Describe a biological engineering application or tool you want to develop and why. One application that might be worth exploring would be in the realm of data storage for an imagined, hypothetical semi-exposed media. For this context special proteins could be designed in applications that bind to sequences that code for hidden malware or faulty sequences and or act for preserving media. I imagine that both this media design and this protein use would be niche but nonetheless fun to design for. Modalities could be as a gentle spray or as a settled solution that could be extracted.
Homework Part 0: Basics of Gel Electrophoresis
[This was a pure watch session. Thus there’s nothing to add here.]
Part 1: Benchling & In-silico Gel Art Restriction Enzymes Simulated on Lambda_NEB: EcoRI, HindIII, BamHI, KpnI, EcoRV, SacI, and SalI
Simple Art produced by way of Paul Vanouse’s Latent Figure Protocol artworks through the use of RC Donovan’s Gel Art Iteration Tool (https://rcdonovan.com/gel-art):
This lab, we were tasked with creating a design that could be generated by an OpenTrons Liquid Handling Robot.
Assignment: Python Script for Opentrons Artwork Review this week’s recitation and this week’s lab for details on the Opentrons and programming it. Done.
Generate an artistic design using the GUI at opentrons-art.rcdonovan.com. As a remote participant, I prototyped a design using the GUI at opentrons-art.rcdonovan.com.
Part A: Conceptual Questions We were allowed answer 9 out of 11 of the following questions:
How many molecules of amino acids do you take with a piece of 500 grams of meat? (on average an amino acid is ~100 Daltons) I expect answers to vary since meat composition vary.
Water composition of meat is estimated to be about 65-80% meat. Some estimates have proteins make up around 1/5 to less than 1/3 of overall muscle tissue.
This week’s homework was divided into 3 parts.
Part A involved SOD1 Binder Peptide Design. That was broken into 3 parts: Part 1: Part 1: Generate Binders with PepMLM
A) Begin by retrieving the human SOD1 sequence from UniProt (P00441) and introducing the A4V mutation. Done B) Using the PepMLM Colab linked from the HuggingFace PepMLM-650M model card: Done C) Generate four peptides of length 12 amino acids conditioned on the mutant SOD1 sequence.
Part 1: DNA Assembly What are some components in the Phusion High-Fidelity PCR Master Mix and what is their purpose? Some of the components include the Phusion DNA Polymerease (key to PCR), dNTPs (these are used to synthesize new DNA), and buffering materials to stabilize the environmental conditions during the raction.
What are some factors that determine primer annealing temperature during PCR? Some of these include buffer conditions, primer melting temperature, primer length, and GC content.
Assignment Part 1: Intracellular Artificial Neural Networks (IANNs) What advantages do IANNs have over traditional genetic circuits, whose input/output behaviors are Boolean functions? IANNs are great for analog computation, while also being scalable in deployment and useful for approximating functions.
Describe a useful application for an IANN; include a detailed description of input/output behavior, as well as any limitations an IANN might face to achieve your goal. Disease diagnostics. Inputs would be expressions levels of biomarkers and signals deemed harmful. Outputs could be in the form of a gene with a strong signal or that is therapetic in nature. Strong limitations could come from unintended interactions with the output, as well as cross-talk generated. Noise within the biological system could affected outputs as well.
General homework questions Explain the main advantages of cell-free protein synthesis over traditional in vivo methods, specifically in terms of flexibility and control over experimental variables. Name at least two cases where cell-free expression is more beneficial than cell production. Cell free expression allows teams to conduct biomanufacturing without living cells and operate beyond the constraints of productions in living systems.
Homework: Final Project Measurement Draft The main aspect of my project that I will measure is the functional activity of a mutated InaZ construct, specifically whether it increases ice nucleation efficiency relative to a control InaZ construct. I will perform this measurement using a controlled freezing assay in which replicate samples are cooled gradually and monitored for the onset of ice formation. The primary data collected will be the temperature at which freezing begins in each sample. Ice formation will be detected through optical observation of crystal formation and through temperature sensors that record the freezing point. In addition, I would verify the identity of the mutated construct using PCR, gel electrophoresis, and DNA sequencing. PCR and gel electrophoresis would be used to confirm the presence and approximate size of the inserted DNA, while DNA sequencing would confirm that the engineered inaZ mutation is correct. Together, these measurements allow me to confirm both that the construct was built properly and that it produces the intended increase in ice nucleation activity.
Part A: The 1,536 Pixel Artwork Canvas | Collective Artwork Contribute at least one pixel to this global artwork experiment before the editing ends on Sunday 4/19 at 11:59 PM EST! A personalized URL was sent to the email address associated with your Discourse account, and you can discuss the artwork on the Discourse! If you did not have a chance to contribute, it’s okay, just make sure you become a TA this fall! 😉
Homework: This homework was a reference to A) the acknowledhing the updated Week 11 and B) direction to make progress on our Final Project. This is expressed in the image below.
Progress: Done.
A) Contributed pixels to the Global Artwork Experiment and Followed up with mastermix concentrations as per part 3 of Homework 11C.
B) The project was refined through constructive class critique and further reflection.
Homework: Work on your Final Project Present it May 12 (MIT/Harvard) or May 13 (Committed Listeners)
Progress: Done. I further developed and practiced my presentation with my study group, classmates, and via some solo practice.
Homework: Homework: Finish your Final Project Present it May 12 (MIT/Harvard) or May 13 (Committed Listeners
Progress: Done. Final preparations were had during this week by which I practiced my presentation with my study group, classmates, and via some solo practice.
As an additional note: my study group held multiple sessions to ensure that each of our projects were further sharpened.
My Final Project, Project Z Freeze was successfully presented. My timeslot was 8:10PM on May 13.
Subsections of Homework
Week 1 HW: Principles and Practices
Describe a biological engineering application or tool you want to develop and why.
One application that might be worth exploring would be in the realm of data storage for an imagined, hypothetical semi-exposed media. For this context special proteins could be designed in applications that bind to sequences that code for hidden malware or faulty sequences and or act for preserving media. I imagine that both this media design and this protein use would be niche but nonetheless fun to design for. Modalities could be as a gentle spray or as a settled solution that could be extracted.
Describe one or more governance/policy goals related to ensuring that this application or tool contributes to an “ethical” future, like ensuring non-malfeasance (preventing harm).
A policy goal to ensure that this application contributes to an ethical future could be ensuring transparent design and standardization of these proteins (and their stats per context) so there is a way to validate their production and application prior to use. This could reduce the chance of bad actors sabotaging batches or distributing lower quality batches.
Describe at least three different potential governance “actions” by considering the four aspects below (Purpose, Design, Assumptions, Risks of Failure & “Success”)
Governance Action
Purpose
Design
Assumptions
Failure and Success Risks
Standardized Libraries
Educates communities on proteins available and safe designs
Create a library to hold these designs, as well as protocols for their manufacture to spec
That communities will agree on these standards and that there are not unnecessary inequities holding these standards
Success means that manufacture paths are straightforward.
Failure means that design paths are too numerous to account for and fragmentation possibilities are higher
Blue-teaming Design
To build a community line of protection of designs and applications
Design blue teaming frameworks and recruit educators to practice
That there is sufficient interest for funding
Success means that it is easier for production and share. Failure means that this pipeline has an established line of people who
Red-teaming Design
To identify means of sabotaging to proteins to protect libraries and distributors
Design blue-teaming framework and recruit educators to practice
That there is sufficient interest for funding
Success means that it is easier for production and share. Failure means that this pipeline has less visibility on sabotage routes
Next, score (from 1-3 with, 1 as the best, or n/a) each of your governance actions against your rubric of policy goals.
Does the option:
Option 1
Option 2
Option 3
Enhance Biosecurity
3
3
3
• By preventing incidents
3
3
3
• By helping respond
3
3
3
Foster Lab Safety
3
3
3
• By preventing incident
3
3
3
• By helping respond
3
3
3
Protect the environment
3
3
3
• By preventing incidents
3
3
3
• By helping respond
3
3
3
Other considerations
3
3
3
• Minimizing costs and burdens to stakeholders
3
2
2
• Feasibility?
3
3
3
• Not impede research
3
3
3
• Promote constructive applications
3
3
3
Last, drawing upon this scoring, describe which governance option, or combination of options, you would prioritize, and why. Outline any trade-offs you considered as well as assumptions and uncertainties.
I would prioritize library standardization. All operations, from that educators to users, can be facilitated from having a common reference frame. Further, among the options, this appears the most neutral. Overall, proper security requires practitioners from both sides of the spectrum: training in protecting and disrupting systems, and being willing to document for the good of the community.
Homework
Answers for Homework Questions from Professor Jabson:
Error rate - 1: 10^6. This is dwarfed by the length of the human genome. The body has numerous selectivity and repair mechanisms to deal with mitigate issues from mutations.
64 / Some codes might not work due to chemical incompatibility and structural reasons.
Answers for Homework Questions from Dr. LeProust:
Phosphoramidite DNA Synthesis Cycle
Increasing inefficiency of reactions with greater length
I’m still forming my thoughts on how this affects my view of the Lysine Contingency.
Week 2 HW: DNA Read, Write, and Edit
Homework
Part 0: Basics of Gel Electrophoresis
[This was a pure watch session. Thus there’s nothing to add here.]
Part 1: Benchling & In-silico Gel Art
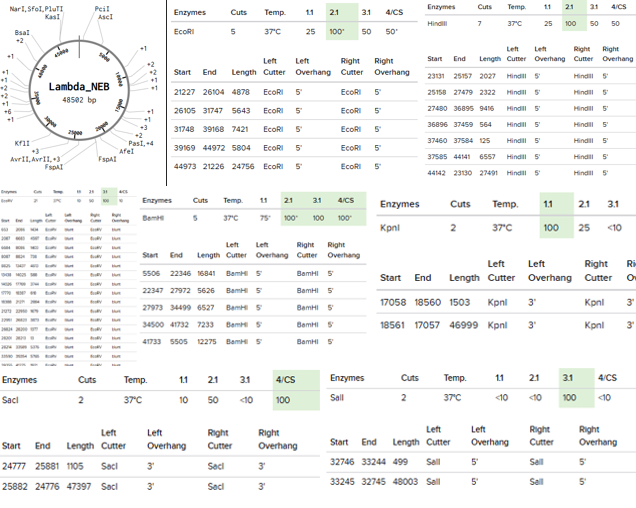
Restriction Enzymes Simulated on Lambda_NEB: EcoRI, HindIII, BamHI, KpnI, EcoRV, SacI, and SalI
Simple Art produced by way of Paul Vanouse’s Latent Figure Protocol artworks through the use of RC Donovan’s Gel Art Iteration Tool (https://rcdonovan.com/gel-art):
“4 corners”, using EcoRI and SalI in Lanes 1 and 10.
Part 2: Gel Art - Restriction Digests and Gel Electrophoresis
[This leaned on Wetlab Access. As a virtual student, this was not required.]
Part 3: DNA Design Challenge
3.1. Choose your protein.
Regarding proteins, I chose U-box domain-containing protein 12, also known as Plant U-box protein 12 or RING-type E3 ubiquitin transferase PUB12. I wanted to start with something and keep things relatively simple.
3.2. Reverse Translate: Protein (amino acid) sequence to DNA (nucleotide) sequence.
Reverse Translation Tool – BCCM – GeneCorner (https://www.genecorner.ugent.be/rev_trans.html)
PUB12, a plant U-box–type E3 ubiquitin ligase DNA sequence
(1) >reverse translation of sp|Q9ZV31|PUB12_ARATH U-box domain-containing protein 12 OS=Arabidopsis thaliana OX=3702 GN=PUB12 PE=2 SV=1 to a 1962 base sequence of most likely codons.
atggcgaaaagcgaaaaacataaactggcgcagaccctgattgatagcattaacgaaatt
gcgagcattagcgatagcgtgaccccgatgaaaaaacattgcgcgaacctgagccgccgc
ctgagcctgctgctgccgatgctggaagaaattcgcgataaccaggaaagcagcagcgaa
gtggtgaacgcgctgctgagcgtgaaacagagcctgctgcatgcgaaagatctgctgagc
tttgtgagccatgtgagcaaaatttatctggtgctggaacgcgatcaggtgatggtgaaa
tttcagaaagtgaccagcctgctggaacaggcgctgagcattattccgtatgaaaacctg
gaaattagcgatgaactgaaagaacaggtggaactggtgctggtgcagctgcgccgcagc
ctgggcaaacgcggcggcgatgtgtatgatgatgaactgtataaagatgtgctgagcctg
tatagcggccgcggcagcgtgatggaaagcgatatggtgcgccgcgtggcggaaaaactg
cagctgatgaccattaccgatctgacccaggaaagcctggcgctgctggatatggtgagc
agcagcggcggcgatgatccgggcgaaagctttgaaaaaatgagcatggtgctgaaaaaa
attaaagattttgtgcagacctataacccgaacctggatgatgcgccgctgcgcctgaaa
agcagcctgccgaaaagccgcgatgatgatcgcgatatgctgattccgccggaagaattt
cgctgcccgattagcctggaactgatgaccgatccggtgattgtgagcagcggccagacc
tatgaacgcgaatgcattaaaaaatggctggaaggcggccatctgacctgcccgaaaacc
caggaaaccctgaccagcgatattatgaccccgaactatgtgctgcgcagcctgattgcg
cagtggtgcgaaagcaacggcattgaaccgccgaaacgcccgaacattagccagccgagc
agcaaagcgagcagcagcagcagcgcgccggatgatgaacataacaaaattgaagaactg
ctgctgaaactgaccagccagcagccggaagatcgccgcagcgcggcgggcgaaattcgc
ctgctggcgaaacagaacaaccataaccgcgtggcgattgcggcgagcggcgcgattccg
ctgctggtgaacctgctgaccattagcaacgatagccgcacccaggaacatgcggtgacc
agcattctgaacctgagcatttgccaggaaaacaaaggcaaaattgtgtatagcagcggc
gcggtgccgggcattgtgcatgtgctgcagaaaggcagcatggaagcgcgcgaaaacgcg
gcggcgaccctgtttagcctgagcgtgattgatgaaaacaaagtgaccattggcgcggcg
ggcgcgattccgccgctggtgaccctgctgagcgaaggcagccagcgcggcaaaaaagat
gcggcgaccgcgctgtttaacctgtgcatttttcagggcaacaaaggcaaagcggtgcgc
gcgggcctggtgccggtgctgatgcgcctgctgaccgaaccggaaagcggcatggtggat
gaaagcctgagcattctggcgattctgagcagccatccggatggcaaaagcgaagtgggc
gcggcggatgcggtgccggtgctggtggattttattcgcagcggcagcccgcgcaacaaa
gaaaacagcgcggcggtgctggtgcatctgtgcagctggaaccagcagcatctgattgaa
gcgcagaaactgggcattatggatctgctgattgaaatggcggaaaacggcaccgatcgc
ggcaaacgcaaagcggcgcagctgctgaaccgctttagccgctttaacgatcagcagaaa
cagcatagcggcctgggcctggaagatcagattagcctgatt
(2) >reverse translation of sp|Q9ZV31|PUB12_ARATH U-box domain-containing protein 12 OS=Arabidopsis thaliana OX=3702 GN=PUB12 PE=2 SV=1 to a 1962 base sequence of consensus codons.
