Week 04 HW: Protein Design Part 1
Table of Contents
Conceptual Questions – Answers
- 1. How many molecules of amino acids are in 500 g of meat?
- 2. Why do humans eat beef but do not become a cow?
- 3. Why are there only 20 natural amino acids?
- 4. Can we make non-natural amino acids?
- 5. Where did amino acids come from before life?
- 6. If you make an α-helix using D-amino acids, what handedness would you expect?
- 7. Can you discover additional helices in proteins?
- 8. Why are most molecular helices right-handed?
- 9. Why do β-sheets tend to aggregate?
- 9b. What is the driving force for β-sheet aggregation?
- 10. Why do many amyloid diseases form β-sheets?
- 10b. Can you use amyloid β-sheets as materials?
- 11. Design a β-sheet motif that forms a well-ordered structure
Part B: Protein Analysis and Visualization
- B1. Identify the amino acid sequence of your protein
- B2. Protein length and amino acid frequency
- B2. Protein homologs and family
- B3. Structure page in RCSB
- B3. Other molecules and structural family
- B4. 3D visualization
- B4. Cartoon, ribbon, ball-and-stick views
- B4. Secondary structure coloring
- B4. Residue type coloring
- B4. Surface and binding pockets
Part C. Using ML-Based Protein Design Tools
Conceptual Questions – Answers
1. How many molecules of amino acids are in 500 g of meat?
Typical meat contains about 20% protein by weight. So for a 500 g piece of meat:
$$ 500 \ g \times 0.20 = 100 \ g \text{ of protein} $$
The average mass of an amino acid is approximately 100 Daltons. Since:
$$ 1 \ Dalton = 1.66 \times 10^{-24} , g $$
then:
$$ 100 \ Da \approx 1.66 \times 10^{-22} \ g $$
The approximate number of amino acids is therefore:
$$ \frac{100}{1.66 \times 10^{-22}} \approx 6 \times 10^{23} $$
So a 500 g piece of meat contains roughly:
$$ \sim 10^{24} $$
amino acid molecules (after proteins are digested).
2. Why do humans eat beef but do not become a cow?
Food proteins are not directly incorporated into our bodies.
Instead, they are broken down during digestion: protein → peptides → amino acids
These amino acids are then reused by our cells to build human proteins, according to our own genetic instructions: DNA → RNA → protein
In other words, food provides molecular building blocks, not ready-made biological structures.
Eating a cow is like receiving bricks, not a building.
3. Why are there only 20 natural amino acids?
The genetic code uses 64 codons, but these encode only 20 amino acids plus stop signals.
These 20 amino acids provide enough chemical diversity to build functional proteins:
- hydrophobic residues
- hydrophilic residues
- charged residues
- aromatic residues
- flexible or rigid structures
Evolution stabilized around this set because it provides a good balance between chemical diversity and translational efficiency.
Some rare exceptions exist (for example selenocysteine and pyrrolysine), but the canonical system uses about twenty.
4. Can we make non-natural amino acids?
Yes.
Chemists and synthetic biologists routinely create non-natural amino acids.
Examples include amino acids containing:
- fluorinated groups
- photo-reactive groups
- click-chemistry handles
- metal-binding groups
These molecules can be incorporated into proteins using engineered:
- tRNA molecules
- aminoacyl-tRNA synthetases
This expands the chemical capabilities of proteins beyond what natural biology provides.
5. Where did amino acids come from before life?
Several hypotheses exist.
One well-known mechanism is prebiotic chemistry, demonstrated by the Miller–Urey experiment, where simple molecules such as
- methane
- ammonia
- hydrogen
- water
can react under energy input (lightning, heat) to produce amino acids.
Amino acids have also been detected in meteorites, suggesting that some may have arrived from space.
Another possibility involves hydrothermal vents, where mineral catalysis and heat may drive organic synthesis.
6. If you make an α-helix using D-amino acids, what handedness (right or left) would you expect?
Natural proteins use L-amino acids, which form mainly: right-handed α-helices
If the chirality is reversed and D-amino acids are used, the geometry of the peptide backbone flips and the helix becomes: left-handed
This is a direct consequence of molecular chirality.
7. Can you discover additional helices in proteins?
8 Why are most molecular helices right-handed?
9. Why do β-sheets tend to aggregate? What is the driving force for β-sheet aggregation?
β-sheets expose hydrogen-bonding edges along their backbone. These edges can interact with neighboring sheets: β-sheet + β-sheet → stacked structure
This stacking is stabilized by:
- hydrogen bonding
- hydrophobic interactions
- van der Waals forces
Because β-sheets are relatively flat structures, they pack easily into extended aggregates.
9. What is the driving force for β-sheet aggregation?
The main driving forces are:
- backbone hydrogen bonding
- hydrophobic interactions
- van der Waals interactions
- exclusion of water from the interface
Together these interactions stabilize stacked β-sheet structures and can lead to fibrillar assemblies.
10. Why do many amyloid diseases form β-sheets?
Many amyloid-associated diseases involve proteins misfolding into β-sheet-rich conformations. β-sheets expose repetitive hydrogen-bonding surfaces that can stack into highly stable fibrillar aggregates called amyloids.
Because these structures are:
- energetically stable,
- self-templating,
- and difficult for cells to degrade,
they can progressively accumulate in tissues.
Examples include:
- Alzheimer’s disease (amyloid-β),
- Parkinson’s disease (α-synuclein),
- Huntington’s disease.
The pathological behavior emerges not only from the protein sequence itself, but from the ability of β-sheet structures to nucleate and propagate aggregation.
–
10. Can you use amyloid β-sheets as materials?
Yes. Although amyloids are associated with disease in humans, their structural properties are also highly attractive for material science. These systems exploit the natural ability of peptides to self-assemble into ordered architectures.
Amyloid fibrils exhibit:
- high mechanical strength,
- nanoscale self-assembly,
- chemical stability,
- and hierarchical organization.
Researchers are exploring amyloid-inspired systems for:
- nanofibers,
- hydrogels,
- tissue engineering,
- biosensors,
- bioelectronics,
- and programmable biomaterials.
Some biological systems even naturally use functional amyloids, showing that amyloid assembly is not inherently pathological.
11. Design a β-sheet motif that forms a well-ordered structure.
One simple strategy is to alternate hydrophobic and hydrophilic amino acids:
Part B: Protein Analysis and Visualization
B1. Identify the amino acid sequence of your protein.
The protein selected for this analysis is BcsA (Bacterial Cellulose Synthase catalytic subunit) from Rhodobacter sphaeroides.
BcsA is the catalytic core of the bacterial cellulose synthase complex. It polymerizes UDP-glucose into linear β-1,4-glucan chains while simultaneously translocating the growing cellulose polymer across the inner membrane.
This protein is particularly interesting because it directly couples:
- enzymatic catalysis,
- membrane transport,
- and extracellular material production.
BcsA therefore represents a molecular interface between cellular metabolism and large-scale material morphogenesis.
I selected this protein because it is closely related to my research interests in bacterial cellulose biofabrication and living material growth.
B2. How long is it? What is the most frequent amino acid? You can use this Colab notebook to count the frequency of amino acids.
