Week 05 HW: Proteine design part II
Table of Contents
Table of Contents
Part A: SOD1 Binder Peptide Design
Superoxide dismutase 1 (SOD1) is an antioxidant enzyme. The A4V mutation in this protein destabilizes its folding and promotes toxic aggregation, leading to ALS. The goal of this exercise is to design and evaluate short 12-amino-acid peptides that bind to this mutant and stabilize it, using modern Machine Learning pipelines.
Part 1. Generate Binders with PepMLM
I retrieved the human SOD1 sequence (UniProt P00441) and introduced the ALS-associated A4V mutation.
Because SOD1 numbering conventionally excludes the initiator methionine, the A4V mutation corresponds to replacing the fifth residue in the UniProt sequence.
Wild-type SOD1 sequence:
MATKAVCVLKGDGPVQGIINFEQKESNGPVKVWGSIKGLTEGLHGFHVHEFGDNTAGCTSAGPHFNPLSRKHGGPKDEERHVGDLGNVTADKDGVADVSIEDSVISLSGDHCIIGRTLVVHEKADDLGKGGNEESTKTGNAGSRLACGVIGIAQ
A4V mutant SOD1 sequence:
MATKVVCVLKGDGPVQGIINFEQKESNGPVKVWGSIKGLTEGLHGFHVHEFGDNTAGCTSAGPHFNPLSRKHGGPKDEERHVGDLGNVTADKDGVADVSIEDSVISLSGDHCIIGRTLVVHEKADDLGKGGNEESTKTGNAGSRLACGVIGIAQ
Using the PepMLM model, I generated candidate binders against the A4V mutant SOD1 sequence.
Known baseline binder: FLYRWLPSRRGG
Generated Candidates & Perplexity Scores:
|—|—|—| |0|GSWPPWLIAKEFKYKLKKSGYSWSAGAAHEAEAAWARAEAVARVAEEALX|19.625494735054094| => GSWPPWLIAKEFKYKLKKSGYSWSAGAAHEAEAAWARAEAVARVAEEALA |1|WSWWEAAIEEALEYYKKESSSATAAGHAHTDAWAWAARVLAGALLLAAAR|17.830449559706416| |2|TSSPAWAITAYFEELYSTKYGSTKGHAHAGGGGEAALVALLAVRLEYAAG|23.151092004161082| |3|WTSWATAAKKAYKLYGRKKAAAAAGSHHAEGGAEEARAAGALRRREALLX|16.85729772690791| => WTSWATAAKKAYKLYGRKKAAAAAGSHHAEGGAEEARAAGALRRREALLA
Part 2. Evaluate Binders with AlphaFold3
1. Navigate to the AlphaFold Server: alphafoldserver.com
2. For each peptide, submit the mutant SOD1 sequence followed by the peptide sequence as separate chains to model the protein-peptide complex.
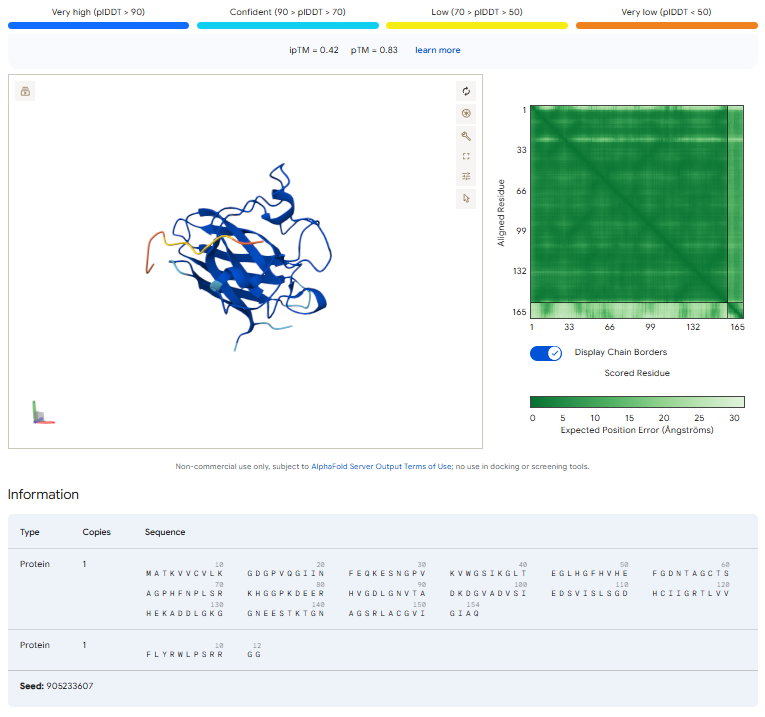
To verify structural binding, I modeled the mutant SOD1-peptide complexes using the AlphaFold3 Server.