atggcnaarwsngaraarcayaarytngcncaracnytnathgaywsnathaaygarath
gcnwsnathwsngaywsngtnacnccnatgaaraarcaytgygcnaayytnwsnmgnmgn
ytnwsnytnytnytnccnatgytngargarathmgngayaaycargarwsnwsnwsngar
gtngtnaaygcnytnytnwsngtnaarcarwsnytnytncaygcnaargayytnytnwsn
ttygtnwsncaygtnwsnaarathtayytngtnytngarmgngaycargtnatggtnaar
ttycaraargtnacnwsnytnytngarcargcnytnwsnathathccntaygaraayytn
garathwsngaygarytnaargarcargtngarytngtnytngtncarytnmgnmgnwsn
ytnggnaarmgnggnggngaygtntaygaygaygarytntayaargaygtnytnwsnytn
taywsnggnmgnggnwsngtnatggarwsngayatggtnmgnmgngtngcngaraarytn
carytnatgacnathacngayytnacncargarwsnytngcnytnytngayatggtnwsn
wsnwsnggnggngaygayccnggngarwsnttygaraaratgwsnatggtnytnaaraar
athaargayttygtncaracntayaayccnaayytngaygaygcnccnytnmgnytnaar
wsnwsnytnccnaarwsnmgngaygaygaymgngayatgytnathccnccngargartty
mgntgyccnathwsnytngarytnatgacngayccngtnathgtnwsnwsnggncaracn
taygarmgngartgyathaaraartggytngarggnggncayytnacntgyccnaaracn
cargaracnytnacnwsngayathatgacnccnaaytaygtnytnmgnwsnytnathgcn
cartggtgygarwsnaayggnathgarccnccnaarmgnccnaayathwsncarccnwsn
wsnaargcnwsnwsnwsnwsnwsngcnccngaygaygarcayaayaarathgargarytn
ytnytnaarytnacnwsncarcarccngargaymgnmgnwsngcngcnggngarathmgn
ytnytngcnaarcaraayaaycayaaymgngtngcnathgcngcnwsnggngcnathccn
ytnytngtnaayytnytnacnathwsnaaygaywsnmgnacncargarcaygcngtnacn
wsnathytnaayytnwsnathtgycargaraayaarggnaarathgtntaywsnwsnggn
gcngtnccnggnathgtncaygtnytncaraarggnwsnatggargcnmgngaraaygcn
gcngcnacnytnttywsnytnwsngtnathgaygaraayaargtnacnathggngcngcn
ggngcnathccnccnytngtnacnytnytnwsngarggnwsncarmgnggnaaraargay
gcngcnacngcnytnttyaayytntgyathttycarggnaayaarggnaargcngtnmgn
gcnggnytngtnccngtnytnatgmgnytnytnacngarccngarwsnggnatggtngay
garwsnytnwsnathytngcnathytnwsnwsncayccngayggnaarwsngargtnggn
gcngcngaygcngtnccngtnytngtngayttyathmgnwsnggnwsnccnmgnaayaar
garaaywsngcngcngtnytngtncayytntgywsntggaaycarcarcayytnathgar
gcncaraarytnggnathatggayytnytnathgaratggcngaraayggnacngaymgn
ggnaarmgnaargcngcncarytnytnaaymgnttywsnmgnttyaaygaycarcaraar
carcaywsnggnytnggnytngargaycarathwsnytnath
3.3. Codon optimization.
Once a nucleotide sequence of your protein is determined, you need to codon optimize your sequence. You may, once again, utilize google for a “codon optimization tool”. In your own words, describe why you need to optimize codon usage.
Optimizing codon usage can help account for host bias as well as reduce the pool of downstream errors to account for, as well as save materials and time.
Which organism have you chosen to optimize the codon sequence for and why?
Which: Arabidopsis thaliana
Why: I wanted to start somewhere, and this organism is familiar.
Resulting Sequence is as follows:
PUB12, a plant U-box–type E3 ubiquitin ligase DNA sequence
ATGGCTAAATCTGAGAAGCATAAGTTGGCTCAGACTCTCATCGATTCTATAAATGAAATTGCTTCTATCTCAGATTCAGTTACTCCAATGAAGAAGCATTGTGCAAATTTGTCTAGGAGATTGTCACTTCTTCTTCCAATGTTGGAAGAGATTAGAGATAATCAAGAGTCTAGCTCTGAAGTCGTGAACGCTTTGCTCTCAGTTAAACAATCTTTATTACATGCTAAGGATCTCTTGTCTTTCGTCAGTCATGTGAGCAAGATATATCTTGTTCTTGAGAGAGATCAAGTGATGGTTAAGTTTCAAAAAGTTACTAGCCTTCTTGAGCAAGCTCTTTCTATAATCCCTTATGAGAATCTTGAAATTTCTGATGAATTGAAAGAACAAGTTGAATTGGTTCTTGTTCAACTTAGAAGATCTTTGGGTAAGAGAGGTGGTGATGTTTACGATGATGAACTTTATAAGGATGTTCTTTCACTTTACAGTGGAAGAGGATCAGTTATGGAGAGTGATATGGTTCGTCGAGTTGCCGAGAAATTGCAACTAATGACTATCACTGATTTGACACAAGAGTCTCTTGCTCTTCTTGATATGGTTTCTTCTAGTGGTGGAGATGATCCTGGAGAGTCATTCGAAAAGATGTCTATGGTTCTTAAGAAAATTAAGGATTTCGTTCAAACCTATAATCCTAACCTAGATGACGCTCCCCTTAGACTTAAATCATCATTGCCTAAATCGAGAGATGATGATCGTGATATGCTTATTCCACCTGAAGAATTCCGTTGTCCTATTTCGCTTGAGCTTATGACTGATCCTGTAATCGTTTCTTCAGGTCAAACCTATGAAAGAGAGTGTATTAAGAAGTGGCTTGAAGGAGGACATTTGACATGTCCTAAGACTCAAGAAACTTTGACATCTGATATCATGACCCCTAATTATGTTCTTAGATCTTTGATCGCTCAATGGTGTGAGTCGAATGGAATCGAGCCTCCAAAGAGGCCAAACATAAGTCAGCCTTCTAGTAAGGCTTCTTCATCATCTAGTGCTCCTGATGACGAACATAATAAGATCGAAGAATTGCTCTTGAAGTTGACTTCTCAGCAACCTGAAGATAGAAGATCCGCTGCTGGAGAGATCAGACTTTTGGCCAAACAAAACAACCATAACAGAGTTGCTATCGCTGCTTCAGGAGCTATTCCACTCTTGGTGAACCTTTTGACTATCTCAAACGATTCCAGAACACAAGAGCATGCTGTTACGTCTATCCTCAACCTTTCTATCTGCCAAGAAAATAAAGGTAAGATCGTTTATTCTAGTGGTGCAGTGCCTGGTATTGTTCATGTTTTGCAGAAGGGATCAATGGAGGCTAGAGAAAACGCTGCTGCTACTCTTTTCTCTCTTTCCGTTATAGATGAGAATAAGGTTACTATTGGAGCTGCTGGAGCAATTCCACCTTTGGTTACTCTCCTTTCTGAAGGATCACAGCGTGGAAAGAAGGATGCTGCTACTGCACTCTTCAACCTTTGTATCTTTCAGGGTAATAAAGGTAAGGCAGTTAGAGCAGGACTTGTGCCTGTGCTTATGAGGCTTTTGACTGAACCTGAATCTGGAATGGTTGATGAGAGCCTTTCTATTCTTGCTATTCTTTCTTCTCATCCAGACGGAAAGTCTGAAGTTGGAGCTGCTGATGCAGTTCCTGTTCTTGTTGATTTCATCAGATCTGGATCTCCTAGAAATAAGGAGAATTCTGCTGCAGTTCTTGTTCACTTGTGTTCATGGAATCAACAACATCTTATCGAAGCACAGAAGCTTGGAATCATGGATCTTCTCATCGAGATGGCTGAAAACGGAACTGATCGTGGTAAGAGAAAGGCCGCACAATTGCTTAATAGATTTTCTAGATTTAACGATCAGCAGAAGCAACACAGTGGTCTTGGTCTTGAAGATCAAATTTCATTGATT
Below is a printout contrasting the two:
3.4. You have a sequence! Now what?
Recombinant DNA technologies could be utilized to make this protein from the DNA (Cell-free or Cell-dependent). Respectively they involve either special mix that can take place in a test tube or through using a live cell’s machinery to produce the protein.
Part 4: Prepare a Twist DNA Synthesis Order
Build Your DNA Insert Sequence
I prepared the above improved sequence as a test order. Below is the initial step through the creation of the DNA/RNA Sequence in Benchling with a Linear Topology.
It was annotated below as such within Benchling before a linear map and file was constructed that could be uploaded to Twist Bio.
Start Codon: ATG
Coding Sequence:
ATGGCTAAATCTGAGAAGCATAAGTTGGCTCAGACTCTCATCGATTCTATAAATGAAATTGCTTCTATCTCAGATTCAGTTACTCCAATGAAGAAGCATTGTGCAAATTTGTCTAGGAGATTGTCACTTCTTCTTCCAATGTTGGAAGAGATTAGAGATAATCAAGAGTCTAGCTCTGAAGTCGTGAACGCTTTGCTCTCAGTTAAACAATCTTTATTACATGCTAAGGATCTCTTGTCTTTCGTCAGTCATGTGAGCAAGATATATCTTGTTCTTGAGAGAGATCAAGTGATGGTTAAGTTTCAAAAAGTTACTAGCCTTCTTGAGCAAGCTCTTTCTATAATCCCTTATGAGAATCTTGAAATTTCTGATGAATTGAAAGAACAAGTTGAATTGGTTCTTGTTCAACTTAGAAGATCTTTGGGTAAGAGAGGTGGTGATGTTTACGATGATGAACTTTATAAGGATGTTCTTTCACTTTACAGTGGAAGAGGATCAGTTATGGAGAGTGATATGGTTCGTCGAGTTGCCGAGAAATTGCAACTAATGACTATCACTGATTTGACACAAGAGTCTCTTGCTCTTCTTGATATGGTTTCTTCTAGTGGTGGAGATGATCCTGGAGAGTCATTCGAAAAGATGTCTATGGTTCTTAAGAAAATTAAGGATTTCGTTCAAACCTATAATCCTAACCTAGATGACGCTCCCCTTAGACTTAAATCATCATTGCCTAAATCGAGAGATGATGATCGTGATATGCTTATTCCACCTGAAGAATTCCGTTGTCCTATTTCGCTTGAGCTTATGACTGATCCTGTAATCGTTTCTTCAGGTCAAACCTATGAAAGAGAGTGTATTAAGAAGTGGCTTGAAGGAGGACATTTGACATGTCCTAAGACTCAAGAAACTTTGACATCTGATATCATGACCCCTAATTATGTTCTTAGATCTTTGATCGCTCAATGGTGTGAGTCGAATGGAATCGAGCCTCCAAAGAGGCCAAACATAAGTCAGCCTTCTAGTAAGGCTTCTTCATCATCTAGTGCTCCTGATGACGAACATAATAAGATCGAAGAATTGCTCTTGAAGTTGACTTCTCAGCAACCTGAAGATAGAAGATCCGCTGCTGGAGAGATCAGACTTTTGGCCAAACAAAACAACCATAACAGAGTTGCTATCGCTGCTTCAGGAGCTATTCCACTCTTGGTGAACCTTTTGACTATCTCAAACGATTCCAGAACACAAGAGCATGCTGTTACGTCTATCCTCAACCTTTCTATCTGCCAAGAAAATAAAGGTAAGATCGTTTATTCTAGTGGTGCAGTGCCTGGTATTGTTCATGTTTTGCAGAAGGGATCAATGGAGGCTAGAGAAAACGCTGCTGCTACTCTTTTCTCTCTTTCCGTTATAGATGAGAATAAGGTTACTATTGGAGCTGCTGGAGCAATTCCACCTTTGGTTACTCTCCTTTCTGAAGGATCACAGCGTGGAAAGAAGGATGCTGCTACTGCACTCTTCAACCTTTGTATCTTTCAGGGTAATAAAGGTAAGGCAGTTAGAGCAGGACTTGTGCCTGTGCTTATGAGGCTTTTGACTGAACCTGAATCTGGAATGGTTGATGAGAGCCTTTCTATTCTTGCTATTCTTTCTTCTCATCCAGACGGAAAGTCTGAAGTTGGAGCTGCTGATGCAGTTCCTGTTCTTGTTGATTTCATCAGATCTGGATCTCCTAGAAATAAGGAGAATTCTGCTGCAGTTCTTGTTCACTTGTGTTCATGGAATCAACAACATCTTATCGAAGCACAGAAGCTTGGAATCATGGATCTTCTCATCGAGATGGCTGAAAACGGAACTGATCGTGGTAAGAGAAAGGCCGCACAATTGCTTAATAGATTTTCTAG
Stop Codon: TAG
Linear Map:
Here’s an example of what you just annotated in Benchling:
Sequence Import and Quote Obtained
The pTwist Amp High Copy: pTwist Amp Vector was chosen after the Clonal Gene Choice was pursued. The quote is to the left.. The annotated sequence page from TwistBio from which a GenBank construct file was downloaded is to the right.
The construct was imported into Benchling to yield the plasmid below.
Part 5: DNA Read/Write/Edit
5.1 DNA Read
(i) What DNA would you want to sequence (e.g., read) and why?
I might want to sequence large viruses, particularly out of sheer curiosity for the instructions that allow them to exist in their current forms.
Sources of interest:
Piacente, F., De Castro, C., Jeudy, S., Molinaro, A., Salis, A., Damonte, G., Bernardi, C., Abergel, C. and Tonetti, M.G., 2014. Giant virus Megavirus chilensis encodes the biosynthetic pathway for uncommon acetamido sugars. Journal of Biological Chemistry, 289(35), pp.24428-24439.
Legendre, M., Arslan, D., Abergel, C. and Claverie, J.M., 2012. Genomics of Megavirus and the elusive fourth domain of Life. Communicative & integrative biology, 5(1), pp.102-106.
Arslan, D., Legendre, M., Seltzer, V., Abergel, C. and Claverie, J.M., 2011. Distant Mimivirus relative with a larger genome highlights the fundamental features of Megaviridae. Proceedings of the National Academy of Sciences, 108(42), pp.17486-17491.
(ii) In lecture, a variety of sequencing technologies were mentioned. What technology or technologies would you use to perform sequencing on your DNA and why?
I would borrow from the methods used in the aforementioned literature, particularly “454-titanium and Illumina HiSeq approaches”. These methods appear adequate. Lack of a priori knowledge of the genome or genomic features not being required is helpful, in addition to single-nucleotide resolution, higher dynamic range, and less DNA/RNA needed.
What is the output of your chosen sequencing technology?
Numerous sequence reads
5.2 DNA Write
(i) What DNA would you want to synthesize (e.g., write) and why?
I am not sure yet, but I am leaning towards DNA origami art to experiment with the medium and explore versatility of applications.