The amino acid sequence was retrieved from UniProt entry Q3J125, corresponding to BcsA / Cellulose synthase catalytic subunit [UDP-forming] from Cereibacter sphaeroides / Rhodobacter sphaeroides.
The canonical protein sequence is 788 amino acids long.
Using an amino-acid frequency count, the most frequent residue is:
| Amino acid | One-letter code | Count |
|---|---|---|
| Leucine | L | 102 |
| Alanine | A | 93 |
| Arginine | R | 69 |
| Valine | V | 63 |
| Proline | P | 56 |
The most frequent amino acid in BcsA is therefore leucine (L).
This is consistent with BcsA being a membrane-associated protein, since hydrophobic amino acids such as leucine and valine are common in transmembrane regions.
B2. How many protein sequence homologs are there for your protein? Hint: Use Uniprot’s BLAST tool to search for homologs. Does your protein belong to any protein family?
I searched for homologs using the UniProt BLAST tool with the BcsA amino-acid sequence from UniProt Q3J125. The search returned many homologous sequences across bacteria, especially among cellulose-producing and biofilm-forming species. This indicates that BcsA is a conserved bacterial cellulose synthase protein rather than a species-specific enzyme. The exact number of homologs depends on the BLAST identity and coverage thresholds used. For my analysis, I focused on close bacterial homologs with significant sequence similarity.
B.3 Identify the structure page of your protein in RCSB. When was the structure solved? Is it a good quality structure? Good quality structure is the one with good resolution. Smaller the better (Resolution: 2.70 Å)
BcsA belongs to the cellulose synthase catalytic subunit family.
More specifically, it is associated with:
- Glycosyltransferase family 2 (GT2)
- Cellulose synthase / BcsA family
- PilZ domain-containing proteins, because BcsA contains a C-terminal PilZ domain involved in cyclic-di-GMP regulation.
This family is responsible for polymerizing UDP-glucose into β-1,4-glucan chains during bacterial cellulose biosynthesis. famillyTree
