. Unlike normal restriction enzymes, BsaI cuts outside of its recognition site, which allows custom overhangs to be generated. I first entered the DNA sequences into Benchling, making the backbone circular and the insert linear. I then checked the restriction sites and used the assembly tool to simulate how the fragments would join. At first, the assembly did not work because some of the sticky ends did not match, so the fragment sequences had to be corrected and reset. After adjusting the design so that the overhangs were compatible, the assembly worked and Benchling produced a final construct map. This showed how Golden Gate Assembly can join DNA fragments in a specific order and orientation.
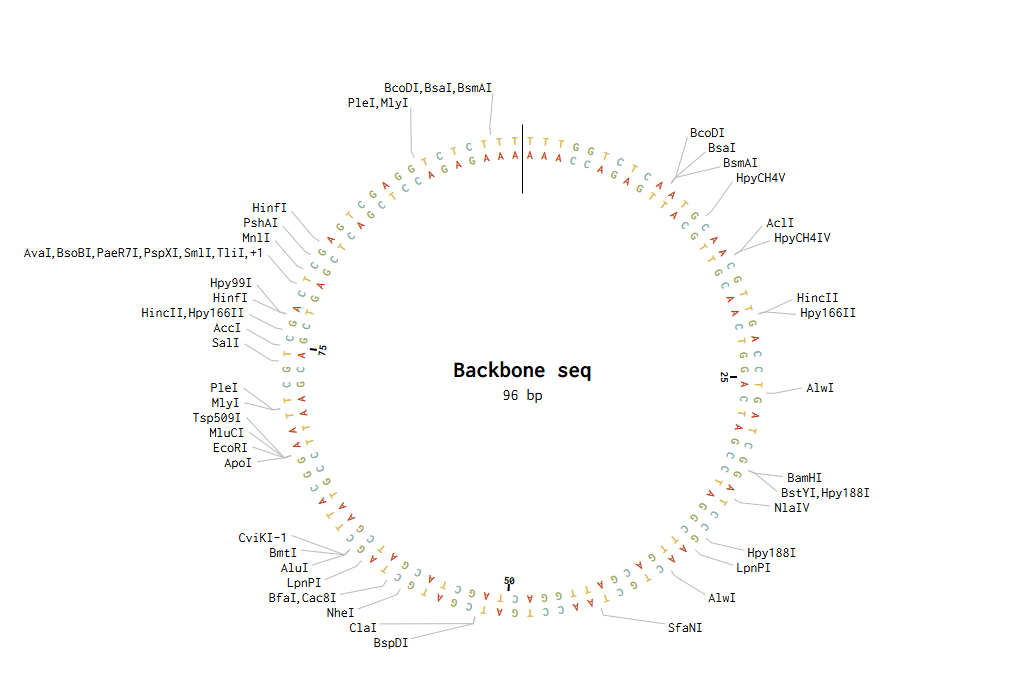
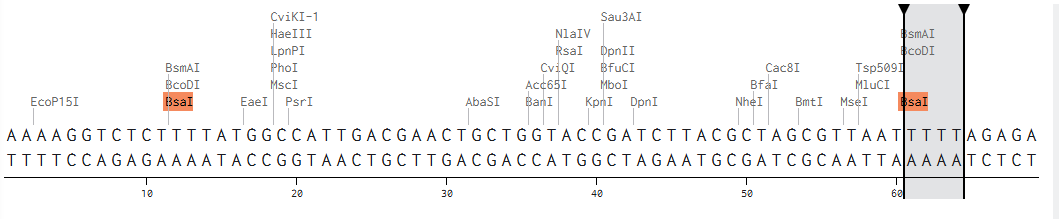
Backbone and insert fragment structures:
And this was the final product after running the Golden Gate Assembly simulation via benchling:
Assignment: Asimov Kernel (Not Done Yet, waiting for Node Access to the Software!)
week-07-hw-genetic-circuits-part-ii
Assignment Part 1: Intracellular Artificial Neural Networks (IANNs)
What advantages do IANNs have over traditional genetic circuits, whose input/output behaviors are Boolean functions?
Advantages include the fact that since IANNs connections between nodes can have varying continuous weightages this is very important when it comes to modelling the effect a gene has on a biological outcome as its very likely that they hold a non-linear relationship and so being able to model for this discrepancy is important as boolean functions assume two discrete states which isnt the case.
Describe a useful application for an IANN; include a detailed description of input/output behavior, as well as any limitations an IANN might face to achieve your goal.
An intracellular artificial neural network (IANN) can be applied in precision cancer therapy, where engineered cells classify disease states and trigger drug delivery only under specific conditions. Inputs include continuous biological signals such as oncogene expression, microRNA profiles, and hypoxia levels, which are integrated through a multilayer gene regulatory network. Each input is weighted, and a nonlinear response (e.g. sigmoidal gene activation) enables pattern recognition rather than simple Boolean logic. If a threshold is reached, the output may be the expression of a therapeutic protein or induction of apoptosis, ensuring high specificity and minimal off-target effects.
However, IANNs face limitations including biological noise affecting reliability, scalability challenges due to circuit complexity and crosstalk, slow response times from gene expression dynamics, evolutionary instability of engineered systems, and practical constraints in safely delivering large genetic circuits into target cells. And so this may hint that they still need to be optimised and improved before actual clinical usage.
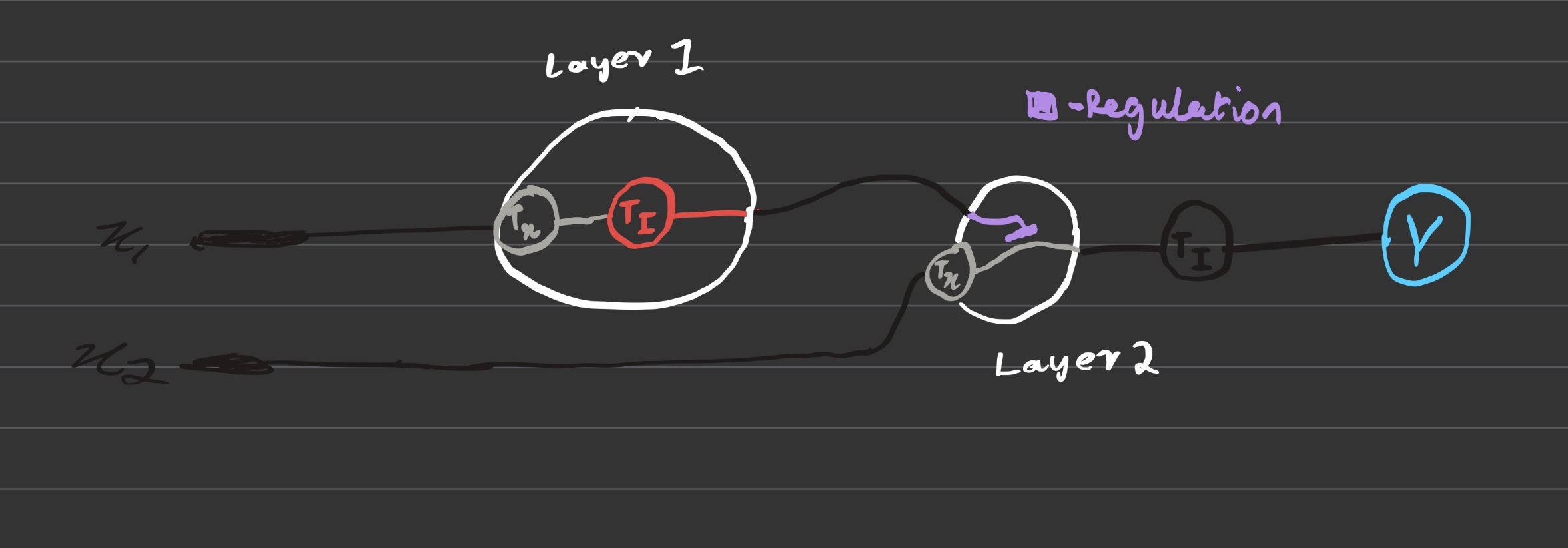
I made the given process a multi layer IANN, where x1 = input DNA coding for CSy4 endoribonuclease, x2 = DNA encoding for flourescent output.