Sources of interest:
Bush, J., Singh, S., Vargas, M., Oktay, E., Hu, C.H. and Veneziano, R., 2020. Synthesis of DNA origami scaffolds: Current and emerging strategies. Molecules, 25(15), p.3386.
Weck, J.M. and Heuer-Jungemann, A., 2025. Fully addressable designer superstructures assembled from one single modular DNA origami. Nature communications, 16(1), p.1556.
DNA origami by Paul W. K. Rothemund, California Institute of Technology, 2004. 100 nanometers in diameter.
(ii) What technology or technologies would you use to perform this DNA synthesis and why?
For validating the structures, if cost didn’t matter, I would consider using next generation sequencing (Illumina, for both sequences of the staples and scaffold) and Atomic Force Microscopy (Visual, especially confirming folds)
Also answer the following questions:
The essential steps of the chosen sequencing methods would be:
Library Prep (DNA Fragmentation and Adapter ligation methods)
Cluster Generation via amplification
Sequencing and base calling
What are the limitations of your sequencing method (if any) in terms of speed, accuracy, scalability?
The major limitations would be cost, error rates, and short read lengths. This would not be efficient to scale as is.
Improvements would involve:
-Hand design of patterns (for ideating improvements)
-Computer design and optimization of material usage
-Production of material and strand-routing precision
5.3 DNA Edit
(i) What DNA would you want to edit and why?
DNA edits that I would like to perform would be those that allow for the minimization and or elimination of metabolic disease states. The why comes down to the quality-of-life improvements for all involved.
(ii) What technology or technologies would you use to perform these DNA edits and why?
I can see myself engaging CRISPR-Cas9 and base editing for their precision. Of the two, especially the latter, in order to reduce off-target based effects.
Also answer the following questions:
How does your technology of choice edit DNA? What are the essential steps?
It uses guide RNA to direct modified Cas proteins to DNA sequences and convert nucleotides.
The major steps woud involve (i) Designing guide RNA that compliment the target gene(s); (ii) deliver said RNA and its editor protein to the cells of interest (iii) waiting as the guide RNA mediated editor binds to the target DNA site (iv) waiting as the deaminase converts targeted base paits (v) waiting as the edits are incoporated (vi) assaying for confirmation via suquencing and functional assay applications
Ethics meditations no doubt should accompany each step.
What preparation do you need to do (e.g. design steps) and what is the input (e.g. DNA template, enzymes, plasmids, primers, guides, cells) for the editing?
Preparation involves (i) identifying seuquences associated with disease states in select organisms, (ii) computationally designing guide RNAs to target them, (iii) computationally optimizing said guide RNAs and selecting a properly paired base editing system (iv) modeling the application of guide RNAs and base editor asystem applyed to DNA sequences highly associated with disease states in the bodies of select organisms (v) validating said results and modifying as needed (vi) developing in vitro cellular models capable of testing editing efficiency, safety, and functional outcomes (vii) Organoid and higher models may follow
What are the limitations of your editing methods (if any) in terms of efficiency or precision?
Avoiding off-target edits is not a given. Validation and optimization steps remain necessary.
Week 3 HW: Lab Automation
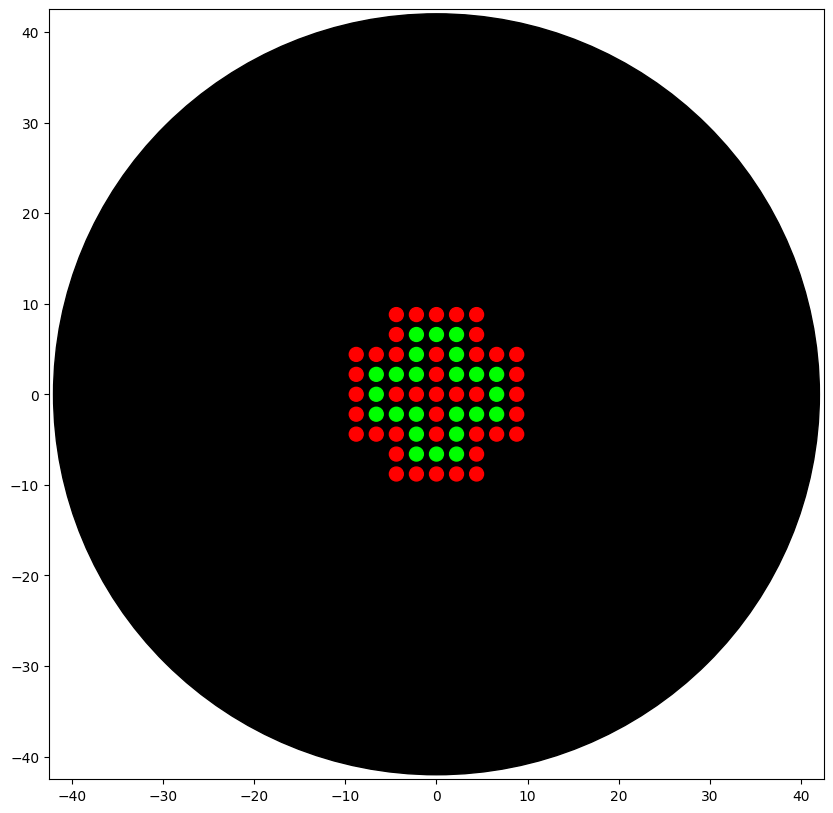
This lab, we were tasked with creating a design that could be generated by an OpenTrons Liquid Handling Robot.
Assignment: Python Script for Opentrons Artwork
Review this week’s recitation and this week’s lab for details on the Opentrons and programming it.
Done.
Generate an artistic design using the GUI at opentrons-art.rcdonovan.com.
As a remote participant, I prototyped a design using the GUI at opentrons-art.rcdonovan.com.
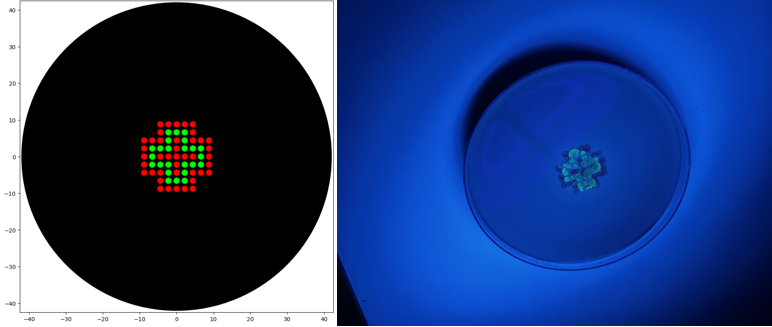
This resulted in a layered plus symbol shown below.
Using the coordinates from the GUI, follow the instructions in the HTGAA26 Opentrons Colab to write your own Python script which draws your design using the Opentrons.
You may use AI assistance for this coding — Google Gemini is integrated into Colab (see the stylized star bottom center); it will do a good job writing functional Python, while you probably need to take charge of the art concept.
If you’re a proficient programmer and you’d rather code something mathematical or algorithmic instead of using your GUI coordinates, you may do that instead.
Acknowledged
The coordinates for generating such can be found here, courtesy of RC Donovan’s tool:
If the Python component is proving too problematic even with AI and human assistance, download the full Python script from the GUI website and submit that:
Not needed, but appreciated.
If you use AI to help complete this homework or lab, document how you used AI and which models made contributions.
For my node, in order to work with their printer which had two colors, a modified version was created. Gemini within was tested and employed to deliver the following result.
Sign up for a robot time slot if you are at MIT/Harvard/Wellesley or at a Node offering Opentrons automation.
The Python script you created will be run on the robot to produce your work of art!
At MIT/Harvard? Lab times are on Thursday Feb.19 between 10AM and 6PM.
At other Nodes? Please coordinate with your Node.
I was added to the William and Mary Node. I coordinated with Margaret and Kate for OpenTrons code submission. My code was submitted to Kate and who was then able to faciliate the printing of my design. Please see below.
Submit your Python file via this form.
DONE.
Post-LAb Questions:
Find and describe a published paper that utilizes the Opentrons or an automation tool to achieve novel biological applications.
DeRoo, J.B., Jones, A.A., Slaughter, C.K., Ahr, T.W., Stroup, S.M., Thompson, G.B. and Snow, C.D., 2025. Automation of protein crystallization scaleup via Opentrons-2 liquid handling. SLAS technology, 32, p.100268.
This work describes an approach by which an Opentrons-2 liquid handling robot was used for automating sitting drop protein crystallization trials. This ability also improve comparability of products produced, improving studies that depend on their proper manufacture. An important detail is how the Opentrons-2 can prove a cost-effective option for laboratory operations. For example, at the time of writing, the Opentrons-2 can be purchased for around 13.5K USD vs that of a Gryphon machine at around 65K USD.
Write a description about what you intend to do with automation tools for your final project.
I’m still forming my thoughts about how I want to effectively use automation tools for my final project.
So far, I am interested in branching off from example #2 given in the Homework and the above example, regarding screening an array of designed biosensor constructs.
One idea had in mind was towards a digital tracing project that revolves around said constructs used to track known entities.
Simply, products are given a unique ID with stored parameters. These are linked within a automation run so that each product is trackable as they are processed. One application that is probably already in use but would be fun to adapt towards something not already applied would be swappable combined wearable crystallized biosensors that are traded in daily for workers that are liable to be exposed to a particular organism and pollutant pairs.
I could use an Echo for transfer of nano-scale components.
The Bravo or Opentrons-2 could be used for precise, automated pipetting ,especially of the crystals.
The multiflow would be used to dispense the larger scale volume components.
The PlateLoc would be helful for sealing the plates.
The inheco could be used for controlled incubation.
The Xpeel would be used for careful desealing of the plates.
Finally, the PHERAstar could be used for reading fluorescence outputs.
Still developing this out from this branch.
Final Project Ideas
Done. My intitial Project Ideas were added:
The initial candidates were:
Project Name: Aptly
Aptamer-Based Sensor for Endocrine Disruptors
Problem: Endocrine-disrupting chemicals can persist in water at concentrations that are difficult to monitor in real time.
Hypothesis: If we engineer high-affinity aptamers that selectively bind hormone-mimicking pollutants, we can detect these contaminants at environmentally relevant levels.
Solution: Develop a portable biosensor that binds engineered aptamers to a measurable fluorescent or electrochemical signal for field-based detection.
Real-world Literature and Examples of Problem:
Bertram, M.G., Gore, A.C., Tyler, C.R. and Brodin, T., 2022. Endocrine-disrupting chemicals. Current Biology, 32(13), pp.R727-R730.
Pironti, C., Ricciardi, M., Proto, A., Bianco, P.M., Montano, L. and Motta, O., 2021. Endocrine-disrupting compounds: An overview on their occurrence in the aquatic environment and human exposure. Water, 13(10), p.1347.
Project Name: NF-Lamp Lighter
Deployable LAMP-Based Sensor for Naegleria fowleri
Problem:
Testing for Naegleria fowleri currently requires sending water samples to a lab, which delays results and limits real-time monitoring.
Hypothesis:
If we combine on-site water filtration with LAMP DNA amplification, we can detect N. fowleri quickly without a full laboratory.
Solution:
Build a deployable mini-station that filters water, runs a LAMP test in a sealed cartridge, and sends a simple detection alert to public health officials.
Real-world Literature and Examples of Problem:
Grace, E., Asbill, S. and Virga, K., 2015. Naegleria fowleri: pathogenesis, diagnosis, and treatment options. Antimicrobial agents and chemotherapy, 59(11), pp.6677-6681.
Maciver, S.K., Piñero, J.E. and Lorenzo-Morales, J., 2020. Is Naegleria fowleri an emerging parasite?. Trends in parasitology, 36(1), pp.19-28.
Project Name: LP Alert
Deployable Biosensor for Legionella pneumophila in Cooling Towers
Problem:
Cooling towers can grow Legionella pneumophila, and detection could be made more quickly.
Hypothesis:
If we combine on-site DNA amplification with CRISPR detection and an electrochemical readout, we can quickly and accurately detect L. pneumophila and avoid having to send samples to a lab.
Solution:
Build a deployable unit that samples cooling tower water, amplifies L. pneumophila DNA, converts detection into an electrical signal, and sends an alert to facility managers.
Real-world Literature and Examples of Problem:
Wéry, N., Bru-Adan, V., Minervini, C., Delgénes, J.P., Garrelly, L. and Godon, J.J., 2008. Dynamics of Legionella spp. and bacterial populations during the proliferation of L. pneumophila in a cooling tower facility. Applied and environmental microbiology, 74(10), pp.3030-3037.
Brigmon, R.L., Turick, C.E., Knox, A.S. and Burckhalter, C.E., 2020. The impact of storms on Legionella pneumophila in cooling tower water, implications for human health. Frontiers in Microbiology, 11, p.543589.
We were allowed answer 9 out of 11 of the following questions:
How many molecules of amino acids do you take with a piece of 500 grams of meat? (on average an amino acid is ~100 Daltons)
I expect answers to vary since meat composition vary.
Water composition of meat is estimated to be about 65-80% meat.
Some estimates have proteins make up around 1/5 to less than 1/3 of overall muscle tissue.
We’ll work with 20% and 30% for upper and lower bounds for simplicity.
Assuming that 100 Daltons roughly equals 100 g/mo
Lower: 500 g × 0.2 = 100 g protein = 1 mol. 6.02 × 10²³ molecules
Upper: 500 g × 0.3 = 150 g protein = 1.5 mol. ~9 × 10²³ molecules
For simplicity, let’s work with Taking the average, 500 grams of meat may yield
Why do humans eat beef but do not become a cow, eat fish but do not become fish?
These products are broken down through digestion and recycled. Our genetic material largely informs the building process of new material.
Why are there only 20 natural amino acids?
I’m not convinced that those are the only ones, in so far of what we have observed on Earth it is likely that the developmental “cost” as per our development over time was too high. 20 reflects optimization, no?
Can you make other non-natural amino acids? Design some new amino acids.
Yes. These can be designed through new side chains, although stability and inclusion may be a problem.
Where did amino acids come from before enzymes that make them, and before life started?
Abiotic processes. Some sources list thermal vents and delivery through meteorites.
Ehrenfreund, P., Bernstein, M.P., Dworkin, J.P., Sandford, S.A. and Allamandola, L.J., 2001. The photostability of amino acids in space. The Astrophysical Journal Letters, 550(1), pp.L95-L99.
Kobayashi, K., Mita, H., Kebukawa, Y., Nakagawa, K., Kaneko, T., Obayashi, Y., Sato, T., Yokoo, T., Minematsu, S., Fukuda, H. and Oguri, Y., 2021. Space exposure of amino acids and their precursors during the Tanpopo Mission. Astrobiology, 21(12), pp.1479-1493.
If you make an α-helix using D-amino acids, what handedness (right or left) would you expect?
Left-handed
Can you discover additional helices in proteins?
Yes
Why are most molecular helices right-handed?
The stereochemistry of L-amino acids constrains backbone geometry in a way that right-handed helices are lower in energy. Right-handed chirality is favored and chirality from the bottom influences chirality at higher structures.
Why do β-sheets tend to aggregate?
What is the driving force for β-sheet aggregation?
[Skipped]
Why do many amyloid diseases form β-sheets?
β-Sheet structures are stable and can easy to produce from misfolding.