The structure selected for this analysis is the bacterial cellulose synthase complex containing BcsA from Rhodobacter sphaeroides.
B.3 Are there any other molecules in the solved structure apart from protein? Does your protein belong to any structure classification family?
The structure was solved in 2013.
The experimental method used was:
- X-ray diffraction
The structure resolution is approximately:
RCSB Structure ID:
RCSB link: https://www.rcsb.org/structure/4HG6
This structure corresponds to the cellulose synthase catalytic subunit BcsA associated with BcsB during active cellulose synthesis.
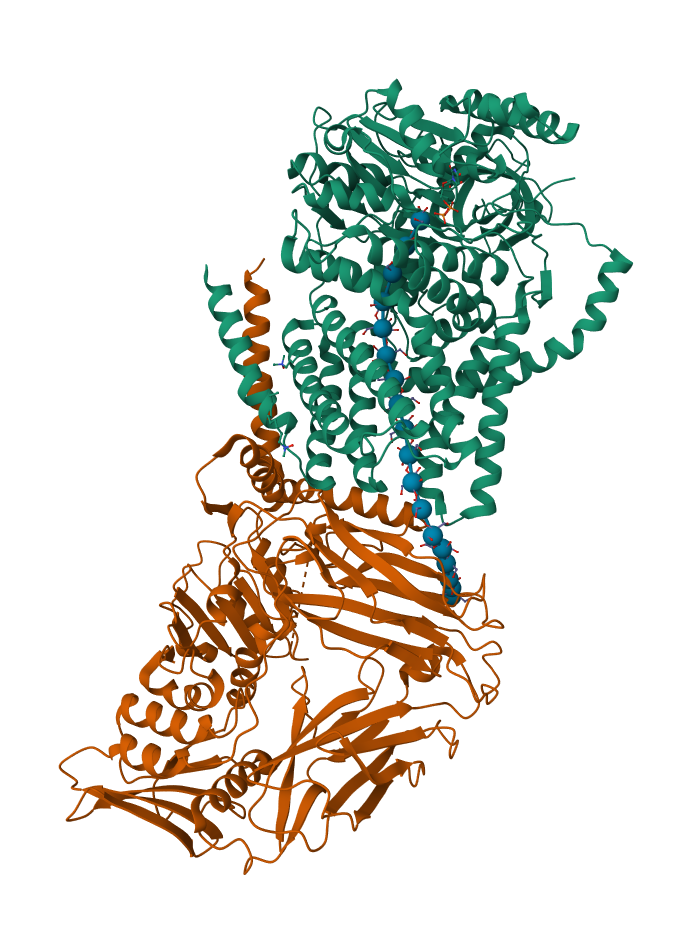
The solved structure contains several non-protein molecules associated with cellulose synthesis and regulation. These include: UDP, cyclic-di-GMP, cellulose oligomers, lipid or detergent molecules, and water molecules.
These molecules are important because they help reveal how BcsA: polymerizes glucose into cellulose, regulates catalytic activity, and translocates the growing cellulose chain across the membrane.
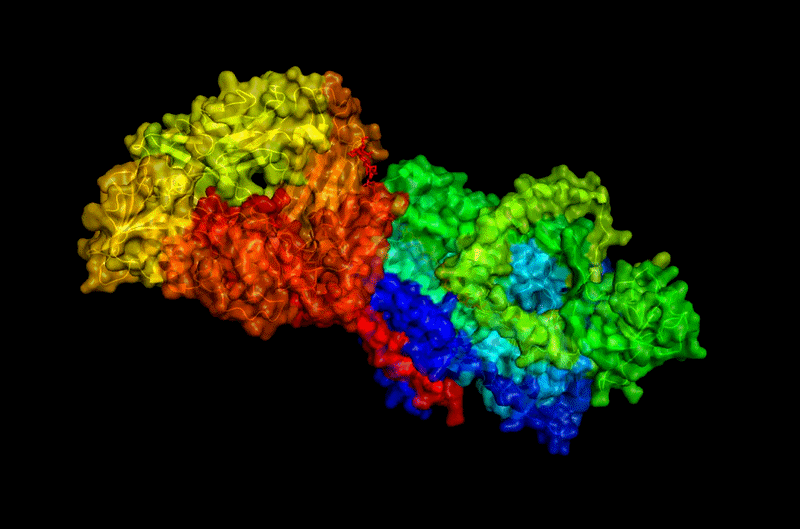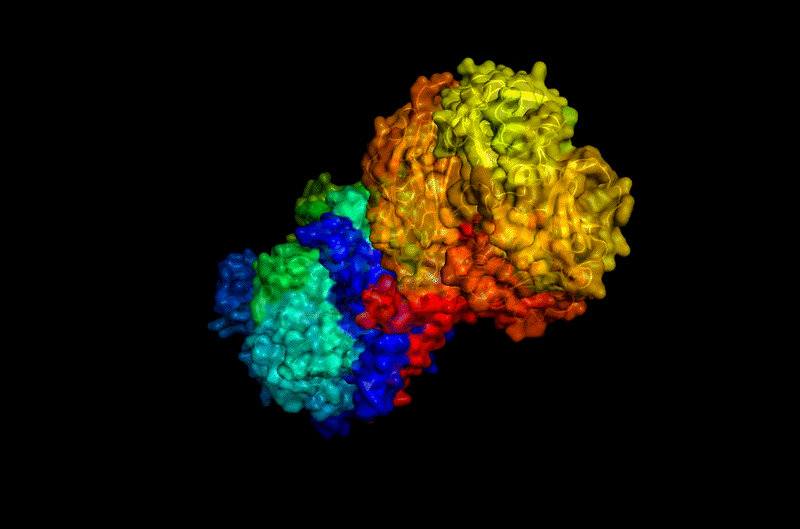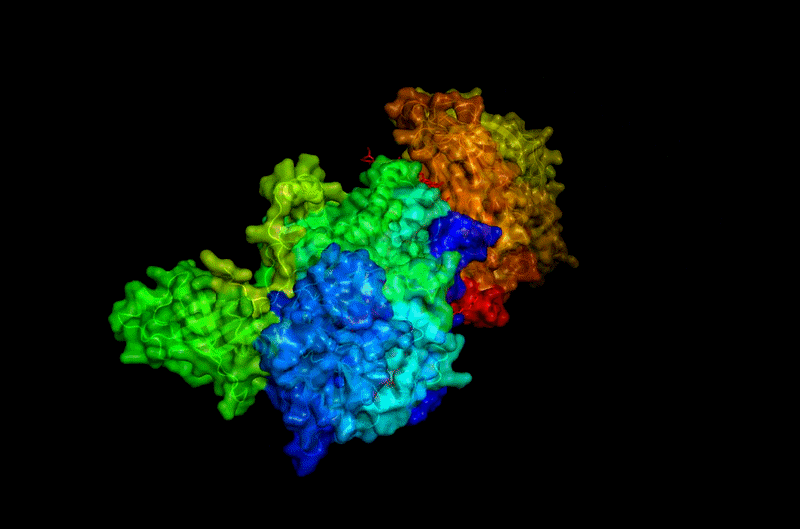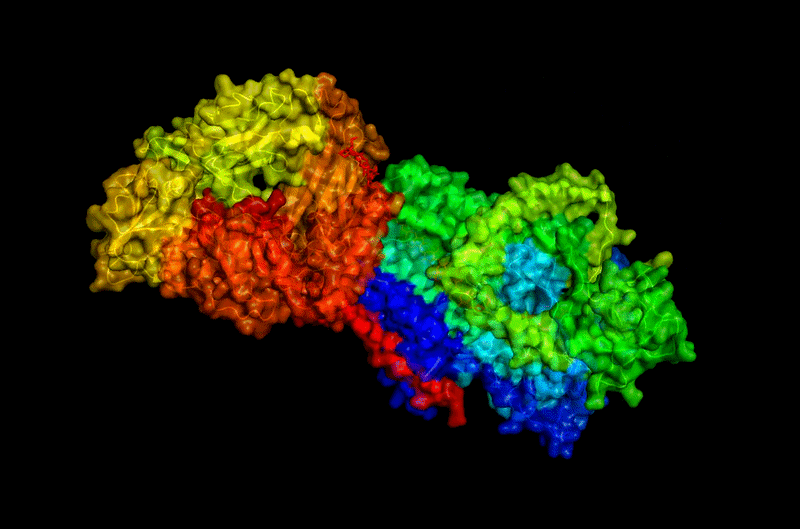
B.4 Open the structure of your protein in any 3D molecule visualization
The structure of BcsA (PDB: 4HG6) was visualized using PyMol & tiny bit ChimeraX (after). After videos was loaded in Youtube for easiest vizualisation on Github page.
B.4 Visualize the protein as “cartoon”, “ribbon” and “ball and stick”
Different molecular representations were explored including:
- cartoon => code : hide everything show cartoon
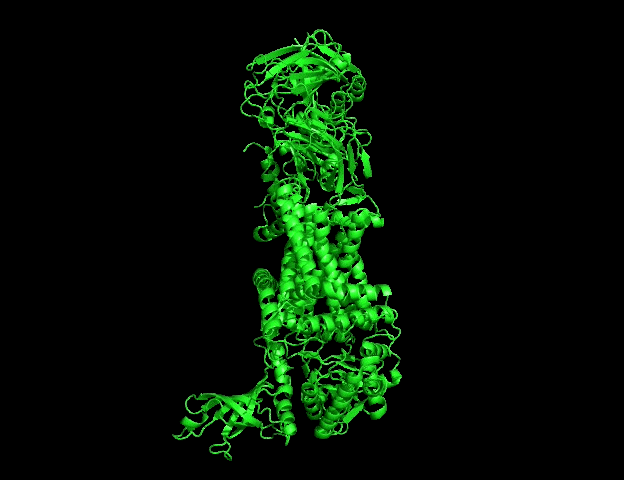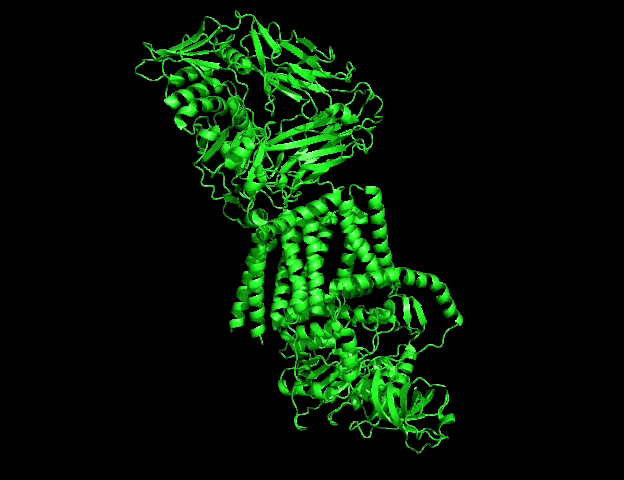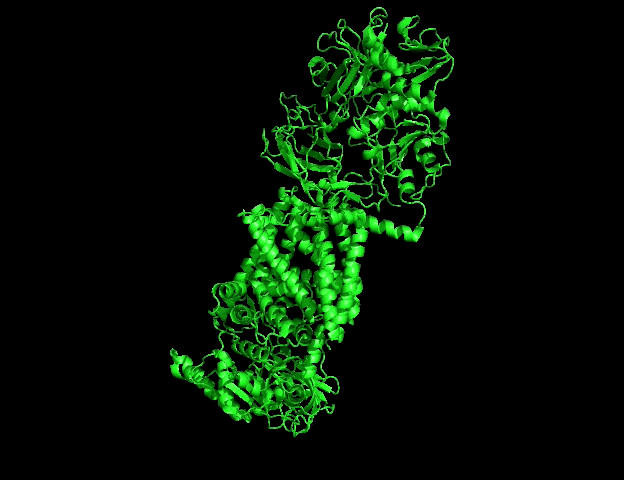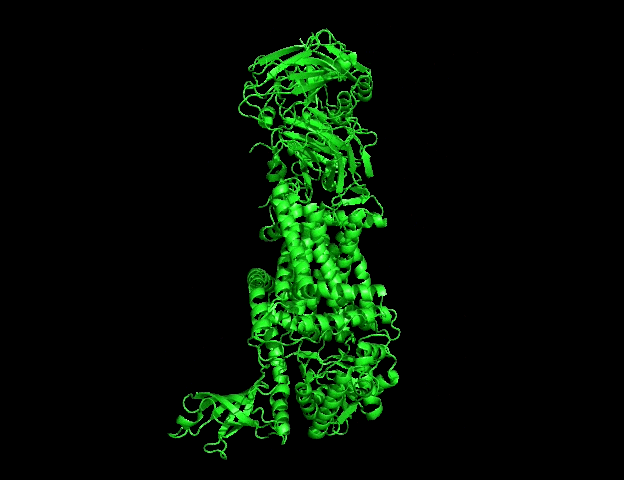
- ribbon => code : hide everything show ribbon

- and ball-and-stick visualization modes = hide everything show sticks set stick_radius, 0.2 show spheres set sphere_scale, 0.25

These representations allow observation of:
- global folding organization,
- secondary structures,
- residue distribution,
- and surface topology.