I thought to make x2 pass through the second node as the input as it hadnt really a function in the first one, not too sure if that is correct but I then proceeded to pass the output of the endoribonuclease translated via X1 to act as a regulator indicated in purple for the expression of the GFP of the final protein which is then transcribed and translated as the final output.
Assignment Part 2: Fungal Materials
What are some examples of existing fungal materials and what are they used for? What are their advantages and disadvantages over traditional counterparts?
Mycelium is an example of a fungal material with a wide range of applications. It is used to produce sustainable materials such as insulation panels and biodegradable packaging, as well as leather alternatives for products like shoes and bags. It is also utilised in food production as mycoprotein, a protein-rich alternative to meat. Additionally, mycelium plays a role in environmental cleanup through mycoremediation, where it can break down plastics and toxic chemicals, acting as a natural recycling system.
Fungal materials are sustainable, biodegradable, and low-energy to produce, making them environmentally friendly compared to plastics and leather. However, they generally have lower mechanical strength, durability, and moisture resistance than traditional materials.
Additionally, they face scalability and consistency challenges, as production is less standardised than conventional manufacturing.
What might you want to genetically engineer fungi to do and why? What are the advantages of doing synthetic biology in fungi as opposed to bacteria?
Compared to bacteria, fungi offer several advantages as they are eukaryotic systems, like humans, enabling correct protein folding and post-translational modifications required for many therapeutic proteins. Combined with their efficient secretion pathways, this makes them highly suitable for producing pharmaceuticals and enzymes for drug discovery. However, there are limitations, as fungi are slower growing, which can impact scalability, and are generally more complex to genetically engineer than bacteria, increasing the barrier to entry.
cell-free-systems
General Homework Questions
Explain the main advantages of cell-free protein synthesis over traditional in vivo methods, specifically in terms of flexibility and control over experimental variables. Name at least two cases where cell-free expression is more beneficial than cell production.
One advantage includes the fact that cell-free protein synthesis gives direct control over reaction composition which means we can precisely control the exact concenctrations of factors like DNA, amino acids and modified nucleotides.
No cell membrane barrier means everything is immediately accessible and modifiable
The speed at which you can cycle designs and tests is much faster thant that if you’d have to do the usual process of cloning then transform then grow
You can precisely and exactly control the specific environments its in, setting the exact pH, temperature and also be able to eliminate toxic constraints.
The bread and butter for cell free systems is the cell extract which includes the ribosomes that are the machinery for protein synthesis, tRNAs to deliver amino acids, translation factors and also the enzymes required for and involved in metabolism.
Energy regeneration is critical in cell-free protein synthesis because ATP is rapidly consumed during transcription and translation, and unlike in living cells, there are no metabolic pathways to replenish it, causing protein synthesis to quickly stop if energy is depleted. Simply adding ATP is insufficient due to rapid consumption and accumulation of inhibitory byproducts, so regeneration systems are required to sustain reactions and improve yield. One common method is the phosphoenolpyruvate (PEP) system, in which PEP donates a phosphate group to ADP via enzymes such as pyruvate kinase present in extracts from Escherichia coli, continuously regenerating ATP and allowing protein synthesis to proceed for longer durations.
Compare prokaryotic versus eukaryotic cell-free expression systems. Choose a protein to produce in each system and explain why.
Prokaryotic cell-free systems, typically derived from Escherichia coli, are fast, cost-effective, and produce high yields, but lack the machinery for complex post-translational modifications such as glycosylation or proper disulfide bond formation.
In contrast, eukaryotic systems (e.g., wheat germ or rabbit reticulocyte extracts) are slower and more expensive but support proper folding and modifications required for many eukaryotic proteins.
For example, a simple enzyme such as Green Fluorescent Protein can be efficiently produced in a prokaryotic system because it does not require extensive post-translational modifications, making it ideal for rapid, high-yield expression. Conversely, a therapeutic protein like Insulin is better suited to a eukaryotic system, as it requires correct folding and disulfide bond formation to be biologically active.
How would you design a cell-free experiment to optimize the expression of a membrane protein? Discuss the challenges and how you would address them in your setup
To optimise expression of a membrane protein in a cell-free system, I would design the experiment as a small screening setup in which the same DNA template is expressed across multiple reaction conditions while varying factors that most strongly affect membrane protein yield and solubility
This could most likely be due to a low concentration of the DNA fragment thats coding for the target protein we can fxi this by increasing the DNA fragment concentration
this could also be due to an enzyme thats accidentally been released into our cell free system thats cleaving our target protein and so we can fix this by checking and ensuring no said enzymes are produced.
This could also be due to a lack of amino acid building blocks to actually synthesise the protein and so we can add a better nutrient medium to ensure this is not in limiting.
Kate Adamala’s HW
My synthetic minimal cell would be a sense-and-respond therapeutic cell that detects joint degradation, the input would be lower levels of cartilage or even higher levels of inflammation in those regions and the output would be release of chemicals that provide lubrication around the said joint.
It could probably not be realised by cell-free Tx/TI alone.
Yes, a genetically modified natural cell would make more sense as it would be able to effectively respond within the organisms cells.
A mammalian cell-free system (HeLa or CHO cell extract is most common) is probably necessary here, primarily because:
Therapeutic protein functionality depends on correct PTMs
NF-κB or similar mammalian response elements are the natural way to sense the arthritic environment
Immunogenicity in the joint space makes bacterial components a serious liability
The desired outcome would be that during times of discomfort or too much inflammation in joints for those with arthritis, the synthetic cell operation would allow to alleviate for some of this.