Can you use amyloid β-sheets as materials? Design a β-sheet motif that forms a well-ordered structure.
[Skipped]
Part B: Protein Analysis and Visualization
The protein that I selected is U-box domain-containing protein 12.Possesses E3 ubiquitin-protein ligase in vitro. I was not too particular on its selection and wanted to start somewhere.
The length of the protein is: 611 aminoacids.
The most common amino acid is: L, which appears 72 times.
This protein does not share 100% homology with any other although there are many with 50-90% homology, per UniProt. Via the Blast Tool, a lot of homologs were found.
Query= EMBOSS_001
Length=611
Score E
Sequences producing significant alignments: (Bits) Value
This can be accessed online, and free. They also provide an illustration service, which is pretty cool. It can be found below. This program did not have the “cartoon”, “ribbon”, and “ball and stick”, but it did have the “Real”, “Outline”, and “Goodsell” (cartoon-like) view modes where are shown from a “Sphere” mode of viewing. See image below.
To visualize it differently, I looked at it from a “tube” viewing mode. It has more helices than sheets. See image below.
Hydrophobic residues are inside the protein core, and hydrophilic residues are on the surface. See image below.
Labeling by residue it has many charged and hydrophobic residues, with the latter being more focused inward. See image below.
Visualizing the surface, this structure appears compact, without deep binding cavities. See image below.
Part C
To be added with images
Part D
This part involved several of us students brainstorming for a lab according to the following instructions:
Find a group of ~3–4 students
Read through the Phage Reading material.
Review the Bacteriophage Final Project Goals for engineering the L Protein:
Choose one or two main goals from the list that you think you can address computationally (e.g., “We’ll try to stabilize the lysis protein,” or “We’ll attempt to disrupt its interaction with E. coli DnaJ.”).
Write a 1-page proposal (bullet points or short paragraphs) describing:
Each individually put your plan on your HTGAA website
Our initial proposal can be found below:
Week 5 HW: Protein Design Part II
This week’s homework was divided into 3 parts.
Part A involved SOD1 Binder Peptide Design.
That was broken into 3 parts:
Part 1: Part 1: Generate Binders with PepMLM
A) Begin by retrieving the human SOD1 sequence from UniProt (P00441) and introducing the A4V mutation.
Done
B) Using the PepMLM Colab linked from the HuggingFace PepMLM-650M model card:
Done
C) Generate four peptides of length 12 amino acids conditioned on the mutant SOD1 sequence.
C/D) Record the ipTM score and briefly describe where the peptide appears to bind. Does it localize near the N-terminus where A4V sits? Does it engage the β-barrel region or approach the dimer interface? Does it appear surface-bound or partially buried? D) In a short paragraph, describe the ipTM values you observe and whether any PepMLM-generated peptide matches or exceeds the known binder.
The IpTM score is .18. This does not appear to indicate high confidence in binding. The lack of clear localiztion, strong β-barrel engagement, meaningful Dimer interfacing, combined with surface bounding of the peptides does not suggest strong binding interactions.
Part 3: Evaluate Properties of Generated Peptides in the PeptiVerse
Paste the peptide sequence:
Paste the A4V mutant SOD1 sequence in the target field.
Check the boxes
Predicted binding affinity
Solubility
Hemolysis probability
Net charge (pH 7)
Molecular weight
Data for such were pasted as pictures below, in this order:
Open the moPPit Colab linked from the HuggingFace moPPIt model card
Done
Make a copy and switch to a GPU runtime.
Done
In the notebook:
Paste your A4V mutant SOD1 sequence.
Choose specific residue indices on SOD1 that you want your peptide to bind (for example, residues near position 4, the dimer interface, or another surface patch).
Set peptide length to 12 amino acids.
Enable motif and affinity guidance (and solubility/hemolysis guidance if available). Generate peptides.
Done
After generation, briefly describe how these moPPit peptides differ from your PepMLM peptides. How would you evaluate these peptides before advancing them to clinical studies?
moPPit peptides appear better to develop. I would evaluate these peptides further computationally before advancing them to clinical studies. This would involve ranking by various qualities and further optimization. If successful, in vitro studies would follow.
Part B: BRD4 Drug Discovery Platform Tutorial (Gabriele)
This assignment was optional. For the sake of priorities, it will not be posted here.
Part C: Final Project: L-Protein Mutants
This was held between of 5 members, 3 of whom were able to provide their results jointly. We persued the Option 1: Mutagenesis.
To save space, given the large volume of images, a link to the inputs can be found in this google doc:
Week 6 HW: Genetic Circuits Part I: Assembly Technologies
Part 1: DNA Assembly
What are some components in the Phusion High-Fidelity PCR Master Mix and what is their purpose?
Some of the components include the Phusion DNA Polymerease (key to PCR), dNTPs (these are used to synthesize new DNA), and buffering materials to stabilize the environmental conditions during the raction.
What are some factors that determine primer annealing temperature during PCR?
Some of these include buffer conditions, primer melting temperature, primer length, and GC content.
There are two methods from this class that create linear fragments of DNA: PCR, and restriction enzyme digests. Compare and contrast these two methods, both in terms of protocol as well as when one may be preferable to use over the other.
PCR uses primers, dNTPs, buffers and polymerase to amplify DNA.
Restriction enzyme digests are very precise in that they cut genomic material at precise locations, but they require restriction sites.
How can you ensure that the DNA sequences that you have digested and PCR-ed will be appropriate for Gibson cloning?
They need to be optimally designed for the system. This requires special attention primer design and their parameters.
How does the plasmid DNA enter the E. coli cells during transformation?
After a shock is induced, plasmid DNA can enter E. Coli cells through pores that emerge in their membrane.
Describe another assembly method in detail (such as Golden Gate Assembly)
6.1 Explain the other method in 5 - 7 sentences plus diagrams (either handmade or online).
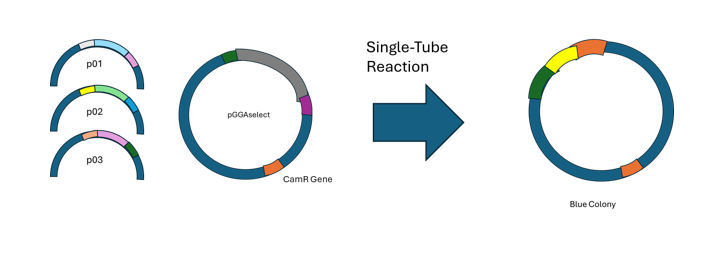
Golden gate assembly is a method for efficient, seamless assembly that uses Type IIS restriction endonucleases to cleave DNA that exists outside of recognition sequences. Designs for inserts and cloning vectors place the Type IIS recognition sites distal to the cleavage site, and this allows efficient removal. NEB states 3 main advantages for GGA. One is that no scar sequence is introduced. Another is that the overhangs can allow for multiple simulaneous assemblies. Further, ligation and digestion can be done at the same time. A truncated example of a successful case is diagrammed below, with inspiration from a figure on NEB’s website.
6.2 Model this assembly method with Benchling or Asimov Kernel!
Route chosen: Asimov Kernel
Owing to the brilliance of the Kernal from Asimov, redesigning the wheel was unnecessary.
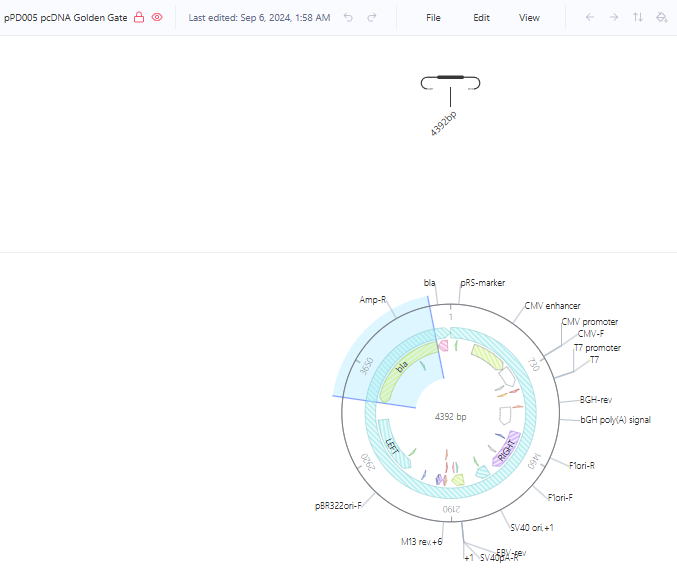
I found the “pPD005 pcDNA Golden Gate” under the Addgene Plasmid Repository detailing a construct that was already created It is shown below in circular form.
Part 2: Asimov Kernel
Create a Repository for your work
Done - Refer to W&M_Whale_Test_Rep_Notebook_Entry in Kernel
Create a blank Notebook entry to document the homework and save it to that Repository
Done - Refer to W&M_Whale_Test_Rep_Notebook_Entry in Kernel
Explore the devices in the Bacterial Demos Repo to understand how the parts work together by running the Simulator on various examples, following the instructions for the simulator found in the “Info” panel (click the “i” icon on the right to open the Info panel)
Done - Refer to W&M_Whale_Test_Rep_Notebook_Entry in Kernel
Create a blank Construct and save it to your Repository
Done - Refer to W&M_Whale_Test_Rep_Notebook_Entry in Kernel
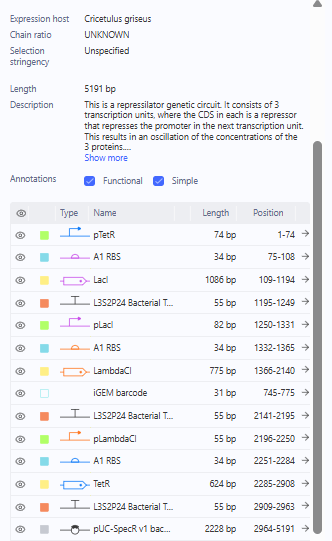
Recreate the Repressilator in that empty Construct by using parts from the Characterized Bacterial Parts repository
Done - Refer to W&M_Whale_Test_Rep_Notebook_Entry in Kernel
Search the parts using the Search function in the right menu
Done - Refer to W&M_Whale_Test_Rep_Notebook_Entry in Kernel
Drag and drop the parts into the Construct
Done - Refer to W&M_Whale_Test_Rep_Notebook_Entry in Kernel
Confirm it works as expected by running the Simulator (“play” button) and compare your results with the Repressilator Construct found in the Bacterial Demos repository
Done - Refer to W&M_Whale_Test_Rep_Notebook_Entry in Kernel
Document all of this work in your Notebook entry - you can copy the glyph image and the simulator graphs, and paste them into your Notebook
Done - Refer to W&M_Whale_Test_Rep_Notebook_Entry in Kernel
Build three of your own Constructs using the parts in the Characterized Bacterials Parts Repo.
10.1. Explain in the Notebook Entry how you think each of the Constructs should function
10.2. Run the simulator and share your results in the Notebook Entry
10.3. If the results don’t match your expectations, speculate on why and see if you can adjust the simulator settings to get the expected outcome
10.1-10.3 These actions were all performed. I exported the images and markdown as below. However, they can be found under W&M_Whale_Test_Rep_Notebook_Entry
To explain constructs – these were recreations and thus were expected to act as the demos, prior to the run of their simulations. The success is in part a testament to effect design of the Kernal software.
What Follows is a markdown export of my Notebook Entry in Ansimov Kernal under W&M Whale Test Rep:
This entry starts with a general summary of the use of Asimov’s Kernel thus far. The user interface takes a little getting used to but is easy to learn.
I started by using the Bacterial Demos Repo as per the homework to understand how parts work together.
A simulation was ran which ran similarly to others other before it. I did not expect difference given that these were demos.
The Repressilator was the first, which was selected by starting from a blank construct using parts from the Characterized Bacterial Parts.
Characterized Bacterial Parts Examination for Repressilator
Represillator Recreation
This was then simulated with results expected from a Demo.
Represillator Recreation Simulation Start
Represillator Recreation Preview
For the Homework, we were asked to create 3 more constructs. I chose recreations of the J23117 Promoter, Self-Replicating Promoter, and Circuit 3. Simulations accompanied them. I expected them to all run similar to the demos, which they did.
J23117 Promoter
J23117 Promoter Simulation Preview
Self-Replicating Promoter
Self-Replicating Promoter Preview
Circuit 3
Circuit 3 Preview
Week 7 HW: Genetic Circuits Part II: Neuromorphic Circuits
Assignment Part 1: Intracellular Artificial Neural Networks (IANNs)
What advantages do IANNs have over traditional genetic circuits, whose input/output behaviors are Boolean functions?
IANNs are great for analog computation, while also being scalable in deployment and useful for approximating functions.
Describe a useful application for an IANN; include a detailed description of input/output behavior, as well as any limitations an IANN might face to achieve your goal.
Disease diagnostics. Inputs would be expressions levels of biomarkers and signals deemed harmful. Outputs could be in the form of a gene with a strong signal or that is therapetic in nature. Strong limitations could come from unintended interactions with the output, as well as cross-talk generated. Noise within the biological system could affected outputs as well.
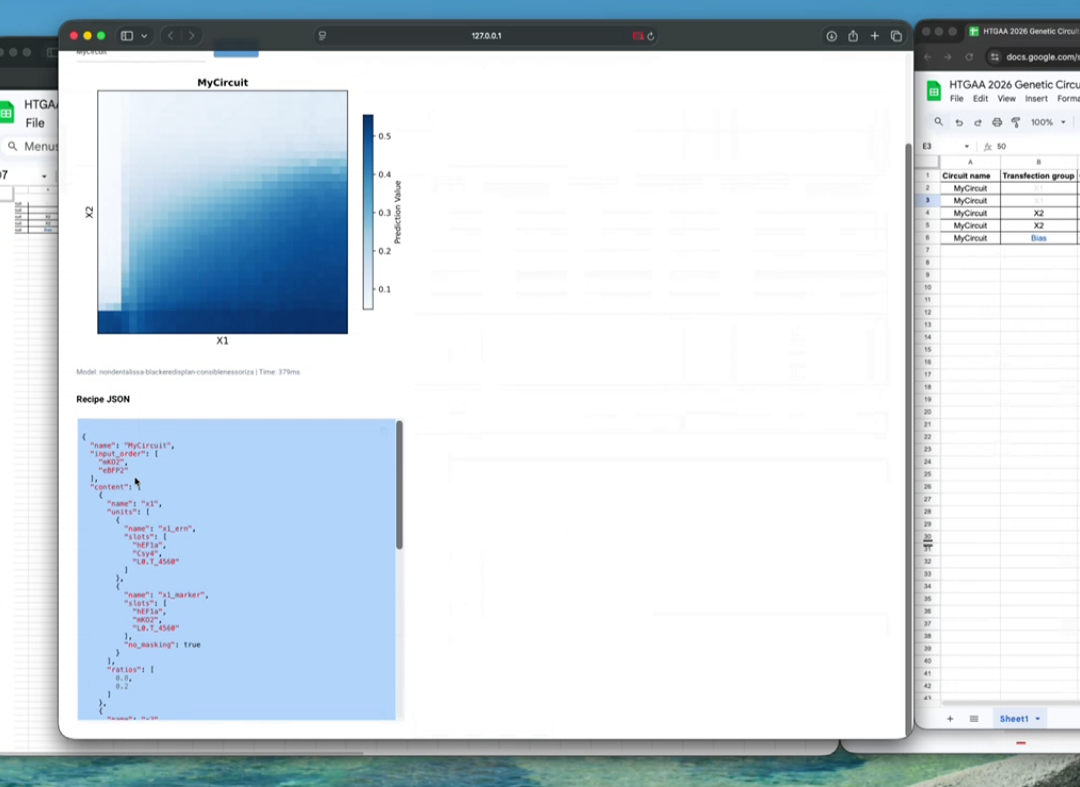
Below is a diagram depicting an intracellular single-layer perceptron where the X1 input is DNA encoding for the Csy4 endoribonuclease and the X2 input is DNA encoding for a fluorescent protein output whose mRNA is regulated by Csy4. Tx: transcription; Tl: translation.Draw a diagram for an intracellular multilayer perceptron where layer 1 outputs an endoribonuclease that regulates a fluorescent protein output in layer 2.