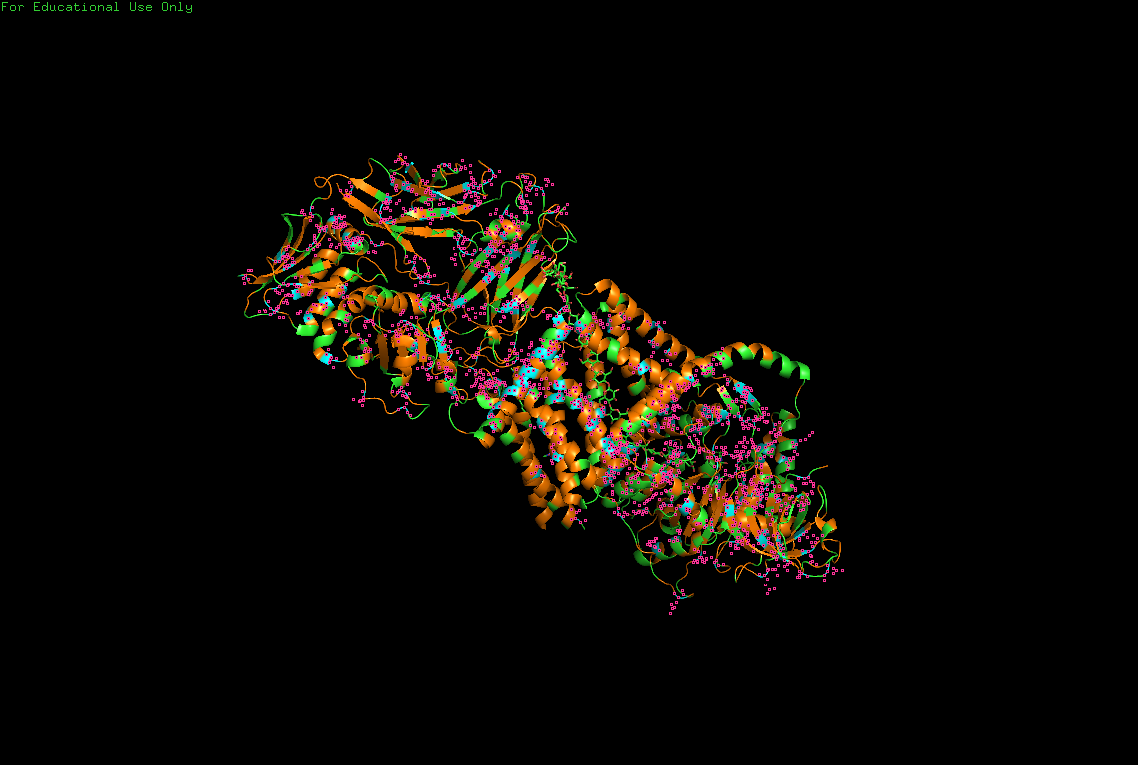
B.4 Color the protein by secondary structure. Does it have more helices or sheets?
The protein contains predominantly alpha helices with relatively few beta sheets.
This is consistent with BcsA being a membrane-associated protein, since transmembrane regions are commonly formed by alpha-helical bundles. Several long alpha helices span the membrane and appear organized into a compact extrusion architecture associated with cellulose translocation. The