Peter Nguyen’s homework questions:
One-sentence pitch
A building wall system embedded with freeze-dried cell-free biosensors that detect seasonal temperature shifts and autonomously trigger the germination of different plant species — turning architecture into a living calendar.
How it works
The wall panels contain a layered substrate of freeze-dried cell-free systems encapsulated in a hydrogel matrix alongside dormant seeds from different plant species. Each CFS unit is engineered with a temperature-sensitive riboswitch or cold-shock promoter tuned to a specific threshold — say, sub-10°C for winter species, 18–22°C for spring/summer. When the trigger temperature is reached, the CFS activates, expressing germination-promoting enzymes (e.g. gibberellin-related proteins or cell-wall loosening factors) that are secreted locally into the seed microenvironment, initiating growth of that season’s designated plant. Different zones of the wall cycle through moss, herbs, climbing flowers, or ornamental grasses depending on the external climate — with no human input.
Societal challenge / market need
Urban spaces are increasingly disconnected from natural seasonal rhythms, contributing to biophilic deficit in dense cities. The indoor plant and living wall market is growing rapidly but remains static — the same wall looks identical year-round. This system makes buildings genuinely responsive organisms, with applications in hospitality, retail, wellbeing spaces, and sustainable architecture.
Addressing CFS limitations
The hydrogel encapsulation keeps the freeze-dried CFS stable and dormant until ambient humidity or a deliberate water-activation event (rainfall ingress, irrigation pulse) rehydrates the matrix. The one-time-use constraint is turned into a feature — each CFS pod is a single-season unit, designed to be swapped out in modular panel cartridges at the start of each season, making maintenance a ritual rather than a problem. Long-term stability is extended by storing unactivated pods at the panel’s rear in a desiccated micro-chamber until the thermal trigger is met.
Ally Huang’s homework Questions
In outer space exploration the most VITAL step and building of infrastructure has to be initializing sustainable crop growth to not only kick-start O2 production to make the new planets/area habitable but to also allow for one of the most underlooked factors which is aggriculture! Since its a given in the world we live in now, we underestimate that itll take alot to produce fertile plants that produce edible crops to allow for common food to be produced later into space exploration.
Nitrogenase complex genes (nifH, nifD, nifK) and ATP-regeneration pathways, engineered into a cell-free synthetic biology platform for on-site biological nitrogen fixation and soil fertilization in extraterrestrial agricultural systems.
Nitrogen is the most limiting nutrient for crop production, yet no extraterrestrial environment has biologically available nitrogen in soil. The nitrogenase complex genes (nifH, nifD, nifK) encode the only known enzymatic machinery capable of converting atmospheric N₂ into bioavailable ammonia — the foundation of all plant protein and growth. Deploying these genes within a cell-free platform eliminates dependence on Earth-supplied fertilizers, removes the need for living microorganisms that pose contamination risks in sterile extraterrestrial soils, and enables programmable, on-demand nutrient production. Solving this bottleneck directly unlocks sustainable crop growth, oxygen generation, and long-term human habitability beyond Earth.
We hypothesize that a cell-free synthetic biology system expressing nifH, nifD, and nifK with an integrated ATP-regeneration cascade can fix atmospheric N₂ into bioavailable ammonia at rates sufficient to fertilize extraterrestrial soil simulants, enabling measurable crop germination and growth without Earth-supplied nitrogen fertilizers — establishing a foundational, contamination-free nutrient production platform for space agriculture.
Experimental Plan:
Samples: Cell-free reactions with purified nitrogenase complex tested against Mars/lunar soil simulants, with and without ATP-regeneration systems.
Controls: Boiled/inactive enzyme controls, Earth soil with standard fertilizer, and unfertilized simulant.
Measurements: Ammonia output (colorimetric assay), plant germination rates, biomass yield, chlorophyll content, and O₂ production over 30-day growth trials.
It can be seen that the molecular weight is 28007.60 when the entire protein genome code is considered, slightly less when the His-purification tag ad the linker are accounted for at 26,941.15 Daltons.
I found this question a bit confusing but got the idea that because different molecules may be protonated to differing extents this means that they will have different mass:charge ratios and so what were calculating for is the most abundantly-averaged mass of the eGFP?
I chose the peaks 875.4421 and 903.7148 as they seemed most abundant and were adjacent to one another and substituting their values into the equation given gave the following molecular weight Z:
(m/z)n = 875.4421
(m/z)n+1 = 903.7148
Z= 30.97 == 31
The peak at 903.7148 is the 31+ charge state
The peak at 875.4421 is the 32+ charge state
So then using the 31+ peak
M = z((m/z)n - 1.0073)
M = 31(903.7148 -1.0073)
M = 31(902.7075)
M=27983.93 Da
Accuracy = (27983.93 - 28006.26 ) / 28006.26
= -0.000797
= -0.08%
OPTIONAL PART II DO
Waters Part III - Peptide mapping
Lysine (K): 20
Arginine(R): 6
There are 19 total peptide sequences achieved from the cleavage via trypsin:
There are 19 peaks over 10%.
Yes, both end up at 19!
Using the peak values from figure 5b of 526.25918 & 525.76712to calculate the charge state.
526.25918 - 525.76712 = .49206
1/.49206 = 2.0322
Therefore charge state = 2
From the comparison of charge states, the peptide was identified as having a single charge state of 1050. Therefore, the peptide is likely FEGDTLVNR, which has a mass of 1050.5214 and corresponds to positions 115–123.
The mass accuracy was calculated using the following equation:
This gives a mass accuracy value of 2.83 ppm.
Waters Part IV - Oligomers
The 8FU Didecamer at 8.33 MDa is the dominant peak — by far the most abundant species in solution, which makes sense as it’s the canonical native assembly state of KLH
The small offsets between expected and measured masses (e.g. 8.0 vs 8.33 MDa) are real and expected — they reflect glycosylation, post-translational modifications, and bound cofactors like copper that aren’t captured in the bare polypeptide masses in Table 1
The 4.013 MDa peak is close to but not exactly the 7FU decamer — it likely corresponds to an 8FU half-decamer (10 × 400 kDa = 4.0 MDa), which is a known dissociation product
The small peaks below 2 MDa (0.1982, 0.79, 1.52) are likely individual subunits or small subcomplexes
Waters Part V
The theoretical molecular weight of the protein is 27,875.41 Da (excluding the start codon, average MW)
The intact LC-MS measurement came in at 27,919.93 Da
That gives a mass error of ~1,597 ppm — which is extremely large; anything above ~10-20 ppm is considered a poor match in intact mass analysis
Based on this, the protein is very unlikely to be eGFP
Unfortunately I wasn't able to contribute to the 1536 pixel canvas artwork — but I would be absolutely thrilled to join as a TA for the next intake!
Part B
Cell-Free Reagent Breakdown
E. Coli Lysate
The broken-down remains of E. coli cells — containing DNA, RNA, and critically the protein machinery like T7 RNA Polymerase that performs the transcription and translation of our desired protein.