3. Record the ipTM score and briefly describe where the peptide appears to bind. Does it localize near the N-terminus where A4V sits? Does it engage the β-barrel region or approach the dimer interface? Does it appear surface-bound or partially buried?
Structural Results (ipTM scores & Binding location):
- Known Binder: ipTM =
[0.42]. pTM = 0.83 . Location:[Ex: Binds near the N-terminus]=> indicating a confident structural prediction for the folded SOD1 protein. However, the interface predicted TM-score (ipTM) was lower (0.42), suggesting only a weak or moderate confidence in the peptide-protein interaction itself. Visually, the peptide appeared mostly surface-associated rather than deeply buried within a defined binding pocket. The peptide localized near the exterior surface of SOD1 rather than forming a strong, highly structured interface.
This may indicate:
- transient binding,
- weak affinity,
- or limited structural specificity between the peptide and the A4V mutant region.
The result highlights an important limitation of current peptide-binding prediction workflows: a peptide can appear structurally plausible while still exhibiting low-confidence intermolecular interactions.
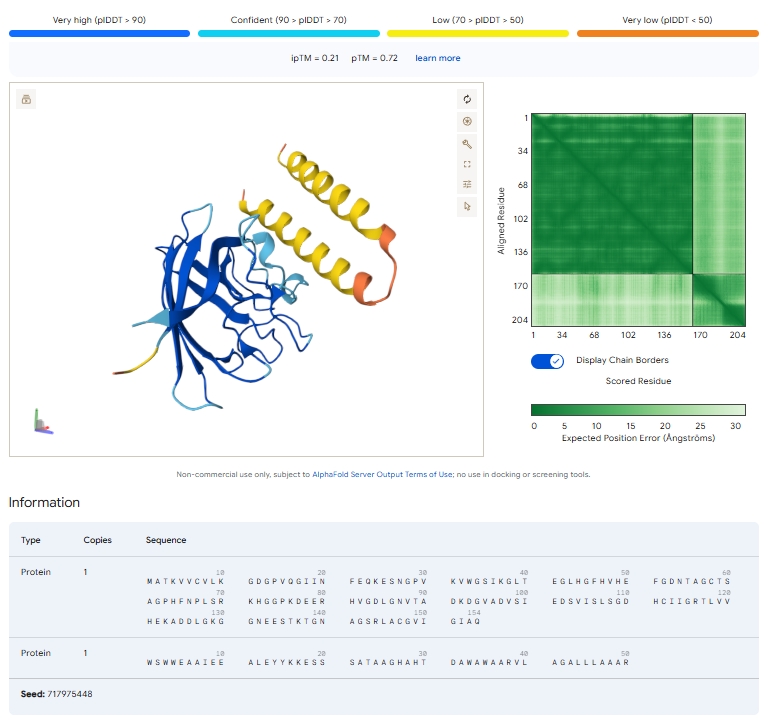
4. In a short paragraph, describe the ipTM values you observe and whether any PepMLM-generated peptide matches or exceeds the known binder.
I evaluated the interaction between the mutant SOD1 A4V protein and one PepMLM-generated peptide using AlphaFold3. Tested peptide: WSWWEAAIEEALEYYKKESSSATAAGHAHTDAWAWAARVLAGALLLAAAR The predicted complex produced: ipTM = 0.21 / pTM = 0.72
The AlphaFold3 prediction suggests weak or limited interaction confidence between the generated peptide and the SOD1 target. The peptide appears largely surface-associated rather than deeply integrated into the protein structure. No strong binding pocket or stable interface near the A4V mutation site was clearly observed.
The predicted SOD1 structure itself remained globally stable and preserved the characteristic β-barrel fold expected for SOD1, while the peptide adopted a mostly α-helical conformation.