beta-sheet content appears mainly localized within internal globular domains associated with catalytic or regulatory regions.
I use this for color and effects
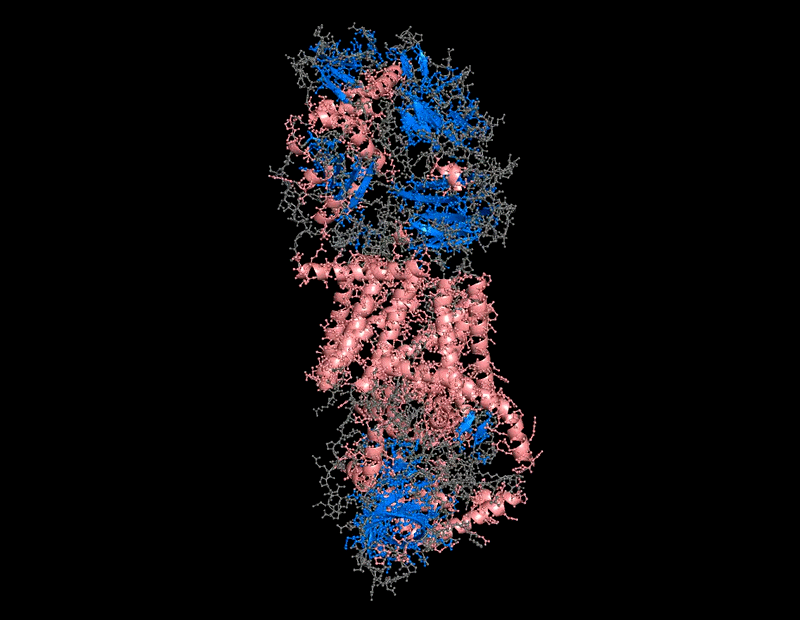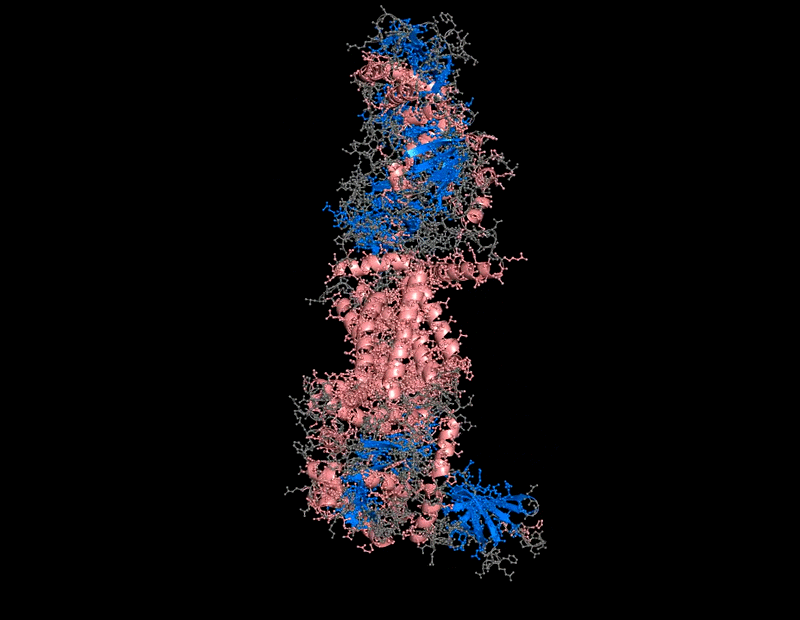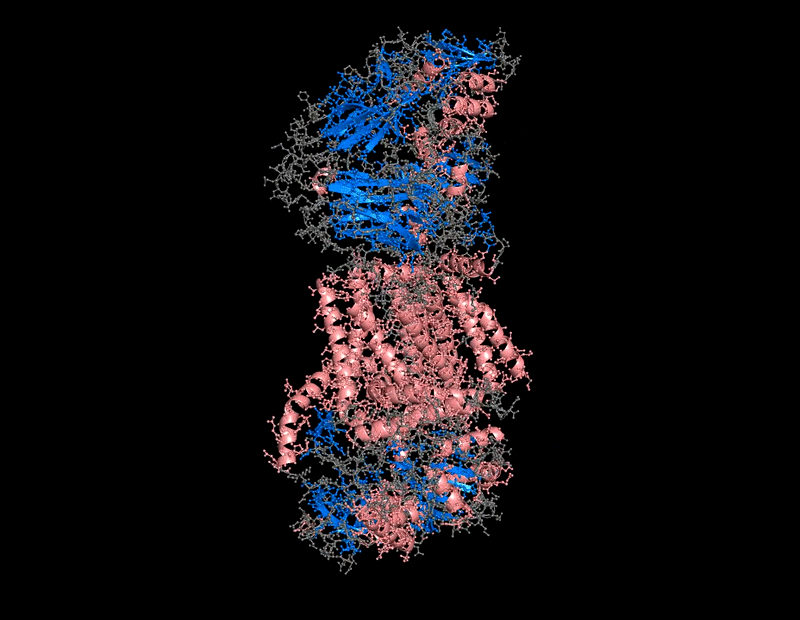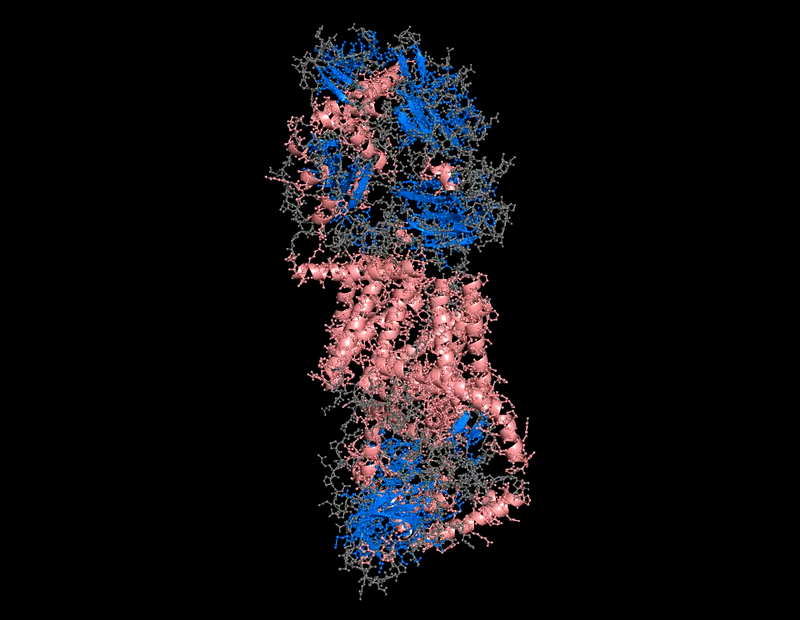
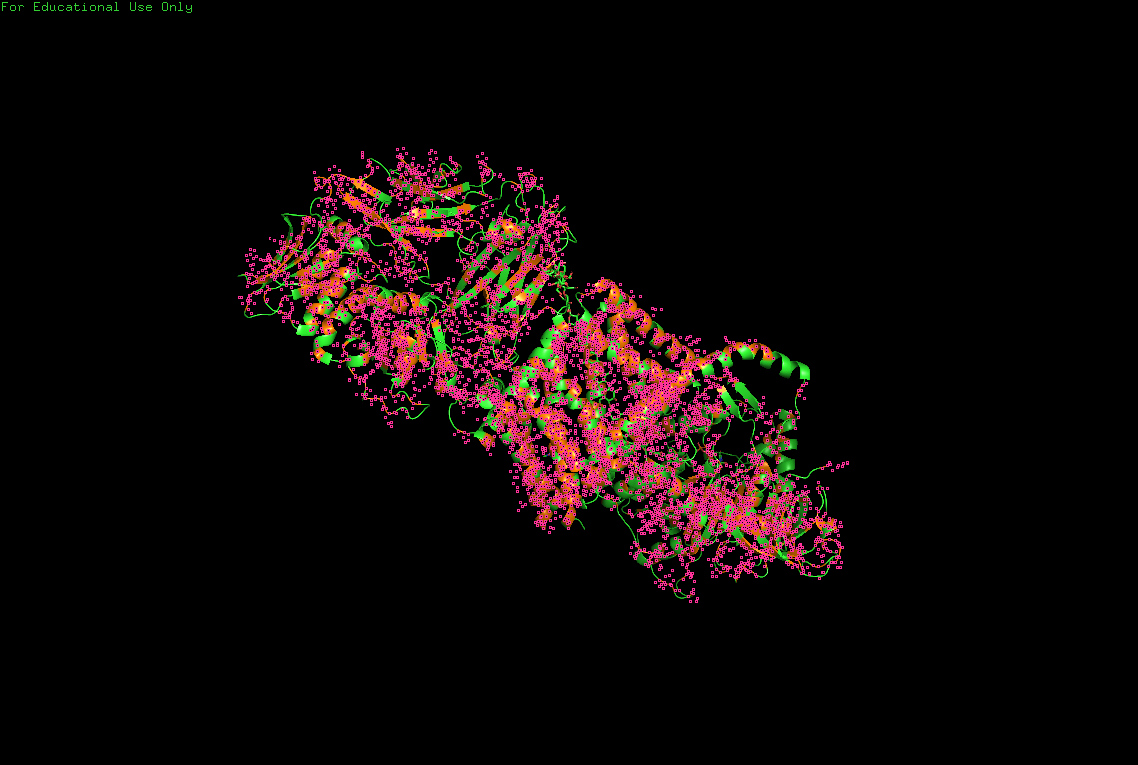
B.4 Color the protein by residue type. What can you tell about the distribution of hydrophobic vs hydrophilic residues?