The diagram can be found below:
Assignment Part 2: Fungal Materials
What are some examples of existing fungal materials and what are they used for? What are their advantages and disadvantages over traditional counterparts?
Some examples of fungal materials are mycelium packaging and fungal leather.
Mycelium packaging can be regrown, replace styrofoam, is biodegradable, and can be sustainably generation. Disadvantages can come in resistance to degrdation under moisture.
Myceliucum leather can be produeced animal free and has the earlier mentioned advantages. Mycelium leather may not be as durable as animal counterparts, but it may be engineered to one day meet or exceed that duability.
What might you want to genetically engineer fungi to do and why? What are the advantages of doing synthetic biology in fungi as opposed to bacteria?
I might genetically engineer fungi to eat waste products or grow as shielding or building material. In either case, the fungi can serve as a meaningful redesinging of the environment. Advantages of doing synthethic biology with fungi are at least three fold: A) you can perform unique protein modications and syntheses that you can’t yet easily do with bacteria, you can take advantage of more complex molecular regulatory systems within fungi, in some cases they may be more safely scalable, and you get easy “Last of Us” Jokes.
Assignment Part 3: First DNA Twist Order
Objectives of this were as follows:
Review the Individual Final Project documentation guidelines.
Done
Submit this Google Form with your draft Aim 1, final project summary, HTGAA industry council selections, and shared folder for DNA designs.
Done.
Review Part 3: DNA Design Challenge of the week 2 homework. Design at least 1 insert sequence and place it into the Benchling/Kernel/Other folder you shared in the Google Form above. Document the backbone vector it will be synthesized in on your website.
This was done as well. The backbone chosen was pSB1C3 (High Copy). As a student without lab access, I was allowed to be exempt from the actual order.
Week 8 HW: Break Week
This was a free week and was not obligatory to mark. This week was used to exploration of course materials and Final Project development.
Week 9 HW: Week 9 — Cell-Free Systems
General homework questions
Explain the main advantages of cell-free protein synthesis over traditional in vivo methods, specifically in terms of flexibility and control over experimental variables. Name at least two cases where cell-free expression is more beneficial than cell production.
Cell free expression allows teams to conduct biomanufacturing without living cells and operate beyond the constraints of productions in living systems.
Two cases where cell-free expression is more beneficial than cell production are where:
A) biomanufacturing would kill the cells
B) teams have a desire to rapidly prototype biomanufacturing workflows after computational modeling
Describe the main components of a cell-free expression system and explain the role of each component.
The main components of cell free expression are as follows:
Cell lysate/mix: These provide the vital translation components, in addiiton to ribosomes, other enzymbes, and tRNAs
Genomic Template: These are needed to encode and develop the protein of interest
Salts: Ioinic condition maintenance
Buffer: Maintaining pH
Cofactors/additives: Promoting enzymatic activity
Amino Acids: These supply building blocks for translation
Energy System: These supply energy to power transcription and translation
Why is energy provision regeneration critical in cell-free systems? Describe a method you could use to ensure continuous ATP supply in your cell-free experiment.
Energy provision generation is critical to sustain reactions. Supplying molecular energy packs that can regenerate ATP during a reaction can assist this.
Compare prokaryotic versus eukaryotic cell-free expression systems. Choose a protein to produce in each system and explain why.
Eukaryotic and prokaryotic cell-free expression systems have their own unqiue advanges. Prokaryotic cell-free expression tends to be great for rapid prototyping and is robust. Eukaryotic systems can have advantages with complex products by which post-translational modification may be desired.
I’d possibly consider developing flourescent proteins in prokaryotic systems while focusing on antibodies with eukarytic systems.
How would you design a cell-free experiment to optimize the expression of a membrane protein? Discuss the challenges and how you would address them in your setup.
A) My initial guess would be to examine setups by which I can utilize detergents and or membrane disaggrgating components and trial setups.
B) Challenges come from aggregation of membrane proteins, insolubility of components, and reduced yields. I would possibly consider different spatial component arrangements, release modalities, temperature changes, and experiment with amounts of disaggrgating components.
Imagine you observe a low yield of your target protein in a cell-free system. Describe three possible reasons for this and suggest a troubleshooting strategy for each.
Three possible reasons could be a poor template, poor environmental considtions, and or lack of energy.
Troubleshooting respectively would look like: attempting template optimization, environmental optimization, and trialing suppying more energy sources for the reaction. Each of these would be systematically trialed.
Homework question from Kate Adamala
Design an example of a useful synthetic minimal cell as follows:
Pick a function and describe it.
One function is fluorescence in the presence of a target molecules.
What would your synthetic cell do? What is the input and what is the output?
This cell would fluoresce with response to environmental exposure to pesticides. The input is the pesticide and the output is fluorescence.
Could this function be realized by cell-free Tx/Tl alone, without encapsulation?
This could be.
Could this function be realized by genetically modified natural cell?
Yes.
Describe the desired outcome of your synthetic cell operation.
The desired outcome would be a deployable cell that could be used to find and determine pesticide levels.
Design all components that would need to be part of your synthetic cell.
The needed components would be an array of cell-regulatory components, amino acides, co-factors, salts, an energy regeneration system, NTPs, amino acids, a DNA template, a DNA template encoding reporter, and cell-free transcription and translation machinery all within a liposome.
What would be the membrane made of?
The membrane could be made of a phospolipid lipsome
What would you encapsulate inside? Enzymes, small molecules.
Everything that needs to be encapsulated would be. Enzumes and small molecules alone is insufficient.
Which organism your Tx/Tl system will come from? Is bacterial OK, or do you need a mammalian system for some reason? (hint: for example, if you want to use small molecule modulated promotors, like Tet-ON, you need mammalian)
A bacterial sourrce, like E. Coli could suffice.
How will your synthetic cell communicate with the environment? (hint: are substrates permeable? or do you need to express the membrane channel?)
The synthetic cell could communicate with the environment through membrane bidirectional membrane exhange of elements, followed by fluorescence in the preosence of the pesticides. What is assumed is that the pesticides pass through the membrane and can interact with the cellular internal machinery.
Experimental details
To Be Added
List all lipids and genes. (bonus: find the specific genes; for example, instead of just saying “small molecule membrane channel” pick the actual gene.)
I could measure the flourescene with a plate reader, compatible microscope, and or similiar system.
Homework question from Peter Nguyen
Freeze-dried cell-free systems can be incorporated into all kinds of materials as biological sensors or as inducible enzymes to modify the material itself or the surrounding environment. Choose one application field — Architecture, Textiles/Fashion, or Robotics — and propose an application using cell-free systems that are functionally integrated into the material. Answer each of these key questions for your proposal pitch:
To Be Added
Write a one-sentence summary pitch sentence describing your concept.
This pitch is for a freeze-dried cell-free biosensor that turns blue in the presence of caffeine to help caffeine-sensitive consumers identify highly caffeinated drinks.
How will the idea work, in more detail? Write 3-4 sentences or more.
The system would contain a freeze-dried cell-free transcription/translation mixture and a caffeine-responsive genetic circuit. When added to a beverage, caffeine would activate the sensing system and induce expression of a visible reporter protein such as Electra2 or a colorimetric enzyme output. The reaction would produce a detectable color change within a short time period, allowing users to estimate whether caffeine is present in the drink.
What societal challenge or market need will this address?
This system could help caffeine-sensitive individuals avoid accidental caffeine exposure in beverages where caffeine content is poorly labeled, inconsistent, or unknown. It may also support rapid beverage testing in restaurants, cafes, or consumer safety settings.
How do you envision addressing the limitation of cell-free reactions (e.g., activation with water, stability, one-time use)?
Stability could be improved through lyophilization additives such as trehalose and by storing the reaction mixture in sealed moisture-resistant packaging until use. The system is likely single-use after hydration, so the biosensor could be designed as a disposable low-cost test strip or reagent capsule.
Past that, I could address the limitation by simply listing warnings for ingredients that could complicate the reaction process.
Homework question from Ally Huang
Freeze-dried cell-free reactions have great potential in space, where resources are constrained. As described in my talk, the Genes in Space competition challenges students to consider how biotechnology, including cell-free reactions, can be used to solve biological problems encountered in space. While the competition is limited to only high school students, your assignment will be to develop your own mock Genes in Space proposal to practice thinking about biotech applications in space!
To Be Added
For this particular assignment, your proposal is required to incorporate the BioBits® cell-free protein expression system, but you may also use the other tools in the Genes in Space toolkit (the miniPCR® thermal cycler and the P51 Molecular Fluorescence Viewer). For more inspiration, check out https://www.genesinspace.org/ .
To Be Added
Provide background information that describes the space biology question or challenge you propose to address. Explain why this topic is significant for humanity, relevant for space exploration, and scientifically interesting. (Maximum 100 words)
Pivoting the prior example:
Astronauts experience sleep disruption and fatigue during long-duration space missions. Lightweight biochemical sensors could help monitor stimulant exposure in environments with limited laboratory equipment. Freeze-dried cell-free systems are promising because they are portable, stable, and programmable.
Name the molecular or genetic target that you propose to study. Examples of molecular targets include individual genes and proteins, DNA and RNA sequences, or broader -omics approaches. (Maximum 30 words)
A caffeine-responsive genetic circuit (core possibly being the CYP1A2 gene) linked to a fluorescent or color-changing reporter protein in a freeze-dried cell-free system.
Describe how your molecular or genetic target relates to the space biology question or challenge your proposal addresses. (Maximum 100 words)
A caffeine biosensor could help astronauts monitor stimulant exposure during missions where sleep and alertness are important. Freeze-dried cell-free systems are useful because they are compact, shelf-stable, and easy to activate with water.
Clearly state your hypothesis or research goal and explain the reasoning behind it. (Maximum 150 words)
I hypothesize that a freeze-dried cell-free biosensor can detect caffeine and produce a measurable fluorescent or colorimetric signal after hydration. The goal is to test whether reporter intensity changes with caffeine concentration and whether the system remains stable after storage.
Outline your experimental plan - identify the sample(s) you will test in your experiment, including any necessary controls, the type of data or measurements that will be collected, etc. (Maximum 100 words)
Freeze-dried cell-free reactions would be rehydrated with different caffeine concentrations, under a variety of common environmental conditions normal to the astronauts. Negative controls would contain no caffeine. Reporter output would be measured using fluorescence or visible color change with a flourescence reader. Stability after storage would also be tested.
Homework Part B:
Put your chosen final project slide in the appropriate slide deck following the instructions on slide 1:
MIT/Harvard/Wellesley ONE FINAL PROJECT IDEA
Committed Listener ONE FINAL PROJECT IDEA
Done.
Submit this Final Project selection form if you have not already.
Done.
Begin planning how you will write your final project documentation based on these guidelines
Done.
Prepare your first DNA order and put it in the “Twist (MIT)” or “Twist (Nodes)” tab of the 2026 HTGAA Ordering: DNA, Reagents, Consumables spreadsheet, as appropriate.
Coordinated with Node. Not having lab access, I was excused from needing to order a physical construct
First Twist order deadline for MIT/Harvard/Wellesley students is Friday, April 3 at 11PM ET
First Twist order deadline for Committed Listeners is Friday, April 10 at 11PM ET. (Your Node Lead will place the Twist order, so please work with them to finalize your constructs and ordering decisions.)
Coordinated with Node. Not having lab access, I was exempt from needing to order a physical construct.
The main aspect of my project that I will measure is the functional activity of a mutated InaZ construct, specifically whether it increases ice nucleation efficiency relative to a control InaZ construct. I will perform this measurement using a controlled freezing assay in which replicate samples are cooled gradually and monitored for the onset of ice formation. The primary data collected will be the temperature at which freezing begins in each sample. Ice formation will be detected through optical observation of crystal formation and through temperature sensors that record the freezing point. In addition, I would verify the identity of the mutated construct using PCR, gel electrophoresis, and DNA sequencing. PCR and gel electrophoresis would be used to confirm the presence and approximate size of the inserted DNA, while DNA sequencing would confirm that the engineered inaZ mutation is correct. Together, these measurements allow me to confirm both that the construct was built properly and that it produces the intended increase in ice nucleation activity.
Restated:
Functional Assay: Ice Nucleation
Objective: Measure efficiency relative to a control construct.
Method: Controlled freezing assay with gradual cooling of replicate samples.
Data Points: Freezing onset temperature, recorded via combined optical observation and thermal sensors.
Genetic Verification
PCR & Gel Electrophoresis: Confirm the presence and approximate size of the inserted DNA.
DNA Sequencing: Verify the exact sequence of the engineered inaZ mutation.
Homework: Waters Part I — Molecular Weight
We will analyze an eGFP standard on a Waters Xevo G3 QTof MS system to determine the molecular weight of intact eGFP and observe its charge state distribution in the native and denatured (unfolded) states. The conditions for LC-MS analysis of intact protein cause it to unfold and be detected in its denatured form (due to the solvents and pH used for analysis).
Based on the predicted amino acid sequence of eGFP (see below) and any known modifications, what is the calculated molecular weight? You can use an online calculator like the one at https://web.expasy.org/compute_pi/
eGFP Sequence:
MVSKGEELFTG VVPILVELDG DVNGHKFSVS GEGEGDATYG KLTLKFICTT GKLPVPWPTL VTTLTYGVQC FSRYPDHMKQ HDFFKSAMPE GYVQERTIFF KDDGNYKTRA EVKFEGDTLV NRIELKGIDF KEDGNILGHK LEYNYNSHNV YIMADKQKNG IKVNFKIRHN IEDGSVQLAD HYQQNTPIGD GPVLLPDNHY LSTQSALSKD PNEKRDHMVL LEFVTAAGIT LGMDELYKLE HHHHHH
Note: This contains a His-purification tag (HHHHHH) and a linker (the LE before it).
Using the calculator, I get Theoretical pI/Mw: 5.90 / 28006.60.