Energy Nucleotide System
Provides the building blocks for RNA and DNA synthesis during transcription, drawing on ATP/GTP derived from guanine and glucose. Also supplies the amino acids and energy needed for protein synthesis downstream.
Translation Mix
The raw material for actually building the coded proteins. Notably, cysteine — with its –SH side chain — provides the reducing capability required for the correct functioning of the enzymes involved.
Nicotinamide (NAD⁺/NADH)
The electron carrier system required for NAD-dependent reactions: metabolic processes, oxidative reactions that generate NADH as a byproduct, and as the precursor for many essential enzymes.
Nuclease-Free Water (Backfill)
Brings the solution to the required volume and dilution factor. The nuclease-free part matters — ordinary water can carry nuclease enzymes that will happily degrade the DNA in the reaction.
Question 2PEP-Based vs. Long-Incubation NMP Systems
The key difference is how each system supplies and sustains ATP for transcription and translation. PEP-based systems are fast and high-powered — but short-lived. ATP generation is rapid, which means rapid depletion of the energy source and a drifting pH as pyruvate accumulates.
Long-incubation NMP systems are slower by design, drawing on a broader nutrient mix that takes longer to metabolise into ATP. The payoff is a lower initial protein expression rate, but also lower phosphate accumulation — meaning better pH stability over the course of the reaction.
Question 3Role of GMP
Guanosine Monophosphate (GMP) is simply a guanine molecule bound to a phosphate group. It can be dephosphorylated to release the guanine nucleotide — and this dephosphorylation step also releases useful energy, so it's doing double duty.
Part C
Cell-Free Master Mix Design
1 — Fluorescent Protein Properties
sfGFP
Engineered specifically to resist misfolding when fused to poorly-folded proteins. Folds in under 10 minutes and tolerates the chaperone-free, crowded environment of cell-free lysates far better than earlier GFP variants — its folding robustness is its defining trait.
mRFP1
A relatively slow-maturing monomer with low acid sensitivity. The notable issue: red FPs pass through green chromophore intermediates during maturation — a fraction of mRFP1 may never fully mature past this green state, or becomes trapped as a dead-end product entirely.
mKO2
Complex multi-step chromophore maturation involving multiple oxidation steps — making it significantly slower overall. Fluorescence signal will substantially lag behind protein synthesis, making mKO2 a poor kinetic reporter, though perfectly fine for endpoint measurements.
mTurquoise2
Standout property: an exceptionally high quantum yield combined with acid stability. Its very low pKa of 3.1 means fluorescence is essentially independent of pH across a huge range — a robust reporter in variable-pH environments.
mScarlet-I
A single amino acid substitution — T74I — drives a marked maturation acceleration, making it one of the fastest-maturing red FPs available and an excellent kinetic reporter. The trade-off is a moderate reduction in fluorescence quantum yield.
Electra2
A recently engineered blue FP derived from the Anthozoa protein eqFP611 via mRuby3. Its tendency toward aggregate formation confers resistance to acidity and proteases in mammalian cells — but in a cell-free context, without cell growth diluting things, aggregation could trap a meaningful proportion of translated Electra2 in non-fluorescent or insoluble fractions, reducing effective signal.
2 — Reagent Adjustment Hypothesis
HypothesismKO2
Reagent
Sodium percarbonate (Na₂CO₃·1.5H₂O₂) at 0.5–2 mM as a slow-release oxygen source.
Rationale
mKO2 chromophore maturation requires two sequential oxidation steps — acylimine formation then third-ring cyclisation — giving it a ~135 minute maturation half-time. In a sealed cell-free reaction, oxygen is consumed both by this multi-step chemistry and by the energy regeneration system, creating a hypoxic microenvironment that stalls maturation and traps translated protein in a non-fluorescent intermediate state. Over 36 hours, this oxygen depletion is cumulative and severe.
Expected Effect
Sustained O₂ availability will drive more translated mKO2 through both oxidation steps to the fully mature orange-emitting state, increasing plateau fluorescence at 36 hours. Because mKO2 requires two oxidation steps versus one for GFP-family proteins, it is disproportionately sensitive to O₂ limitation — making this a mechanistically specific intervention rather than a generic tweak. If the reaction is run in an open-well format where O₂ is not limiting, no effect should be observed, which would falsify the oxygen-depletion mechanism.
4 — Reaction Composition Per Well
20 µL Total Volume
E. Coli Lysate
6 µL
2X Optimised Master Mix
10 µL
FP DNA Template
2 µL
Custom Reagent Supplements
2 µL
Total
20 µL
Awaiting fluorescence data
Analysis of fluorescence data from the global experiment will follow approximately one week after data is returned. Date TBD.
Opentrons-Integrated Modular Peptide Production Platform — full project proposal including experimental design, automation workflow, validation, and budget.
Author: Vithushan Varatharaj
Project: Opentrons-Integrated Modular Peptide Production Platform
SECTION 1: ABSTRACT
Peptide therapeutics—spanning antimicrobial peptides, hormones, cytokines, and neuropeptides—hold immense promise across medicine and research. Yet their production remains prohibitively expensive and technically complex, accessible only to well-resourced institutions and limiting their adoption in research and therapeutic development. Current methods lack modularity and scalability, forcing researchers to reinvent production workflows for each new peptide target.
To address this barrier, I am developing a modular, peptide-agnostic expression platform integrated with laboratory automation, treating peptide production not as a bespoke engineering problem, but as a plug-and-play system: a researcher specifies a peptide sequence, receives a standardised plasmid construct, and executes a semi-automated workflow to recover purified peptide—all within a single working day, without specialist expertise or expensive custom synthesis.
SECTION 2: PROJECT AIMS
At a glance I aim to achieve 3 things with this project:
(1) designing, fabricating, and validating the Opentrons Flex gel electrophoresis module;
(2) Designing, expressing, and validating a cecropin-based AMP using the His-SUMO cassette to show off its use ind developing AntiMicrobial Peptides, a class example of how useful synthetic peptides are in the real world, in this instance providing an alternative to and combatting growing antibiotic resistance;
Andfinally for (3), establishing the platform as a fully ‘plug-and-play’ pipeline with upstream computational peptide design integration.
Aim 1 — The Experimental Aim
“The first aim of my final project is to design, fabricate, and validate a miniaturised gel electrophoresis module integrated into the Opentrons Flex platform, by utilising custom CAD-designed hardware, integrated camera vision for AI-assisted band detection, and an automated biopsy-punch excision mechanism.”
For the methods and tools this will require I’ve outlined the following below, and will go into detail of the reasoning for each in the experimental design section:
Custom mini gel electrophoresis board (CAD-designed, Opentrons Flex-compatible)
Camera-guided band detection
Calibrated biopsy-punch excision on Opentrons Flex capable of picking up and dropping gel sample.