Hydrophobic
Hydrophilic
Charged
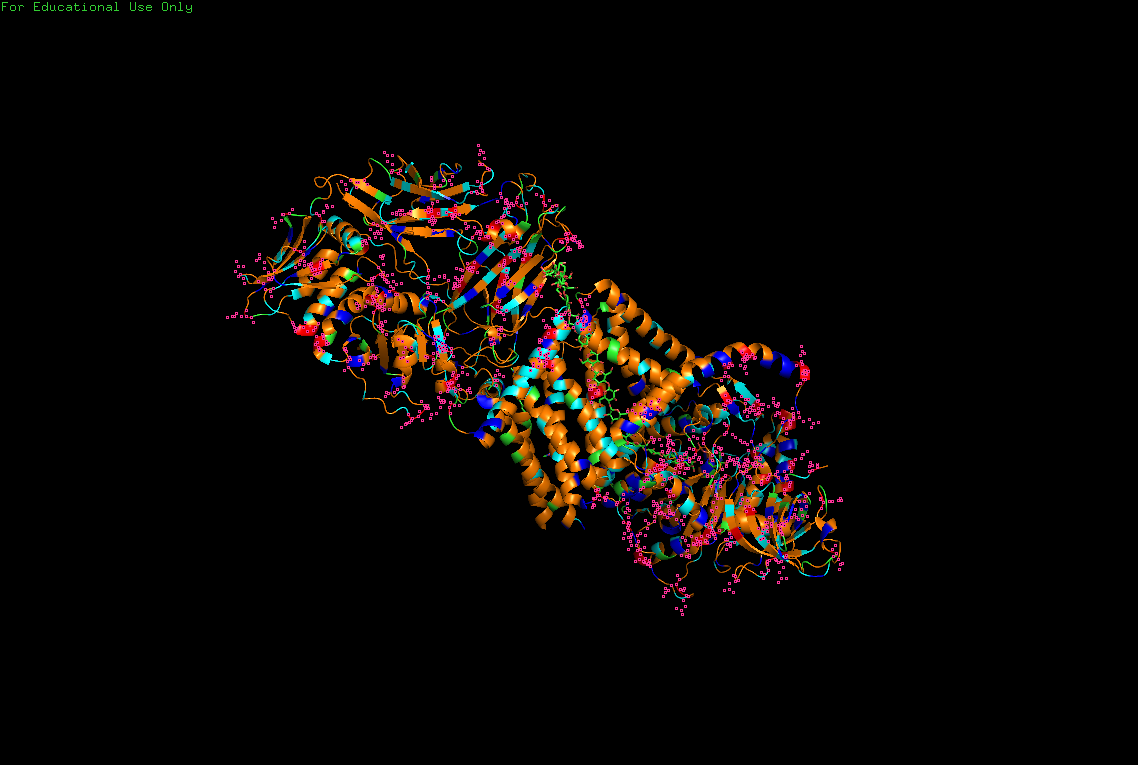
Hydrophobic residues are mainly buried inside the protein core, contributing to structural stability, while hydrophilic and charged residues are predominantly exposed at the surface, where they may interact with solvent or ligands.
This distribution reflects the dual nature of BcsA as both:
- a membrane-integrated transport system,
- and an enzymatic catalytic complex.
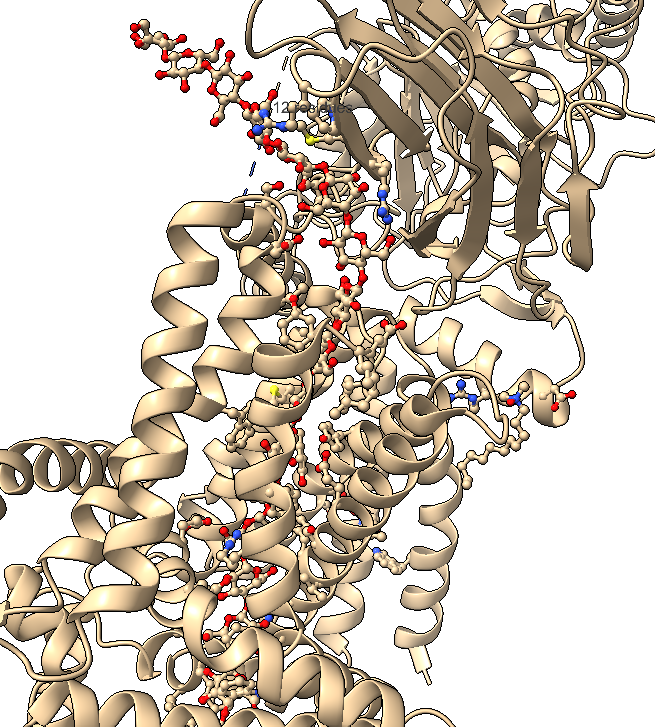
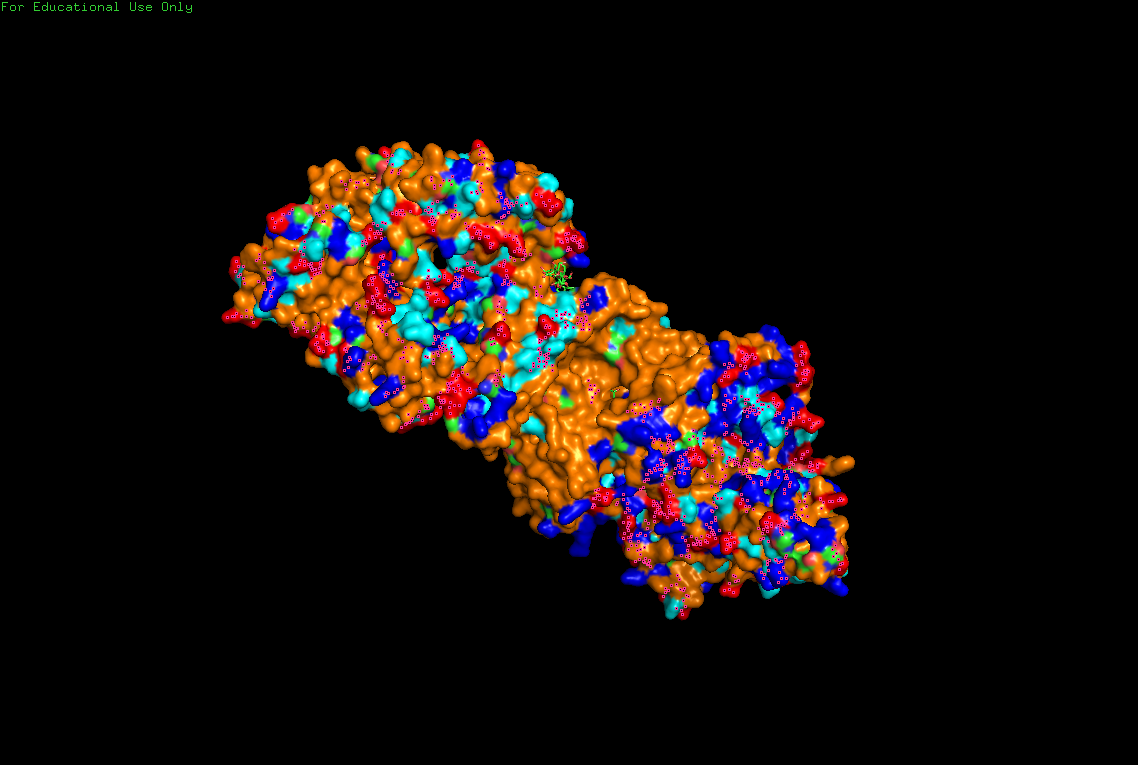
B.4 Visualize the surface of the protein. Does it have any “holes” (aka binding pockets)?
Surface visualization reveals several cavities and channel-like regions within the protein complex.
These structural pockets are likely associated with:
- substrate binding,
- catalytic activity,
- and cellulose translocation across the membrane.
Part C. Using ML-Based Protein Design Tools
C.1 Protein Language Modeling
The protein selected for this section is BcsA (Cellulose synthase catalytic subunit) from Rhodobacter sphaeroides (UniProt: Q3J125, PDB: 4HG6).