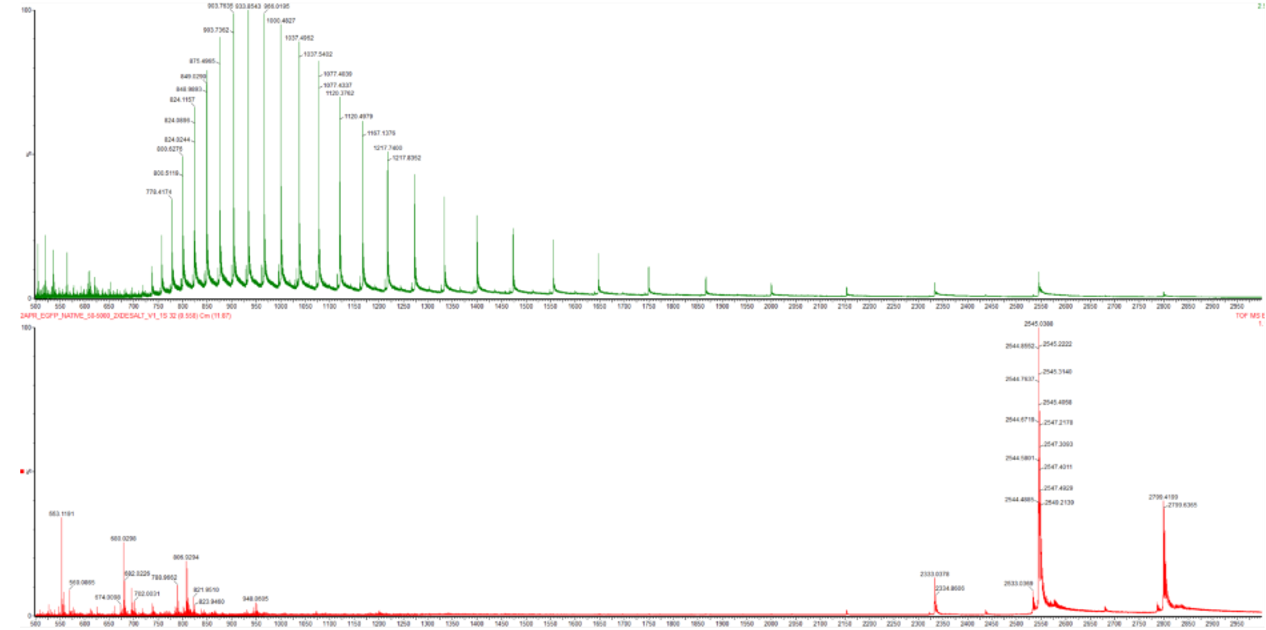
Calculate the molecular weight of the eGFP using the adjacent charge state approach described in the recitation. Select two charge states from the intact LC-MS data (Figure 1) and:
Determine z or each adjacent pair of peaks (n , n+1) using: z = ((m/z_n+1)/(m/z_n) - (m/z_n+1)) = ((m/z_n+1)/(m/z_n) - (m/z_n+1))
z = ((966.0390)/(1000.4302-966.0390)= 28.09
z is roughly 28 (28+)
Determine the MW of the protein using the relationship between
MW = z (m/z-1.0073) = 28 (1000.4302-1.0073) = 27.98384 kDa
Calculate the accuracy of the measurement using the deconvoluted MW from 2.2 and the predicted weight of the protein from 2.1 using:
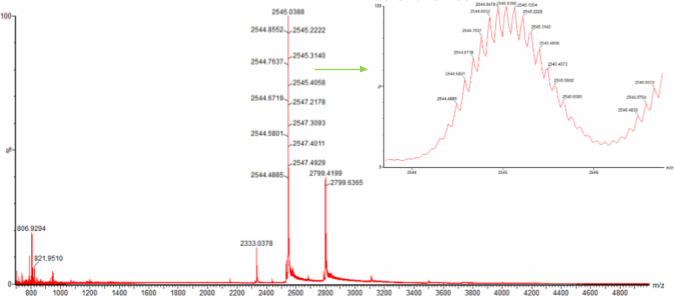
Can you observe the charge state for the zoomed-in peak in the mass spectrum for the intact eGFP? If yes, what is it? If no, why not?
Potentially. Assuming our peak as truly 1473.7420, our z is rougly 19+. I may need more information to answer more conclusively.
Homework: Waters Part II — Secondary/Tertiary structure
This was optional and skipped.
Homework: Waters Part III — Peptide Mapping - primary structure
We will digest the eGFP protein standard into peptides using trypsin (an enzyme that selectively cleaves the peptide bond after Lysine (K) and Arginine (R) residues. The resulting peptides will be analyzed on the Waters BioAccord LC-MS to measure their molecular weights and fragmented to confirm the amino acid sequence within each peptide – generating a “peptide map”. This process is used to confirm the primary structure of the protein.
There are a variety of tools available online to calculate protein molecular weight and predict a list of peptides generated from a tryptic digest. We will be using tools within the online resource Expasy (the bioinformatics resource portal of the Swiss Institute of Bioinformatics (SIB)) to predict a list of tryptic peptides from eGFP.
How many Lysines (K) and Arginines (R) are in eGFP? Please circle or highlight them in the eGFP sequence given in Waters Part I question 1 above. (Note: adding the sequence to Benchling as an amino acid file and clicking biochemical properties tab will show you a count for each amino acid).
Copy/paste the sequence above into the input box in the PeptideMass tool to generate expected list of peptides.
Done
Use Figure 4 below as a guide for the relevant parameters to predict peptides from eGFP.
Done
Click “Perform the Cleavage” button in the PeptideMass tool and report the number of peptides generated when using trypsin to perform the digest.
Done
Based on the LC-MS data for the Peptide Map data generated in lab (please use Figure 5a as a reference) how many chromatographic peaks do you see in the eGFP peptide map between 0.5 and 6 minutes? You may count all peaks that are >10% relative abundance.
I see between 18 and 27.
Assuming all the peaks are peptides, does the number of peaks match the number of peptides predicted from question 2 above? Are there more peaks in the chromatogram or fewer?
Yes, at the upper end
Identify the mass-to-charge (m/z)) of the peptide shown in Figure 5b. What is the charge (z)) of the most abundant charge state of the peptide (use the separation of the isotopes to determine the charge state). Calculate the mass of the singly charged form of the peptide [M+H]+) based on its m/z and z.
A) 525.76712
B) (2*525.76712)-(1.0073)= 1,051.53424 Da
Identify the peptide based on comparison to expected masses in the PeptideMass tool. What is mass accuracy of measurement? Please calculate the error in ppm. (Recall that Accuracy = |MW experiment - MWtheory/MWtheory).
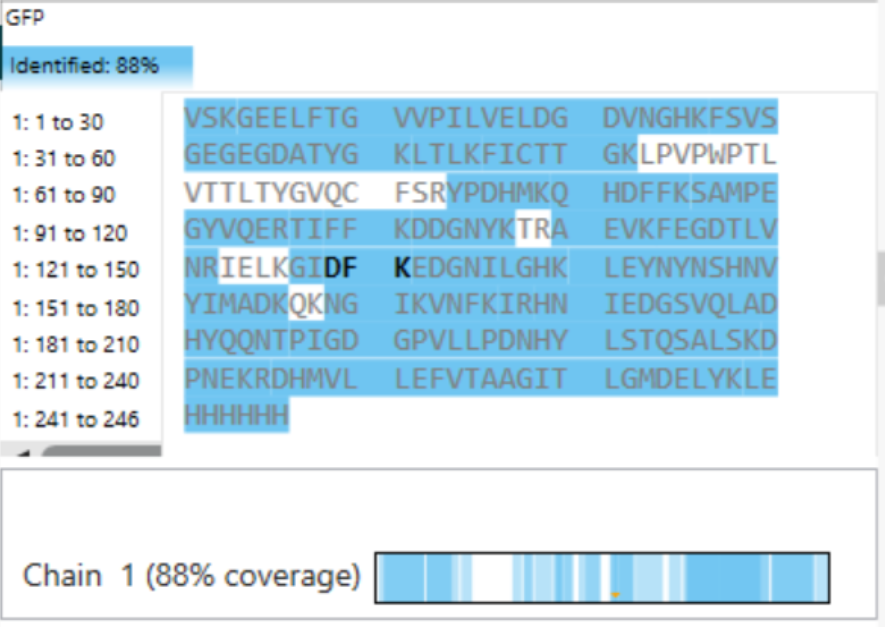
What is the percentage of the sequence that is confirmed by peptide mapping? (see Figure 6).
Chain 1 = 88%
Homework: Waters Part IV — Oligomers
We will determine Keyhole Limpet Hemocyanin (KLH)’s oligomeric states using charge detection mass spectrometry (CDMS). CDMS single-particle measurements of KLH allow us to make direct mass measurements to determine what oligomeric states (that is, how many protein subunits combine) are present in solution. Using the known masses of the polypeptide subunits (Table 1) for KLH, identify where the following oligomeric species are on the spectrum shown below from the CDMS (Figure 7):
7FU Decamer = (340 kDa x 10) = 3.4 MDa
8FU Didecamer = (400 kDa x 20) = 8 MDa
8FU 3-Decamer = (400 kDa x 30) = 12 MDa
8FU 4-Decamer = (400 kDa x 30) = 16 MDa
Homework: Waters Part V — Did I make GFP?
Please fill out this table with the data you acquired from the lab work done at the Waters Immerse Lab in Cambridge, or else the data screenshots in this document if you were unable to have lab work done at Waters.
Screenshots listed on lab page.
Week 11 HW: Bioproduction & Cloud Labs
Part A: The 1,536 Pixel Artwork Canvas | Collective Artwork
Contribute at least one pixel to this global artwork experiment before the editing ends on Sunday 4/19 at 11:59 PM EST!
A personalized URL was sent to the email address associated with your Discourse account, and you can discuss the artwork on the Discourse!
If you did not have a chance to contribute, it’s okay, just make sure you become a TA this fall! 😉
Multiple pixels were contrited to Artwork Canvases. Below the CFPS and Overall Artwork Contributions are given.
CFPS Contributions
Overall Artwork Contributions
Done: Added pixels. See above.
Make a note on your HTGAA webpages including:
Done: I added a total of 14 pixels by the end of the experiment: 12 colored pixels and 2 removal pixels.
what you contributed to the community bioart project (e.g., “I made part of the DNA on the bottom right plate”)
Done: I added a couple small details to subfeatures of some designs. The general idea was to help complete objects in progress or add a small sub-feature, visually. One example was adding a temporary light to one of the spaceships.
Contributions to a follow-up activity at Synbiobeta were done as well as shown below.
what you liked about the project, and
Done: I liked the collaborative and semi-collaborative nature of the project. There was space for those who wanted to add with coordination and space for those who wanted to add individually from their own vision. Seeing what people came up with was great, as well.
what about this collaborative art experiment could be made better for next year.
Done: To improve it, one could widen the color section, plate area, and reduce the painting refresh-time.
Part B: Cell-Free Protein Synthesis | Cell-Free Reagents
Referencing the cell-free protein synthesis reaction composition (the middle box outlined in yellow on the image above, also listed below), provide a 1-2 sentence description of what each component’s role is in the cell-free reaction.
E. coli Lysate:
BL21 (DE3) Star Lysate (includes T7 RNA Polymerase) - This contains the core metabolic components and enyzmes for transcription and translation. The T7 RNA polymerase is needed for eventual gene transcription and mRNA towards eventual protein production.
Salts/Buffer:
Potassium Glutamate - This is a source of potassium and anions for the reaction, providing needed blance for ribosome function.
HEPES-KOH pH 7.5 - This buffer helps to maintain a stable pH, again helpful for ribosome stability.
Magnesium Glutamate - This provides Mg2+m which is helpful for enzymatic activity
Potassium phosphate monobasic - This works with Potassium dibasic as a secondary buffering system for pH stabilization.
Potassium phosphate dibasic - This works with Potassium monobasic as a secondary buffering system for pH stabilization.
Energy / Nucleotide System:
Ribose - This serves along with gluclose as energy and carbon sources to power the reactions.
Glucose - This serves along with ribose as energy and carbon sources to power the reactions.
AMP - This servves as a ATP synthesis precursor. This and the following 3 are required for RNA synthesis.
CMP - This servves as a CTP precursor.
GMP - This servves as a GTP precursor.
UMP - This servves as a UTP precursor.
Guanine - This serves a a purine base and precursor to produce guanine nucleotides.
Translation Mix (Amino Acids):
17 Amino Acid Mix - This mix supplies the amnio acids used for protein synthesis.
Tyrosine - This is key for phosphorylation but specifically vital as a substrate in forming the target protein.
Cysteine - This is separated due to its instability and is required in protein synthesis.
Additives:
Nicotinamide - This functions as the NAD+ biosynthesis precursor.
Backfill:
Nuclease Free Water - This serves as the solvent for the reaction.
Describe the main differences between the 1-hour optimized PEP-NTP master mix and the 20-hour NMP-Ribose-Glucose master mix shown in the Google Slide above. (2-3 sentences)
The main differences betweem the two master mixes can be found in energy regeration, time-spent for reactions, recycling of components, qualities of metabolism engaged, and composition. Both are effective, but they serve different purposes, be it short-term production vs longer production over time. The use comes down to the context of synthesis desired.
Bonus question: How can transcription occur if GMP is not included but Guanine is?
If reaction pathways exist to convert guanine to a product that can facilitate GMP’s role, transcription can reasonably occur.
Part C: Planning the Global Experiment | Cell-Free Master Mix Design
Given the 6 fluorescent proteins we used for our collaborative painting, identify and explain at least one biophysical or functional property of each protein that affects expression or readout in cell-free systems. (Hint: options include maturation time, acid sensitivity, folding, oxygen dependence, etc) (1-2 sentences each)
Create a hypothesis for how adjusting one or more reagents in the cell-free mastermix could improve a specific biophysical or functional property you identified above, in order to maximize fluorescence over a 36-hour incubation. Clearly state the protein, the reagent(s), and the expected effect.
Increasing HEPES-KOH (through improved maintenance of a near neutral pH) may improve the fluorescence from mKO2 over a 36-hour incubation period by stabilizing its reaction to pH and reducing pH dependent fluorescence loss.
The second phase of this lab will be to define the precise reagent concentrations for your cell-free experiment. You will be assigned artwork wells with specific fluorescent proteins and receive an email with instructions this week (by April 24). You can begin composing master mix compositions here.
Done. 3 wells were utilized as indicated by the image below.
The codes for each are found below:
The final phase of this lab will be analyzing the fluorescence data we collect to determine whether we can draw any conclusions about favorable reagent compositions for our fluorescent proteins. This will be due a week after the data is returned (date TBD!). The reaction composition for each well will be as follows:
6 μL of Lysate
10 μL of 2X Optimized Master Mix from above
2 μL of assigned fluorescent protein DNA template
2 μL of your custom reagent supplements
Total: 20 μL reaction
Acknowledged. Reactions were made and compositions were honored.
Part D: Build-A-Cloud-Lab | (optional) Bonus Assignment
Use this simulation tool to create an interesting looking cloud lab out of the Ginkgo Reconfigurable Automation Carts. This is just a minimal implementation so far, but I would love to see some fun designs!
Using the simulation tool, I made a setup that mixed RACs. What follows is a RAC separated by a 1M, folloed by a mix of RACs separated by.25M, followed by another mix of RACs separated by another 1M leading to a final RAC. These are all in one line as shown below.
Week 12 HW: Building Genomes
Homework: This homework was a reference to A) the acknowledhing the updated Week 11 and B) direction to make progress on our Final Project. This is expressed in the image below.