Aim 2 — Developmental Aim
Following the success of Aim 1, I would want to put this into full swing using my twist order and for a very real world use case scenario for this system, the synthesis of Antmicrobial peptides.
In a world where growing resistance for antibiotics is seemingly one of the biggest races humanity and medical scientists are up against, AMPs providing an alternative for more common antibiotics in bacterial inhibition puts them up as a very interesting field of study in the uses of synthetic peptides.
This would mean getting to put the pipeline into action, using the validated module as the centrepiece of a full AMP production pipeline. Providing a scenario where its effectiveness can be modelled for and measured quantitatively via assaying for the level of inhibition, as opposed to having to jump into the other more eye-catching but ethically implicating uses case scenarios for synthetic peptides.
This part would, in addition to what was mentioned in (1), require the following:
Benchling sequence design of His-SUMO-cecropin fusion cassette
Twist Bioscience gene fragment synthesis (insert ordered)
Restriction enzyme digestion and ligation into pET-28a vector
Zone-of-inhibition assay against Bacillus subtilis ATCC 6633
Edit: Upon reassessing I realise this is a bit of mash up with Experimental aim too, as I realise this step is what mainly uses my Benchling order in full swing, but initially to keep things more concentrated I had split it this way. I realise another form of validating the experimental aim would just be to simply see it calibrate fully and succesfully extract our band of choice but I would prefer in practice to have it try to run the entire workflow of Aim 2 itself to test as gettign to work with inhibition readings of the AMPs effects on bacterial inhibition is far more interesting and I feel proves as a real world use for this project!
Aim 3 — Visionary Aim (Long-Term)
The long-term vision is a * fully democratized, AI-enabled ** closed-loop peptide discovery platform - where a researcher anywhere in the world inputs a ** desired biological activity! **
An Opentrons-based workflow to produce, purify, and validate functional peptide within a single working day — no specialist expertise, no expensive synthesis contracts, no institutional gatekeeping. If realised, this platform could catalyse a paradigm shift in how peptide therapeutics are discovered and developed: compressing timelines from months to days, enabling small biotech enterprises and academic labs in resource-limited settings to compete in therapeutic development, and — at the clinical horizon — accelerating the response to emerging multidrug-resistant pathogens in real time.
Now the biological activity can vary (as I have and will mention 100 times being the fanboy I am!) to the wide ranges of uses synthetic peptides have. The researcher may, as this project is determined to demonstrate, test for different Antimicrobial peptides effectiveness against a certain bacteria.But beyond AMPs, the platform could eventually cater to therapeutic peptide development across oncology, neuropeptide-based pain and mood disorders, antiviral defence, accelerated wound healing, and even the commercial cosmetics and skincare industry — essentially anywhere a specific biological activity can be defined and a peptide can be designed to achieve it.
A field that particularly interests me — as I hinted with AI-enabled — is tying the pipeline with ML tools like AlphaFold or pepMLM for peptide drug discovery; similar to how this project works with AMPs, but this time aimed at viruses.
SECTION 3: BACKGROUND
Literature Context
Regarding the actual process of DNA gel band extraction and purification for use in cloning I found the following study that had tried a manual approach to this:
Sánchez-Flores et al. (2025) developed and validated cost-effective protocols for DNA extraction from agarose gels using chaotropic salt dissolution or freeze-thaw cycles, without reliance on commercial silica-column kits. Both methods yielded DNA of sufficient quality for downstream PCR amplification, restriction digestion, and bacterial transformation. Critically, the study demonstrated that gel-based DNA recovery is achievable in resource-constrained settings with minimal specialised equipment, establishing the feasibility of integrating gel extraction into low-cost automated workflows. This directly informs the DNA processing design within this project, supporting the use of miniaturised gel electrophoresis and automated band excision as a viable, scientifically sound approach to construct screening. Nature Scientific Reports
And then in terms of the actual AMP production and testing of antimicrobial activity part of this project, this was a vital experiment that showed how the His-SUMO fusion protein can be used to prevent from self-host toxicity interfering with the desired outcome as the AMP is usually toxic to the E. coli producing it too.
Park et al. (2021) demonstrated that expressing cecropin B as a His-SUMO fusion protein with a three-glycine linker in E. coli BL21(DE3) effectively mitigates host toxicity during intracellular expression, a critical barrier for producing membrane-disrupting AMPs in bacterial systems. The SUMO domain acts as a combined solubility enhancer and steric shield, preventing the cecropin peptide from engaging host membranes during production. Following SUMO protease cleavage and Ni-NTA purification, the released cecropin B exhibited potent and enhanced antimicrobial activity against Bacillus subtilis, validating the fusion architecture as a generalised, recoverable production strategy. This work directly establishes the experimental and architectural precedent for the His-SUMO cassette design at the core of this project. PMC8578067
Innovation
This project is novel and innovative in three distinct ways. First, it develops entirely new hardware — a custom-designed, CAD-fabricated miniaturised gel electrophoresis module integrated into the Opentrons Flex deck with camera-guided AI band detection and automated biopsy-punch excision — automating a step that has remained stubbornly manual and rate-limiting in molecular biology workflows for decades. Second, it establishes a genuinely modular, peptide-agnostic production architecture where only the DNA coding insert changes between targets, fundamentally challenging the current assumption that each new peptide requires bespoke design, expression optimisation, and purification development. Third, by coupling this automation with standardised His-SUMO fusion biology and Ginkgo Bioworks-compatible high-throughput assay infrastructure, the project expands the boundaries of synthetic biology by making sophisticated peptide manufacturing operationally accessible — not just technically possible — for resource-constrained laboratories and researchers without specialist training.
Significance
The Problem: Whilst demand for peptide therapeutics has surged — particularly AMPs as next-generation antibiotics against multidrug-resistant pathogens — the cost and complexity of peptide production remain prohibitively high. Current methods depend on expensive custom synthesis services (typically $50–500/mg for synthetic peptides) or specialised fermentation infrastructure, effectively gatekeeping peptide development to well-funded pharmaceutical companies and elite research institutions.
Importance: This barrier is not merely an efficiency problem — it is a systemic constraint that stalls progress in antimicrobial resistance research, limits therapeutic development for rare and neglected diseases, and perpetuates global health inequities by making potentially life-saving treatments accessible only to wealthy institutions and populations.
Broader Societal Contribution: A modular, automated peptide production platform would lower the barrier to entry for small biotechs, academic labs, and researchers in low- and middle-income countries, democratising access to both the research tools and the therapeutic products that peptide science enables. This has direct implications for global health equity, particularly in regions where AMR burden is highest and research infrastructure is most limited.
Scientific Advancement: Successfully demonstrating that the custom Opentrons gel electrophoresis module can reliably automate band detection and excision contributes novel hardware and open-source methodology to the broader automation community, with applications well beyond peptide production. Integrating computational design tools upstream would establish a complete in silico-to-wet-lab feedback loop, enabling rational iteration cycles currently impossible with conventional approaches.