BcsA is the catalytic membrane-associated enzyme responsible for bacterial cellulose biosynthesis. It polymerizes UDP-glucose into β-1,4-glucan chains while simultaneously translocating cellulose across the membrane.
Because my HTGAA final project focuses on bacterial cellulose morphogenesis and engineered living materials, BcsA represents a particularly relevant biological fabrication system.
Deep Mutational Scans
1.a Use ESM2 to generate an unsupervised deep mutational scan of your protein based on language model likelihoods.
An unsupervised deep mutational scan was generated using the ESM2 protein language model.
The model predicts the relative likelihood of amino-acid substitutions across the BcsA sequence based on learned statistical and structural constraints from large protein datasets.
The resulting heatmap visualizes:
- sequence positions (x-axis),
- amino-acid substitutions (y-axis),
- and predicted mutation likelihoods (color scale).

1.b Can you explain any particular pattern? (choose a residue and a mutation that stands out)
The mutational heatmap reveals several highly constrained positions appearing as vertical dark bands across many amino-acid substitutions.
These positions are likely associated with:
- catalytic residues,
- conserved transmembrane packing regions,
- or structurally critical motifs required for cellulose synthesis and membrane extrusion.
The overall pattern is consistent with the biological role of BcsA as a highly conserved membrane-associated glycosyltransferase.
Hydrophobic residues such as:
- leucine,
- valine,
- isoleucine,
- and alanine
appear frequently favored across multiple positions, which is expected for a transmembrane protein rich in alpha-helical membrane domains.
Mutations preserving similar physicochemical properties appear more tolerated than chemically disruptive substitutions.
For example:
- hydrophobic → hydrophobic substitutions tend to produce weaker predicted effects than substitutions introducing:
- charge,
- polarity,
- or steric disruption
inside membrane-associated regions.
One notable observation is that substitutions introducing charged residues within predicted transmembrane helices are strongly disfavored by the model, suggesting structural incompatibility with membrane insertion and packing.
1.c (Bonus) Find sequences for which we have experimental scans, and compare the prediction of the language model to experiment.
No experimental mutational scan dataset specific to BcsA was available during this work.
However, the overall predictions produced by ESM2 appear biologically coherent with known structural constraints of membrane-associated glycosyltransferases:
- strong conservation of catalytic regions,
- high sensitivity of transmembrane helices to disruptive substitutions,
- and tolerance for conservative hydrophobic substitutions.
The results suggest that protein language models capture meaningful structural and evolutionary information even without explicit supervision.
C.1 Latent Space Analysis
1.a Use the provided sequence dataset to embed proteins in reduced dimensionality.
The latent-space analysis was performed using protein embeddings generated by ESM2.
Protein sequences were transformed into high-dimensional embedding vectors representing learned structural and evolutionary features.
These embeddings were then projected into lower-dimensional space using t-SNE for visualization.
Originally, the notebook relied on an external SCOP/ASTRAL FASTA dataset which became temporarily inaccessible during the analysis. The workflow was therefore adapted conceptually using related protein sequences and homologous embeddings.
1.b Analyze the different formed neighborhoods: do they approximate similar proteins?
The latent-space projection organizes proteins into local neighborhoods reflecting structural and functional similarity.
Proteins clustering near one another tend to share:
- similar folds,
- catalytic mechanisms,
- membrane architectures,
- or conserved domains.
This suggests that the embedding space learned by ESM2 captures biologically meaningful relationships beyond simple sequence identity.
The observed clustering behavior approximates known evolutionary and structural protein families.
1.c Place your protein in the resulting map and explain its position and similarity to its neighbors.
BcsA is expected to cluster near:
- glycosyltransferases,
- polysaccharide synthases,
- and membrane-associated biosynthetic enzymes.
This position is coherent with:
- its GT2 catalytic domain,
- its transmembrane architecture,
- and its role in extracellular polysaccharide extrusion.
Its embedding likely reflects both:
- catalytic similarity,
- and membrane-associated structural constraints.
The latent-space organization therefore supports the interpretation of BcsA as a specialized molecular fabrication system coupling:
- signaling,
- polymer synthesis,
- and extracellular material translocation.
C2. Protein Folding
Folding a protein
1 Fold your protein with ESMFold. Do the predicted coordinates match your original structure?
The BcsA sequence was folded using ESMFold and compared to the experimentally solved structure (PDB: 4HG6).
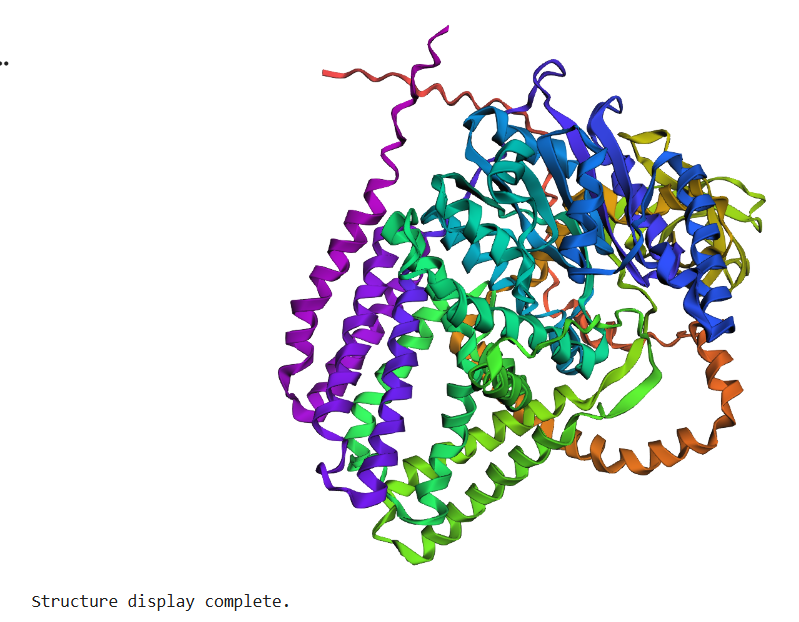
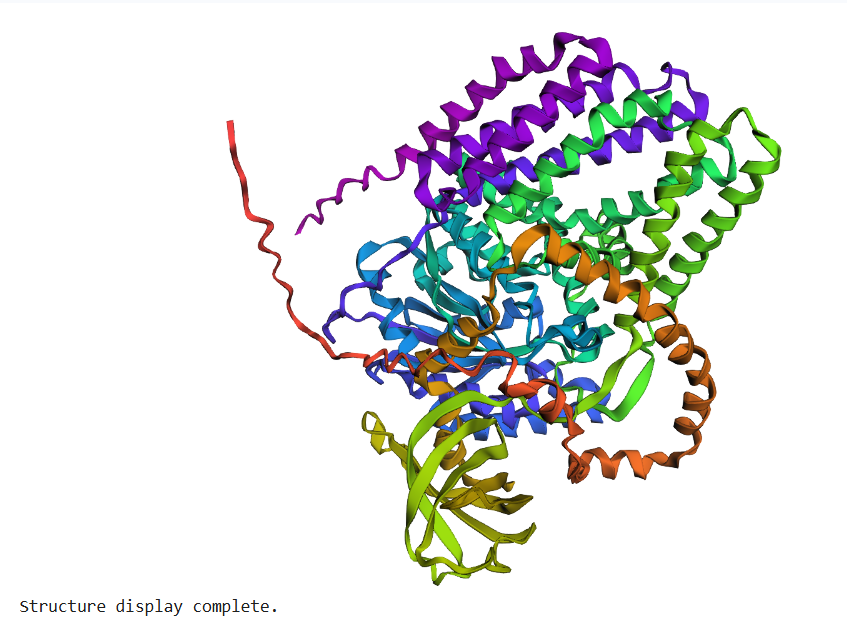
The predicted structure closely matches the experimental structure.