Progress: Done.
A) Contributed pixels to the Global Artwork Experiment and Followed up with mastermix concentrations as per part 3 of Homework 11C.
B) The project was refined through constructive class critique and further reflection.
Week 13 HW: AI, SynBio, and Scaling Health Innovation (ARPA-H)
Homework: Work on your Final Project Present it May 12 (MIT/Harvard) or May 13 (Committed Listeners)
Progress: Done. I further developed and practiced my presentation with my study group, classmates, and via some solo practice.
Week 14 HW: Bio Design & Bio Fabrication
Homework: Homework: Finish your Final Project Present it May 12 (MIT/Harvard) or May 13 (Committed Listeners
Progress: Done. Final preparations were had during this week by which I practiced my presentation with my study group, classmates, and via some solo practice.
As an additional note: my study group held multiple sessions to ensure that each of our projects were further sharpened.
My Final Project, Project Z Freeze was successfully presented. My timeslot was 8:10PM on May 13.
Special Note: As a Committed Listener without lab access, documentation within my page is for purely acknowledgement purposes. There were no explicit instructions for Committed Listeners to submit lab work for this week, and with that, I reviewed the material.
This lab covered standard lab practices and review.
Special Note: As a Committed Listener without lab access, documentation within my page is for purely acknowledgement purposes. There were no explicit instructions for Committed Listeners to submit lab work for this week, and with that, I reviewed the material.
This lab covered gel art through restriction digests and gel electrophoresis.
Special Note: The Homework for this class included the Post-Lab Questions. For ease of interprertation, the Post-Lab is reposted below and to ease documentation. Further, the other parts of the homework were placed after. There were no other explicit instructions for Committed Listeners that were not already within the homework, so other aspects of this lab were relegated to purely review.
In this lab, we were tasked with creating a design that could be generated by an OpenTrons Liquid Handling Robot.
Special Note: As per (https://2026a.htgaa.org/2026a/course-pages/weeks/week-04/index.html), “Lab work this week is contained within the homework assignment below.”.
This week’s Lab work was effectively part of this week’s Homework. This is reflected in Part D of the week 4 homework but is reposted below for ease.
Part D. Group Brainstorm on Bacteriophage Engineering As follows, the assigned work was:
Special Note: This week’s Lab work was effectively part of this week’s Homework. This is reflected in Part C of the week 5 homework.
As per (https://2026a.htgaa.org/2026a/course-pages/weeks/week-05/index.html), “Lab work this week is contained within the homework assignment below.”.
Special Note: As a Committed Listener without lab access, I was excused from this and documentation within my page is for purely acknowledgement purposes. There were no explicit instructions for Committed Listeners to submit lab work for this week, and with that, I reviewed the material.
This week’s lab had a dry and wet component. As a Global Comitted Listener without lab access, I was excused from the wetlab component. I joined for the dry component over which were were allowed to work as a team. That said, what follows are snapshots of our work. The focus was the building of our own IANN.
Pre-Lab | Overview Download Neuromorphic Wizard: download this folder NeuromorphicWizard onto your machine by clicking “Download all” in the upper right. Follow the instructions in ‘README.md’ for a step-by-step installation guide.
Special Note: As a Committed Listener without lab access, documentation within my page is for purely acknowledgement purposes. There were no explicit instructions for Committed Listeners to submit lab work for this week, and with that, I reviewed the material.
Homework, shown on the page of Week 9 is reposted here to ease interpretation and documentation.
General homework questions Explain the main advantages of cell-free protein synthesis over traditional in vivo methods, specifically in terms of flexibility and control over experimental variables. Name at least two cases where cell-free expression is more beneficial than cell production. Cell free expression allows teams to conduct biomanufacturing without living cells and operate beyond the constraints of productions in living systems.
Mass Spectrometry
Special Note: As a Committed Listener without lab access, documentation within my page is for purely acknowledgement purposes. There were no explicit instructions for Committed Listeners to submit lab work for this week, and with that, I reviewed the material.
The appendix listed figures to be used for the homework. These were downloaded and posted below for documentation purposes.
See: “Appendix – Figures from Lab work to be used for Homework” from https://2026a.htgaa.org/2026a/course-pages/weeks/week-10/lab/index.html
Special Note: This assignment was a combined homework and laboratory assignment that was reflected in both the homeowork and lab. What follows is a repost of what was asked from the laboratory page, as answered from the homework page post.
Part A: The 1,536 Pixel Artwork Canvas | Collective Artwork Contribute at least one pixel to this global artwork experiment before the editing ends on Sunday 4/19 at 11:59 PM EST! A personalized URL was sent to the email address associated with your Discourse account, and you can discuss the artwork on the Discourse! If you did not have a chance to contribute, it’s okay, just make sure you become a TA this fall! 😉
Special Note: This lab had both a section mandatory for all students and one especially for committed listeners.
Post Lab Questions | Mandatory for All Students (References supplied from lab:
A) Du W, Song Y, Liu M, Yang H, Zhang Y, Fan Y, Luo X, Li Z, Wang N, He H, Zhou H, Ma W, Zhang T. Gene expression pattern analysis of a recombinant Escherichia coli strain possessing high growth and lycopene production capability when using fructose as carbon source. Biotechnol Lett. 2016 Sep;38(9):1571-7. doi: 10.1007/s10529-016-2133-0. Epub 2016 Jul 5. PMID: 27379652.
Special Note: As a Committed Listener without lab access, documentation within my page is for purely acknowledgement purposes. There were no explicit instructions for Committed Listeners to submit lab work for this week, and with that, I reviewed the material.
This lab covered standard lab practices and review.
Week 2 Lab: Lab DNA Gel Art
Special Note: As a Committed Listener without lab access, documentation within my page is for purely acknowledgement purposes. There were no explicit instructions for Committed Listeners to submit lab work for this week, and with that, I reviewed the material.
This lab covered gel art through restriction digests and gel electrophoresis.
Week 3 Lab: Lab Automation
Special Note: The Homework for this class included the Post-Lab Questions. For ease of interprertation, the Post-Lab is reposted below and to ease documentation. Further, the other parts of the homework were placed after. There were no other explicit instructions for Committed Listeners that were not already within the homework, so other aspects of this lab were relegated to purely review.
In this lab, we were tasked with creating a design that could be generated by an OpenTrons Liquid Handling Robot.
Post-Lab Questions:
Find and describe a published paper that utilizes the Opentrons or an automation tool to achieve novel biological applications.
DeRoo, J.B., Jones, A.A., Slaughter, C.K., Ahr, T.W., Stroup, S.M., Thompson, G.B. and Snow, C.D., 2025. Automation of protein crystallization scaleup via Opentrons-2 liquid handling. SLAS technology, 32, p.100268.
This work describes an approach by which an Opentrons-2 liquid handling robot was used for automating sitting drop protein crystallization trials. This ability also improve comparability of products produced, improving studies that depend on their proper manufacture. An important detail is how the Opentrons-2 can prove a cost-effective option for laboratory operations. For example, at the time of writing, the Opentrons-2 can be purchased for around 13.5K USD vs that of a Gryphon machine at around 65K USD.
Write a description about what you intend to do with automation tools for your final project.
I’m still forming my thoughts about how I want to effectively use automation tools for my final project.
So far, I am interested in branching off from example #2 given in the Homework and the above example, regarding screening an array of designed biosensor constructs.
One idea had in mind was towards a digital tracing project that revolves around said constructs used to track known entities.
Simply, products are given a unique ID with stored parameters. These are linked within a automation run so that each product is trackable as they are processed. One application that is probably already in use but would be fun to adapt towards something not already applied would be swappable combined wearable crystallized biosensors that are traded in daily for workers that are liable to be exposed to a particular organism and pollutant pairs.
I could use an Echo for transfer of nano-scale components.
The Bravo or Opentrons-2 could be used for precise, automated pipetting ,especially of the crystals.
The multiflow would be used to dispense the larger scale volume components.
The PlateLoc would be helful for sealing the plates.
The inheco could be used for controlled incubation.
The Xpeel would be used for careful desealing of the plates.
Finally, the PHERAstar could be used for reading fluorescence outputs.
Still developing this out from this branch.
(Part of Homework 3)
Assignment: Python Script for Opentrons Artwork
Review this week’s recitation and this week’s lab for details on the Opentrons and programming it.
Done.
Generate an artistic design using the GUI at opentrons-art.rcdonovan.com.
As a remote participant, I prototyped a design using the GUI at opentrons-art.rcdonovan.com.
This resulted in a layered plus symbol shown below.
Using the coordinates from the GUI, follow the instructions in the HTGAA26 Opentrons Colab to write your own Python script which draws your design using the Opentrons.
You may use AI assistance for this coding — Google Gemini is integrated into Colab (see the stylized star bottom center); it will do a good job writing functional Python, while you probably need to take charge of the art concept.
If you’re a proficient programmer and you’d rather code something mathematical or algorithmic instead of using your GUI coordinates, you may do that instead.
Acknowledged
The coordinates for generating such can be found here, courtesy of RC Donovan’s tool:
If the Python component is proving too problematic even with AI and human assistance, download the full Python script from the GUI website and submit that:
Not needed, but appreciated.
If you use AI to help complete this homework or lab, document how you used AI and which models made contributions.
For my node, in order to work with their printer which had two colors, a modified version was created. Gemini within was tested and employed to deliver the following result.
Sign up for a robot time slot if you are at MIT/Harvard/Wellesley or at a Node offering Opentrons automation.
The Python script you created will be run on the robot to produce your work of art!
At MIT/Harvard? Lab times are on Thursday Feb.19 between 10AM and 6PM.
At other Nodes? Please coordinate with your Node.
I was added to the William and Mary Node. I coordinated with Margaret and Kate for OpenTrons code submission. My code was submitted to Kate and who was then able to faciliate the printing of my design. Please see below.
This week’s Lab work was effectively part of this week’s Homework. This is reflected in Part D of the week 4 homework but is reposted below for ease.
Part D. Group Brainstorm on Bacteriophage Engineering
As follows, the assigned work was:
1.Find a group of ~3–4 students
Done.
2.Read through the Phage Reading material listed under “Reading & Resources” below.
Done.
3.Review the Bacteriophage Final Project Goals for engineering the L Protein:
*Increased stability (easiest)
*Higher titers (medium)
*Higher toxicity of lysis protein (hard)
Done.
4.Brainstorm Session
Choose one or two main goals from the list that you think you can address computationally (e.g., “We’ll try to stabilize the lysis protein,” or “We’ll attempt to disrupt its interaction with E. coli DnaJ.”).
Increased stability was chosen by my group.
Write a 1-page proposal (bullet points or short paragraphs) describing:
Which tools/approaches from recitation you propose using (e.g., “Use Protein Language Models to do in silico mutagenesis, then AlphaFold-Multimer to check complexes.”).
Why do you think those tools might help solve your chosen sub-problem?
Name one or two potential pitfalls (e.g., “We lack enough training data on phage–bacteria interactions.”).
Include a schematic of your pipeline.
Done. Our Members were: Jason Ross, Jay Handfield, Nana Agyei, Raphael Aca, and Xavier Palmer. Our team’s answers can be be found below:
5.Each individually put your plan on your HTGAA website
Include your group’s short plan for engineering a bacteriophage
Done. See the image above.
Week 5 Lab: Protein Design Part II
Special Note: This week’s Lab work was effectively part of this week’s Homework. This is reflected in Part C of the week 5 homework.
Week 6 Lab : Genetic Circuits Part I: Assembly Technologies
Special Note: As a Committed Listener without lab access, I was excused from this and documentation within my page is for purely acknowledgement purposes. There were no explicit instructions for Committed Listeners to submit lab work for this week, and with that, I reviewed the material.
Week 7 Lab : Genetic Circuits Part II: Neuromorphic Circuits
This week’s lab had a dry and wet component. As a Global Comitted Listener without lab access, I was excused from the wetlab component. I joined for the dry component over which were were allowed to work as a team. That said, what follows are snapshots of our work. The focus was the building of our own IANN.
Pre-Lab | Overview
Download Neuromorphic Wizard: download this folder NeuromorphicWizard onto your machine by clicking “Download all” in the upper right. Follow the instructions in ‘README.md’ for a step-by-step installation guide.
Familiarize yourself with the concepts behind each component of the lab: 1) how endoribonucleases can be used to perform arithmetic inside of cells and 2) how Lipofectamine 3000 works to transfect plasmids into human cells.
Done.
Overview | Background
In this two-day lab, you will design and build your very own IANN using a library of plasmids from the Ron Weiss lab and human embryonic kidney (HEK) 293 cells. IANNs differ from traditional synthetic genetic circuits because IANNs can perform analog computations, rather than being limited to digital computations. IANNs are also universal function approximators–given an adequate number of intracellular artificial neurons, you can use an IANN to achieve any input/output behavior you’d like.
Acknoweledged.
Overview | Concepts Learned & Skills Gained
This is a lab with a dry and wet component. In the dry lab component, you will design a neuromorphic circuit in groups of 3. Once your design has been finalized, you will write instructions for an OT-2 to build your circuit for you. In the wet lab component, a TA will upload your OT-2 instructions and you will observe the OT-2 building and transfecting your IANN into HEK293 cells.
Above is a snapshot of the google form submitted by Jason Ross. His page can be reviewed for more project details and progress.
Week 8 Lab: Break Week
This was a free week and was not obligatory to mark. This week was used to exploration of course materials and Final Project development.
Week 9 Lab: Cell-Free Systems
Special Note: As a Committed Listener without lab access, documentation within my page is for purely acknowledgement purposes. There were no explicit instructions for Committed Listeners to submit lab work for this week, and with that, I reviewed the material.
Homework, shown on the page of Week 9 is reposted here to ease interpretation and documentation.
General homework questions
Explain the main advantages of cell-free protein synthesis over traditional in vivo methods, specifically in terms of flexibility and control over experimental variables. Name at least two cases where cell-free expression is more beneficial than cell production.
Cell free expression allows teams to conduct biomanufacturing without living cells and operate beyond the constraints of productions in living systems.
Two cases where cell-free expression is more beneficial than cell production are where:
A) biomanufacturing would kill the cells
B) teams have a desire to rapidly prototype biomanufacturing workflows after computational modeling
Describe the main components of a cell-free expression system and explain the role of each component.
The main components of cell free expression are as follows:
Cell lysate/mix: These provide the vital translation components, in addiiton to ribosomes, other enzymbes, and tRNAs
Genomic Template: These are needed to encode and develop the protein of interest
Salts: Ioinic condition maintenance
Buffer: Maintaining pH
Cofactors/additives: Promoting enzymatic activity
Amino Acids: These supply building blocks for translation
Energy System: These supply energy to power transcription and translation
Why is energy provision regeneration critical in cell-free systems? Describe a method you could use to ensure continuous ATP supply in your cell-free experiment.
Energy provision generation is critical to sustain reactions. Supplying molecular energy packs that can regenerate ATP during a reaction can assist this.
Compare prokaryotic versus eukaryotic cell-free expression systems. Choose a protein to produce in each system and explain why.
Eukaryotic and prokaryotic cell-free expression systems have their own unqiue advanges. Prokaryotic cell-free expression tends to be great for rapid prototyping and is robust. Eukaryotic systems can have advantages with complex products by which post-translational modification may be desired.