Field-Level Change: If the platform’s aims are fully realised, the field could transition from expensive, bespoke peptide engineering toward a standardised, democratised model — where peptide identity is the only input and purified, validated product is the guaranteed output. This would reshape how peptide-based therapeutics, research reagents, and diagnostic tools are developed, shortening timelines, reducing costs, and broadening participation across the global research community.
Bioethical Considerations
This project invokes multiple foundational bioethical principles that must be carefully considered. The principle of non-maleficence is paramount: if the automated pipeline produces systematic errors — through hardware failure, software bugs, or protocol inconsistencies — falsified results could propagate downstream into clinical or therapeutic development contexts, potentially reaching patients. The principle of responsibility is equally critical: whilst the platform is designed to democratise beneficial research, its accessibility and ease-of-use could reduce barriers for bad actors seeking to synthesise harmful bioactive peptides, toxic agents, or antimicrobial compounds with weaponisable properties. The principle of justice underpins the project’s democratisation goal — equitable access to peptide therapeutics benefits marginalised populations currently excluded from these innovations — but creates a productive ethical tension: can democratisation be achieved without simultaneously enabling harm? Finally, the principle of beneficence demands that the platform’s design actively prioritise the generation of social good, not merely the technical demonstration of capability.
To ensure ethical conduct, I propose the following safeguards: (1) Rigorous validation and quality control — extensive benchmarking of the Opentrons module against validated standards, redundant QC checkpoints at each automated step, and transparent public reporting of error rates, failure modes, and accuracy thresholds before any deployment beyond the research context; (2) Responsible open-access governance — publishing detailed protocols openly whilst simultaneously engaging with synthetic biology governance bodies (iGEM, ABSA International, the UK Biosafety Association) to establish community-agreed guidelines for user competency requirements and institutional oversight; (3) Institutional biosafety integration — recommending that all users implement appropriate biosafety protocols for their target peptides and mandating institutional biosafety review for novel or high-risk sequences; and (4) Adaptive oversight — building governance frameworks that evolve alongside the platform rather than being fixed at time of publication. Potential unintended consequences include overcompliance deterring legitimate researchers in under-resourced settings, or insufficient controls enabling misuse in settings with weak institutional oversight. Key uncertainties include whether computational tools like pepMLM can reliably predict off-target toxicity, and whether open protocols can be structured to minimise misuse without stifling innovation. The alternative — licensing only to vetted institutions — perpetuates the very inequities this project aims to address. The optimal path is community-driven governance, transparent risk communication, and iterative oversight mechanisms that balance openness with responsibility.
SECTION 4: EXPERIMENTAL DESIGN, TECHNIQUES, TOOLS, AND TECHNOLOGY
For the major part of this project relying on the success of the new Opentrons module, I needed to think of a way to integrate a module that satisfied the housing requirements — and limitations — of the Opentrons Flex machine.
Gel Electrophoresis Module Design
To begin, I made sure to keep in mind the maximum housing dimensions of a module slot in the Opentrons Flex. The Flex deck slot has a footprint of 106mm (W) × 194.5mm (L), which set a hard constraint on what the module could physically be. My estimated gel electrophoresis module dimensions (125mm W × 175mm L × 70mm H) sit comfortably within this envelope, confirming that a miniaturised gel board would physically fit.
From there it was a matter of devising a way to actually automate the process of cutting out a DNA gel band and moving it into a purification setup — this was arguably one of the hardest parts, as I spent a long time trying to figure out how to get a blade in there. After more careful thought I came across biopsy hole punchers used in the medical field to take skin samples from patients.
I thought: why not implement something like this to “hole punch” a sample of the desired DNA gel band, instead of trying to cut it out with a blade? This would be something we could implement more easily using the arm of the Opentrons Flex machine, substituting the biopsy punch in place of the usual pipette head (see Fig. 3).
With that solved, the next logistical hurdle was devising a way to pick up the right band — and in theory this one was more straightforward: use a small camera module to aid the movement of the pipette holder (now holding our biopsy punch) to punch out the required band. The camera is mounted directly above the gantry, feeding images to an AI pipeline that identifies the correct band by size relative to the DNA ladder, then the Z-axis descends and the punch fires (see Fig. 3).
The module itself is designed as a three-layer stack:
Bottom layer (125mm × 85mm) — the structural base housing the power input and Opentrons dock interface that locks the module into the deck slot.
Middle layer (70mm × 50mm) — sits beneath the gel tray and contains a UV transilluminator. This illuminates the SYBR-stained gel from below, giving the camera above a high-contrast image of the DNA bands against a dark background.
Top layer / gel tray (~120mm × 80mm) — the electrophoresis tank itself, with a cathode buffer chamber (−) at one end and an anode buffer chamber (+) at the other, flanking a central gel casting area of 50mm × 70mm. DNA migrates from cathode to anode under the applied electric field.
All three layers shown together as an exploded view:
The full system — showing the gantry arm, AI camera, biopsy punch head, gel board slot, and power/USB data connections — is shown in the assembly view below:
Antimicrobial Peptide Synthesis Pipeline Demonstration: Cecropin AMP DNA Construct Architecture
The core idea behind this project is to build a reusable, semi-automated pipeline that takes you from a DNA sequence of interest all the way to a purified, functional peptide — without having to redesign the workflow each time. The central bottleneck I wanted to address was the DNA screening and extraction step: traditionally, after running a gel electrophoresis, you manually identify your band under UV light, cut it out with a blade, and purify it. This is slow, inconsistent, and entirely dependent on operator skill. The gel module described above replaces that step entirely.
The overall pipeline looks like this:
Design your peptide-encoding DNA construct in Benchling and order the gene fragment from Twist Bioscience as being done in this experiment OR as I envisioned it, extracting whatever known sequence of DNA you require by simply adding the genome DNA and the required RE enzymes and then using the known DNA fragment size to match it with its band allowing for extraction of novel peptides in this simple workflow!
Perform a restriction enzyme digest to generate the insert fragment
Run the digest on the custom gel electrophoresis module in the Opentrons Flex
The Opentrons camera detects the correct band; the biopsy punch excises it automatically
Purify the DNA from the gel plug
Ligate the insert into the pET-28a expression vector
Transform into E. coli BL21(DE3) and screen colonies by PCR
Induce expression with IPTG; purify the His-SUMO-cecropin fusion protein via Ni-NTA chromatography
Validate functional peptide production via zone-of-inhibition assay against Bacillus subtilis
Detailed Experimental Plan
DISCLAIMER: As creditted in section 6, I used Ronans instructions to help sharpen the following section up as a few sections were a bit hazy to me, but as mentioned later I did back test myself with peer reviewed research!