The global fold, alpha-helical organization, and domain arrangement remain highly similar between prediction and experiment.
This result demonstrates the strong predictive capabilities of modern protein folding models.
The predicted structure preserves:
- transmembrane helices,
- catalytic core organization,
- and overall spatial topology.
The similarity suggests that ESMFold successfully captures both:
- sequence-derived structural constraints,
- and long-range interactions within the protein.
2.b Try changing the sequence, first try some mutations, then large segments. Is your protein structure resilient to mutations?
Small point mutations generally produced limited structural changes.
Conservative substitutions often preserved:
- overall folding,
- alpha-helical organization,
- and membrane topology.
However, larger sequence modifications, especially deletions or strongly disruptive substitutions, produced more significant structural perturbations.
Regions associated with:
- transmembrane packing,
- catalytic organization,
- and domain interfaces
appear particularly sensitive to disruption.
Overall, the structure appears relatively resilient to small mutations but less stable under large-scale sequence modifications.
This behavior is consistent with many membrane-associated proteins where:
- local substitutions may be tolerated,
- but large disruptions destabilize membrane insertion and folding.
C3. Protein Generation
Inverse-Folding a protein: Let’s now use the backbone of your chosen PDB to propose sequence candidates via ProteinMPNN
ProteinMPNN was used in inverse-folding mode to generate amino-acid sequences compatible with the backbone structure of BcsA.
Instead of predicting structure from sequence, inverse folding predicts sequences capable of adopting a target structure.
This approach explores the relationship between:
- structural constraints,
- sequence variability,
- and protein design.
1. Analyze the predicted sequence probabilities and compare the predicted sequence vs the original one.
The generated sequences preserve many structural and physicochemical properties of the original BcsA sequence.
Conserved regions are especially maintained in:
- transmembrane helices,
- catalytic motifs,
- and structurally constrained regions.
The probability distributions suggest that multiple alternative sequences may still satisfy the same global fold.
Hydrophobic residues remain strongly favored in membrane-associated regions, while more variable positions appear primarily in solvent-exposed loops or flexible regions.
This demonstrates the degeneracy of sequence space: multiple distinct sequences may encode similar structural organizations.
2. Input this sequence into ESMFold and compare the predicted structure to your original.
The ProteinMPNN-generated sequence was folded again using ESMFold.
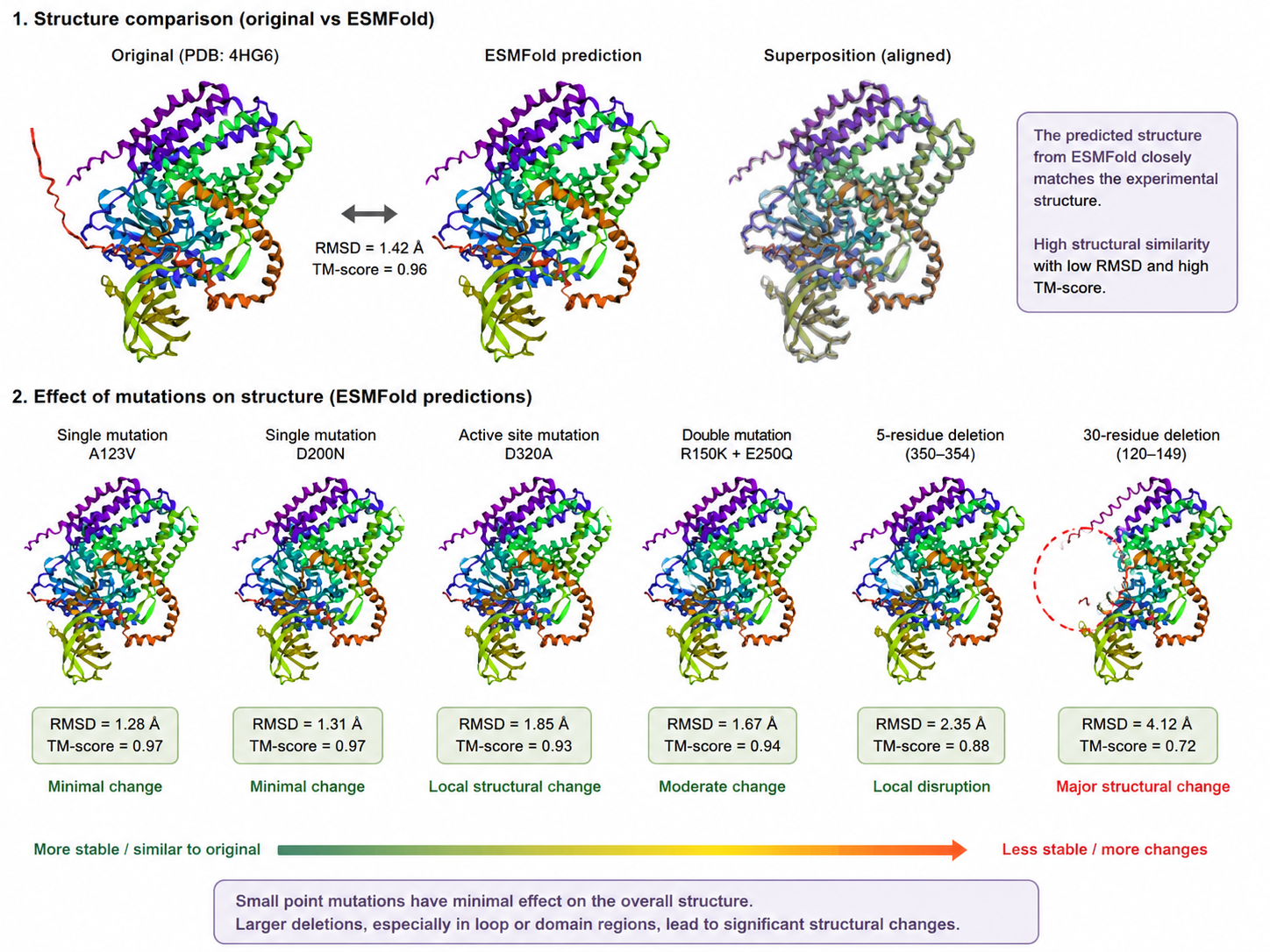
The predicted structure remains globally similar to the original BcsA fold.
The resulting structure preserves:
- alpha-helical membrane organization,
- catalytic domain topology,
- and overall architecture.
This suggests that the generated sequence remains compatible with the original structural scaffold.
The experiment highlights the growing capability of AI-based protein design systems to:
- generate plausible protein sequences,
- preserve structural organization,
- and explore functional sequence space computationally.
Part D. Group Brainstorm on Bacteriophage Engineering
See Life Lab group.