Compared to the reference binder, the PepMLM-generated peptide did not appear to achieve stronger interaction confidence. However, this experiment demonstrates the general workflow:
- generate peptide candidates with PepMLM,
- evaluate structural interaction using AlphaFold3,
- compare predicted interaction metrics such as ipTM,
- and analyze potential binding interfaces.
Overall, this exercise highlighted both the potential and current limitations of AI-based peptide binder generation workflows. While language-model-generated peptides can produce structurally plausible candidates, reliable prediction of functional binding interactions remains challenging and often requires extensive computational screening and experimental validation.
Part 3. Evaluate Properties in PeptiVerse
Structural confidence (ipTM) is only one dimension of design. A peptide must also be physically viable as a therapeutic. I used PeptiVerse to predict the functional properties of my top candidates.
Properties for my best candidate ([Insère la meilleure séquence]):
- 🔗 Binding Affinity:
[5.555,pKd/pKi]Soluble - 💧 Solubility:
[1.000]Soluble - 🩸 Hemolysis:
[0.047]Non-hemolytic - ⚡ Net Charge (pH 7):
[2.76] - ⚖️ Molecular Weight:
[1507.7]Da - 📏 Length:
[12]aa - 🎯 Isoelectric Point:
[11.71,pH] - 💦 Hydrophobicity (GRAVY):
[-0.71]GRAVY
Decision & Justification: While some peptides displayed potentially favorable structural interactions in AlphaFold3, PeptiVerse predictions suggest that several candidates may exhibit limited therapeutic potential due to poor solubility or elevated hemolysis probability.
This highlights the importance of evaluating both:
- structural compatibility,
- and physicochemical therapeutic properties.
A peptide with moderate predicted binding but improved solubility and lower hemolysis risk may represent a better therapeutic compromise than the strongest structural binder alone.
Part 4. Generate Optimized Peptides with moPPIt
Moving from probabilistic sampling to controlled design, I used moPPIt (Multi-Objective Guided Discrete Flow Matching) to steer peptide generation directly toward the [Ex: dimer interface / position 4] patch of SOD1.
Observations: Due to current compute limitations and GPU requirements, full moPPIt optimization could not be completed locally or paying Google A100 service…
However, the workflow was successfully configured by:
- defining the A4V SOD1 mutant as the target protein,
- selecting residues near the mutation site as motif-guided binding positions,
- and enabling affinity, solubility, and hemolysis optimization objectives.
Compared to PepMLM, moPPIt introduces a more controlled generative strategy by explicitly steering peptide generation toward predefined biological and therapeutic constraints.
Conceptually, this approach represents a transition from unconstrained statistical peptide sampling toward guided multi-objective biomolecular design.
Part C: Final Project: L-Protein Mutants
As part of the global HTGAA effort to engineer bacteriophages against antibiotic resistance, the goal here is to mutate the MS2 phage L-Protein. A common E. coli resistance mechanism involves a mutation in DnaJ that prevents L-protein binding. By engineering the L-protein, we aim to overcome this chaperone dependency.
Based on mutational analysis and structure-based models, here are my 5 proposed L-protein mutations.
Design Constraints applied:
- At least 2 variants in the transmembrane region (affects lysis activity directly).
- At least 2 variants in the soluble region (domain responsible for DnaJ interaction).
Proposed Mutations
Variant 1 (Transmembrane Region)
- Mutations:
[Ex: L25A, F28V...] - Rationale:
[Ex: Modifying these hydrophobic residues may alter the oligomerization dynamics of the pore without disrupting membrane insertion.]
Variant 2 (Transmembrane Region)
- Mutations:
[Ex: I20V, L21A...] - Rationale:
[Ex: Derived from positive mutational scores, aiming to create a faster integration into the E. coli membrane.]
Variant 3 (Soluble Region - DnaJ Interaction)
- Mutations:
[Ex: R15A, K16E...] - Rationale:
[Ex: By changing the charge distribution in the soluble tail, we aim to decrease the L-protein's dependency on the DnaJ chaperone for folding.]
Variant 4 (Soluble Region)
- Mutations:
[Ex: D22N, Y24F...] - Rationale:
[Ex: Selected based on sequence alignment (avoiding highly conserved sites) to allow autonomous insertion.]
Variant 5 (Combinatorial / Random)
- Mutations:
[Ex: T10A, P11G...] - Rationale:
[Ex: A broader structural perturbation to test if increased flexibility in the N-terminus accelerates the breakdown of the membrane.]