Design the His-SUMO-CecropinB fusion cassette in Benchling — annotate promoter, RBS, His-tag, SUMO domain, GGG linker, cecropin coding sequence, and terminator. Confirm correct ORF and codon optimisation for E. coli. (Day 0 — ~2 hrs)
Submit Twist Bioscience DNA fragment order — upload the finalised sequence as a clonal gene fragment flanked by NdeI and XhoI restriction sites. (Day 0 — ~30 min, ~10 day turnaround)
Prepare pET-28a vector — midi-prep and sequence-verify the backbone; confirm NdeI and XhoI cut sites are present and functional. (Day 10 — ~4 hrs)
Restriction enzyme digest — digest both the Twist fragment and pET-28a with NdeI + XhoI (NEB) at 37°C for 1 hr; set up reactions using the Echo525 (Ginkgo Bioworks) for precise nanolitre enzyme transfers into a 384-well PCR plate. (Day 11 — ~2 hrs)
Run gel electrophoresis on the custom Opentrons Flex gel module — load digested products onto a 1.5% agarose SYBR Safe gel seated on the custom mini-board fitted into the Opentrons Flex deck. (Day 11 — ~45 min)
Automated band detection — the camera mounted on the Opentrons Flex pipette head captures a gel image; the OpenCV-based AI pipeline identifies the ~543 bp insert band by calibrating against the DNA ladder in Lane 1. (Day 11 — ~5 min)
Automated biopsy-punch band excision — the Opentrons Flex descends a biopsy-punch attachment to the confirmed band coordinates; the gel plug is excised and deposited into a collection tube. (Day 11 — ~10 min)
Gel purification of excised plug — dissolve gel plug in chaotropic salt solution (Sánchez-Flores 2025 protocol); bind, wash, and elute DNA. Quantify by Nanodrop (target ≥5 ng/µL). (Day 11 — ~1 hr)
Ligation — ligate purified insert into NdeI/XhoI-digested pET-28a using T4 DNA Ligase (NEB) at 16°C overnight, 3:1 insert:vector molar ratio; incubate in the Inheco Plate Incubator (Ginkgo). (Day 11–12 — 16 hrs)
Bacterial transformation — heat-shock ligation product into chemically competent E. coli BL21(DE3); plate on LB + kanamycin (50 µg/mL) agar. Incubate overnight at 37°C. Expected: >50 colonies per plate. (Day 12 — ~2 hrs hands-on)
Colony PCR screening — pick 8–12 colonies; amplify with T7 promoter and T7 terminator primers using the ATC Thermal Cycler in a 96-well Armadillo PCR plate. Confirm ~650 bp band for insert-positive clones. (Day 13 — ~3 hrs)
Colony PCR gel confirmation on Opentrons module — run PCR products on the gel module to confirm insert-positive clones; automated band detection confirms correct size. (Day 13 — ~1 hr)
IPTG induction of expression — inoculate a confirmed positive clone into LB + kanamycin; grow to OD600 ~0.6 in the Cytomat shaking incubator (Ginkgo); add 0.5 mM IPTG via Multiflo dispenser; induce at 18°C for 16 hr. (Day 14–15)
Cell harvest and lysis — centrifuge cultures using the HiG Centrifuge (Ginkgo); resuspend in lysis buffer; lyse by freeze-thaw + lysozyme; clarify lysate by centrifugation. (Day 15 — ~3 hrs)
Ni-NTA affinity purification — pass clarified lysate over Ni-NTA agarose column (Qiagen); wash with 20 mM imidazole; elute with 250 mM imidazole. Confirm purity and expected ~15 kDa band by SDS-PAGE. (Day 15 — ~3 hrs)
Optional SUMO protease cleavage — incubate purified fusion protein with Ulp1 SUMO protease (1 hr, RT) to release active cecropin peptide with native N-terminus. (Day 16 — ~2 hrs)
Zone-of-inhibition assay against Bacillus subtilis — spread B. subtilis ATCC 6633 lawn on LB agar; spot purified peptide (and controls: ampicillin positive, PBS negative) onto lawn using Multiflo; seal plates with Plateloc; incubate 18 hr at 37°C. (Day 16–17)
Assay readout and quantification — image plates and measure OD600 in 96-well format using the Spark Plate Reader (Ginkgo); calculate growth inhibition % relative to negative control; determine IC50 or zone diameter. (Day 17 — ~2 hrs)
Data analysis — export Spark data; plot dose-response curves; compare to ampicillin positive control to confirm functional antimicrobial activity. (Day 17 — ~2 hrs)
Document and report — compile gel images, SDS-PAGE results, and antimicrobial assay data; assess platform modularity and identify limitations for Aim 2. (Day 18)
Techniques Checklist
Pipetting
Pipetting
Lab Safety
Bioethical Considerations
DNA
DNA Gel Art
DNA Sequencing
DNA Editing
DNA Construct Design
Restriction Enzyme Digestion
Gel Electrophoresis
DNA Purification From Gel
Databases (GenBank, NCBI, Benchling)
Lab Automation
Creating Code for Laboratory Automation
Using Liquid Handling Robots (Opentrons Flex)
Designing a Twist Order
Creating a plan to use the Autonomous Lab at Ginkgo Bioworks
Protein Design
Protein Design
Use of Boltz or PepMLM (Long term visionary)
Use of Asimov Kernel
Use of Benchling
Models and Notebooks
Databases
Bioproduction
Bioproduction
Chassis Selection (E. coli BL21(DE3))
Registry of Standard Biological Parts
Plasmid Preparation
Bacterial Culturing
Quality Control/Analysis
Bacterial Processing (Centrifugation, Lysis, DNA Purification)
Cell-Free Systems
Cell Free Reactions
Freeze-Dried Cell Free Systems
miniPCR Tools
Protein Purification
Gibson Assembly / Cloning
Primer Design or Selection
PCR Reactions
Gibson Assembly
Other Cloning Methods (Restriction Enzyme Digestion)
CRISPR
CRISPR/Cas9
Designing Prime Editing gRNA
Technique Deep-Dive 1: His-SUMO Fusion Protein Expression
The His-SUMO fusion strategy is a well-established approach for producing short, toxic, or poorly soluble peptides in E. coli. The 6×His tag at the N-terminus provides a high-affinity handle for Ni-NTA affinity purification, enabling single-step recovery of the fusion protein from crude cell lysate with high selectivity. The SUMO domain (Smt3 from Saccharomyces cerevisiae) serves dual roles: as a solubility-enhancing chaperone that promotes correct folding of the attached peptide, and as a steric shield that physically occludes the antimicrobial cecropin sequence from engaging the inner membrane of the E. coli host during expression — directly addressing the host toxicity problem. Following purification, SUMO protease (Ulp1) cleaves specifically at the SUMO domain’s C-terminal diglycine motif, releasing the cecropin peptide with its native N-terminus intact — a critical requirement since N-terminal truncation or modification of cecropins significantly reduces antimicrobial activity. This technique is validated by the Park et al. (2021) precedent and is directly transferable to other peptide targets by simply replacing the cecropin coding sequence.
Technique Deep-Dive 2: Automated Gel Electrophoresis with Opentrons Flex
The custom gel electrophoresis module represents the core hardware innovation of this project, designed as a miniaturised module that occupies a standard Opentrons Flex deck slot whilst maintaining full compatibility with the platform’s other labware and liquid-handling functions. A miniaturised horizontal gel tank (designed to accommodate 1.5% agarose mini-gels) is precisely dimensioned to fit within the Flex’s working envelope, allowing the robot’s pipette head to operate above and around it without obstruction. A small camera mounted on the pipette head carriage captures high-resolution images of SYBR-stained gels; an onboard AI image-processing pipeline (Python-based, OpenCV) identifies band positions by comparing detected pixel intensities against a DNA ladder reference loaded in a defined lane, calculating expected band positions from user-specified fragment sizes. Once the target band is localised in X-Y space, a biopsy-punch attachment replaces the standard pipette tip and descends to the calculated Z-depth to excise a clean cylindrical gel plug — ready for immediate downstream gel purification. This approach eliminates a classically manual, UV-exposure-dependent, operator-skill-dependent step, replacing it with a standardised, hands-free, and reproducible automated operation.
Potential US-based Industry Council Partners
Addgene — pET-28a vector source
ATCC — Bacillus subtilis ATCC 6633 for antimicrobial assay
Twist Biosciences — synthetic His-SUMO-CecropinB gene fragment
SECTION 5: RESULTS & QUANTITATIVE EXPECTATIONS
Validation Choice
The chosen validation experiment is the demonstration of the Opentrons Flex gel electrophoresis module — specifically, its ability to autonomously detect and excise a target DNA band from a restriction-digested sample, with the recovered DNA confirmed as clonable by successful ligation, transformation, and colony PCR screening. This experiment was selected because the gel module is the core novel hardware contribution of the project, and demonstrating that it produces DNA of equivalent quality to manual excision directly validates the foundational step of the entire automated peptide production pipeline.
Validation Protocol
Cast a 1.5% agarose gel in TAE buffer containing 1× SYBR Safe DNA stain; allow to set in the custom mini-board.
Digest the Twist-synthesised His-SUMO-CecropinB gene fragment with NdeI + XhoI (NEB) at 37°C for 1 hr.
Load digested fragment (Lane 2) and 1 kb DNA ladder (Lane 1) onto gel; run at 100V for 30 min.
Seat the gel board in the designated Opentrons Flex deck slot.
Execute Opentrons Python script: camera captures gel image; OpenCV pipeline calibrates against the ladder and localises the ~543 bp insert band.
Opentrons Flex positions the biopsy-punch attachment over the band coordinates and descends to the calibrated gel depth; gel plug is excised and deposited into a collection tube.
Purify excised plug by chaotropic salt dissolution (Sánchez-Flores 2025); elute in 30 µL TE buffer.
Quantify recovered DNA by Nanodrop (target: ≥5 ng/µL).
Set up ligation with NdeI/XhoI-digested pET-28a (T4 DNA Ligase, 16°C overnight).
Transform ligation product into E. coli BL21(DE3); plate on LB + kanamycin.
Screen 8 colonies by colony PCR using T7 primers; run products on the Opentrons gel module.
Success criterion: ≥4/8 colonies screen positive for the ~650 bp insert band.
Techniques Used
The validation draws on four key synthetic biology techniques. Restriction enzyme digestion using NdeI and XhoI (New England Biolabs) generates the diagnostic band pattern that the Opentrons camera system must correctly resolve — making digestion fidelity the upstream prerequisite for all downstream module calibration. Gel electrophoresis is both the object of validation and the analytical platform: the AI pipeline must distinguish the 543 bp insert from vector backbone fragments, requiring robust image processing and reliable band separation. Automated biopsy-punch excision is the novel hardware step under test, demanding sub-millimetre X-Y-Z positional accuracy from the Opentrons Flex to ensure the punch engages the correct gel region without contaminating adjacent bands. Colony PCR serves as the downstream biological readout that translates hardware performance into a concrete molecular biology outcome — confirming that the automatically excised DNA is intact, correctly sized, and fully functional for restriction cloning.
Data & Quantitative Expectations
In terms of quantitative results, the fully-fledged AMP production protocol would involve measuring inhibition zones at varying concentrations of purified cecropin peptide spotted onto a lawn of Bacillus subtilis — with zone diameter serving as a direct readout of antimicrobial activity
The primary quantitative output is DNA recovery yield (ng/µL by Nanodrop), benchmarked against manual gel excision performed in parallel as a control. Based on the Sánchez-Flores (2025) chaotropic dissolution data and the Park et al. (2021) cloning workflow, an automated recovery of ≥5 ng/µL is expected — sufficient for ligation — with no statistically significant difference from manual recovery (unpaired t-test, p > 0.05), as shown in the simulated dataset below.
Challenges & Troubleshooting
The most likely technical challenge is imprecise band detection caused by variability in gel staining intensity, UV illumination uniformity, or band smearing — any of which could lead the AI pipeline to mislocate the target band and result in an off-target or empty biopsy punch. This is mitigated by using a fixed DNA ladder in a defined lane as an absolute spatial reference and implementing a pipeline confidence threshold below which results are flagged for manual review rather than proceeding automatically. A second significant challenge is biopsy-punch depth calibration: insufficient penetration depth recovers inadequate DNA, whilst over-penetration introduces agarose debris that inhibits downstream ligation and transformation — addressed by calibrating gel thickness at casting time and performing a test punch on a sacrificial gel before each experimental run. Gel-to-gel variability in agarose concentration and band migration distance is an ongoing experimental limitation mitigated by strictly standardising gel preparation (fixed agarose mass, buffer volume, run time, and voltage) and designing the AI pipeline to tolerate a defined range of positional uncertainty. Finally, if colony PCR success rates fall below 50%, the workflow will be benchmarked against a commercial silica-column gel extraction kit to quantify the yield gap and identify whether the bottleneck lies in the biopsy-punch excision, the chaotropic dissolution step, or the ligation conditions.
Sánchez-Flores, A. et al. (2025). Cost-effective DNA extraction from agarose gels using chaotropic salts or freeze-thaw cycles. Nature Scientific Reports. https://www.nature.com/articles/s41598-025-87572-w
Navigating this project led me into regions of biotechnology and synthetic biology I had never dealt with before as a 1st year student still building from the basics. Because of this, I frequently used AI (Claude) for explanations and solutions to small hurdles I encountered — though I always back-tested the methods provided against peer-reviewed research online.
After seeing Ronan’s section on how to integrate Claude Code into the GitHub repo (unfortunately much later than I would have preferred!), I also used Claude to help with formatting and write-up — ensuring everything was written in my own words, but using Claude Code to help place those words correctly, handle images, and structure the page. It definitely saved hours of time and made learning this webpage format of project documentation fast enough to get done within the time constraints I’ve been under. Massive thank you Ronan!