I’d possibly consider developing flourescent proteins in prokaryotic systems while focusing on antibodies with eukarytic systems.
How would you design a cell-free experiment to optimize the expression of a membrane protein? Discuss the challenges and how you would address them in your setup.
A) My initial guess would be to examine setups by which I can utilize detergents and or membrane disaggrgating components and trial setups.
B) Challenges come from aggregation of membrane proteins, insolubility of components, and reduced yields. I would possibly consider different spatial component arrangements, release modalities, temperature changes, and experiment with amounts of disaggrgating components.
Imagine you observe a low yield of your target protein in a cell-free system. Describe three possible reasons for this and suggest a troubleshooting strategy for each.
Three possible reasons could be a poor template, poor environmental considtions, and or lack of energy.
Troubleshooting respectively would look like: attempting template optimization, environmental optimization, and trialing suppying more energy sources for the reaction. Each of these would be systematically trialed.
Week 10 Lab: Pipetting
Mass Spectrometry
Special Note: As a Committed Listener without lab access, documentation within my page is for purely acknowledgement purposes. There were no explicit instructions for Committed Listeners to submit lab work for this week, and with that, I reviewed the material.
The appendix listed figures to be used for the homework. These were downloaded and posted below for documentation purposes.
Figure 5. LC-MS chromatogram and MS spectrum of eGFP from the intact MS analysis on Xevo G3 QTof.
Figure 6. Intact MS spectrum for eGFP from Xevo G3 QTof. Inset: Zoom-in of the 10+ charge state.
Figure 7. Native eGFP protein mass spectrum from the Waters Xevo G3 QTof MS. The inset is a zoom-in of the charge state at m/z 2800. Use the spacing between the peaks in the zoom-in to determine the charge state (z) and calculate the molecular weight of the protein.
Figure 8. Native eGFP protein mass spectrum from the Waters Xevo G3 QTof MS. The inset is a zoom-in of the charge state at m/z 2800. Use the spacing between the peaks in the zoom-in to determine the charge state (z) and calculate the molecular weight of the protein.
Figure 9. LC-MS Chromatogram of the eGFP tryptic peptide map from the Waters BioAccord system.
Figure 10. Mass Spectrum of a tryptic peptide from eGFP (at 2.78 minutes from Figure 9) to show different charge states of a selected peptide from the Waters BioAccord LC-MS system
Figure 11. Fragmentation of the same eGFP tryptic peptide from Figure 10, showing the data used to reconstruct its amino acid sequence.
Report 1: Peptide Map Report of eGFP from the Waters BioAccord LC-MS System to show the LC-MS chromatogram, Coverage Map of the eGFP amino acid sequence, and predicted tryptic peptides matched to their MS/MS fragmentation patterns.
*Component name: T# refers to the K or R residue in eGFP (refer to homework or see sequence below); ie, T27 = the peptide resulting after trypsin cleaves the bond after the 27th cleavage site (the 27th K or R in the amino acid sequence). The Observed RT (min) refers to the chromatographic peak’s elution time (see Figure 4). The Observed mass, Expected Mass, and the Mass Error provide a good idea of how well the two values agree (we’re looking for +/- 10 ppm or smaller). The Charge column denotes the charge state at which the peak was observed in the mass spectrum; the Peptide column provides the best tryptic peptide sequence from eGFP as a match.
Figure 12. Amino acid coverage of the peptides detected from eGFP standard from the lab protocol.
Sequences shown in blue denote peptides that were detected by their molecular weight in the MS spectrum, alongside confirmatory fragmentation spectra to identify their amino acid sequence.
Peptides not highlighted were not detected or identified, for a number of potential reasons:
The sequence is too small (<5 amino acids) or too large (>20 amino acids) to provide confident identification
There may be too much “noise” (ie, signal from small molecule ion contaminants in the sample or solvents) to provide a strong signal from the MS data to detect and identify the peptide confidently.
There may be a modification in the amino acid sequence provided by the eGFP protein manufacturer that does not align with the amino acid sequence that was provided in the documentation.
Figure 13. Mass spectrum of Keyhole Limpet Hemocyanin (KLH) acquired on the CDMS.
Week 11 Lab: Introduction to Cloud Laboratories
Special Note: This assignment was a combined homework and laboratory assignment that was reflected in both the homeowork and lab. What follows is a repost of what was asked from the laboratory page, as answered from the homework page post.
Part A: The 1,536 Pixel Artwork Canvas | Collective Artwork
Contribute at least one pixel to this global artwork experiment before the editing ends on Sunday 4/19 at 11:59 PM EST!
A personalized URL was sent to the email address associated with your Discourse account, and you can discuss the artwork on the Discourse!
If you did not have a chance to contribute, it’s okay, just make sure you become a TA this fall! 😉
Multiple pixels were contrited to Artwork Canvases. Below the CFPS and Overall Artwork Contributions are given.
CFPS Contributions
Overall Artwork Contributions
Done: Added pixels. See above.
Make a note on your HTGAA webpages including:
Done: I added a total of 14 pixels by the end of the experiment: 12 colored pixels and 2 removal pixels.
what you contributed to the community bioart project (e.g., “I made part of the DNA on the bottom right plate”)
Done: I added a couple small details to subfeatures of some designs. The general idea was to help complete objects in progress or add a small sub-feature, visually. One example was adding a temporary light to one of the spaceships.
Contributions to a follow-up activity at Synbiobeta were done as well as shown below.
what you liked about the project, and
Done: I liked the collaborative and semi-collaborative nature of the project. There was space for those who wanted to add with coordination and space for those who wanted to add individually from their own vision. Seeing what people came up with was great, as well.
what about this collaborative art experiment could be made better for next year.
Done: To improve it, one could widen the color section, plate area, and reduce the painting refresh-time.
Part B: Cell-Free Protein Synthesis | Cell-Free Reagents
Referencing the cell-free protein synthesis reaction composition (the middle box outlined in yellow on the image above, also listed below), provide a 1-2 sentence description of what each component’s role is in the cell-free reaction.
E. coli Lysate:
BL21 (DE3) Star Lysate (includes T7 RNA Polymerase) - This contains the core metabolic components and enyzmes for transcription and translation. The T7 RNA polymerase is needed for eventual gene transcription and mRNA towards eventual protein production.
Salts/Buffer:
Potassium Glutamate - This is a source of potassium and anions for the reaction, providing needed blance for ribosome function.
HEPES-KOH pH 7.5 - This buffer helps to maintain a stable pH, again helpful for ribosome stability.
Magnesium Glutamate - This provides Mg2+m which is helpful for enzymatic activity
Potassium phosphate monobasic - This works with Potassium dibasic as a secondary buffering system for pH stabilization.
Potassium phosphate dibasic - This works with Potassium monobasic as a secondary buffering system for pH stabilization.
Energy / Nucleotide System:
Ribose - This serves along with gluclose as energy and carbon sources to power the reactions.
Glucose - This serves along with ribose as energy and carbon sources to power the reactions.
AMP - This servves as a ATP synthesis precursor. This and the following 3 are required for RNA synthesis.
CMP - This servves as a CTP precursor.
GMP - This servves as a GTP precursor.
UMP - This servves as a UTP precursor.
Guanine - This serves a a purine base and precursor to produce guanine nucleotides.
Translation Mix (Amino Acids):
17 Amino Acid Mix - This mix supplies the amnio acids used for protein synthesis.
Tyrosine - This is key for phosphorylation but specifically vital as a substrate in forming the target protein.
Cysteine - This is separated due to its instability and is required in protein synthesis.
Additives:
Nicotinamide - This functions as the NAD+ biosynthesis precursor.
Backfill:
Nuclease Free Water - This serves as the solvent for the reaction.
Describe the main differences between the 1-hour optimized PEP-NTP master mix and the 20-hour NMP-Ribose-Glucose master mix shown in the Google Slide above. (2-3 sentences)
The main differences betweem the two master mixes can be found in energy regeration, time-spent for reactions, recycling of components, qualities of metabolism engaged, and composition. Both are effective, but they serve different purposes, be it short-term production vs longer production over time. The use comes down to the context of synthesis desired.
Bonus question: How can transcription occur if GMP is not included but Guanine is?
If reaction pathways exist to convert guanine to a product that can facilitate GMP’s role, transcription can reasonably occur.
Part C: Planning the Global Experiment | Cell-Free Master Mix Design
Given the 6 fluorescent proteins we used for our collaborative painting, identify and explain at least one biophysical or functional property of each protein that affects expression or readout in cell-free systems. (Hint: options include maturation time, acid sensitivity, folding, oxygen dependence, etc) (1-2 sentences each)
Create a hypothesis for how adjusting one or more reagents in the cell-free mastermix could improve a specific biophysical or functional property you identified above, in order to maximize fluorescence over a 36-hour incubation. Clearly state the protein, the reagent(s), and the expected effect.
Increasing HEPES-KOH (through improved maintenance of a near neutral pH) may improve the fluorescence from mKO2 over a 36-hour incubation period by stabilizing its reaction to pH and reducing pH dependent fluorescence loss.
The second phase of this lab will be to define the precise reagent concentrations for your cell-free experiment. You will be assigned artwork wells with specific fluorescent proteins and receive an email with instructions this week (by April 24). You can begin composing master mix compositions here.
Done. 3 wells were utilized as indicated by the image below.
The codes for each are found below:
The final phase of this lab will be analyzing the fluorescence data we collect to determine whether we can draw any conclusions about favorable reagent compositions for our fluorescent proteins. This will be due a week after the data is returned (date TBD!). The reaction composition for each well will be as follows:
6 μL of Lysate
10 μL of 2X Optimized Master Mix from above
2 μL of assigned fluorescent protein DNA template
2 μL of your custom reagent supplements
Total: 20 μL reaction
Acknowledged. Reactions were made and compositions were honored.
Part D: Build-A-Cloud-Lab | (optional) Bonus Assignment
Use this simulation tool to create an interesting looking cloud lab out of the Ginkgo Reconfigurable Automation Carts. This is just a minimal implementation so far, but I would love to see some fun designs!
Using the simulation tool, I made a setup that mixed RACs. What follows is a RAC separated by a 1M, folloed by a mix of RACs separated by.25M, followed by another mix of RACs separated by another 1M leading to a final RAC. These are all in one line as shown below.
Week 12 Lab: Bioproduction of Beta-Carotene and Lycopene
Special Note: This lab had both a section mandatory for all students and one especially for committed listeners.
Post Lab Questions | Mandatory for All Students
(References supplied from lab:
A) Du W, Song Y, Liu M, Yang H, Zhang Y, Fan Y, Luo X, Li Z, Wang N, He H, Zhou H, Ma W, Zhang T. Gene expression pattern analysis of a recombinant Escherichia coli strain possessing high growth and lycopene production capability when using fructose as carbon source. Biotechnol Lett. 2016 Sep;38(9):1571-7. doi: 10.1007/s10529-016-2133-0. Epub 2016 Jul 5. PMID: 27379652.
B) Aristidou, A.A., San, K.-Y. and Bennett, G.N. (1999), Improvement of Biomass Yield and Recombinant Gene Expression in Escherichia coli by Using Fructose as the Primary Carbon Source. Biotechnol Progress, 15: 140-145. https://doi.org/10.1021/bp980115v
Additional used for below:
C) Shumskaya, M., & Wurtzel, E. T. (2013). The carotenoid biosynthetic pathway: thinking in all dimensions. Plant science : an international journal of experimental plant biology, 208, 58–63. https://doi.org/10.1016/j.plantsci.2013.03.012
Which genes when transferred into E. coli will induce the production of lycopene and beta-carotene, respectively?
The genes that when transferred into E.Coli will induce the production of lycopene and beta-carotene are crtB, crtE, crtl, and crtY.
Why do the plasmids that are transferred into the E. coli need to contain an antibiotic resistance gene?
The antibiotic resistance genes allow for selection pressue among E.Coli who have the plasmid vs those who do not.
What outcomes might we expect to see when we vary the media, presence of fructose, and temperature conditions of the overnight cultures?
I’d imagine that we would see changes in bacterial growth and protein expressions.
Generally describe what “OD600” measures and how it can be interpreted in this experiment.
OD600 works to measure cell density via turbidity. You can use this to estimate population growth and overall production of biomass.
What are other experimental setups where we may be able to use acetone to separate cellular matter from a compound we intend to measure?
Applications towards pigment isolation, lipid extraction, and generally any that lean on solvent mixture optimizations may suffice.
Why might we want to engineer E. coli to produce lycopene and beta-carotene pigments when Erwinia herbicola naturally produces them?
We can scale up production and farm E.coli in far more diverse environments. Further, we can edit E.Coli more easily for this process.
Post Lab Questions | For Committed Listeners Only
Let’s get in touch with our metabolic pathway
What are the enzymes of the carotene pathway?
The enzymes of the carotene pathway are Phytoene synthase, GGPP synthase, Phytoene desaturase, and Lycopene ε-cyclase.
Within this pathway, which is the rate determining step (the step that takes the longest)? Which enzyme is responsible for this step?
phytoene synthase (PSY) synthesis
Zhou, X., Rao, S., Wrightstone, E., Sun, T., Lui, A. C. W., Welsch, R., & Li, L. (2022). Phytoene Synthase: The Key Rate-Limiting Enzyme of Carotenoid Biosynthesis in Plants. Frontiers in plant science, 13, 884720. https://doi.org/10.3389/fpls.2022.884720
Notes for design of a DNA construct for bioproduction
The first thing to do is to decide what organism you are going to use for this (E. coli or S. cerevisiae) for production. Which would you choose and why (emphases on production differences)?
I lean on E. Coli for the purpose of rapid prototyping and famailiairity (including easier genetic engineering)
Now choose one of the enzymes and lets outline the parts of the construct for expression
Enzyme chosen: Phytoene synthase
For E. coli lets create a expression vector that works as a plasmid you choose E. coli let’s create a expression vector that works as a plasmids
Now, for making a functional construct there are a variety of biological parts needed for this, like ribosome binding sites, terminators, operators and promoters. The last ones are the most important in terms of enzyme or protein production. Let’s elaborate further on this biopart.
Promoters function to recruit RNA polymerase to kickstart transicription.
What types of promoters do we have?
Eukaryotic, Prokaryotic
If we wanted to turn off the transcription of a gene in response to a metabolite, what type of promoter would be most useful? What if we wanted this to increase in the presence of the metabolite?
A) For turning off transcroption, we would need a promoter that can be repressed (Repressible)
B) To increase transcription, we need an inducible promoter.
Now choose one of the genes of the metabolic pathway previously described (Carotene/lycopene )and choose one enzyme to make an expression construct. What promoter could you use for this? Why did you choose it?
Ptac might be the best for the ability to induce production tightly.
What is the origin of replication?
The origin of replication refers the a DNA sequence that determines plasmid replicability within a host.
What types of origin of replication do we have?
We have many types such as ColE1, pMB1, pSC101, R6K, 15A, and more. Some have relaxed and stringent controls of replication. There are numerous incompatibility groups and varied copy numbers among them. Host compatibility/incompatibility and copy number are common means to categorize them.
(Extra) What are compatibility groups?
Compatibility groups are categories of plasmids that can coexist within the same bacterial cells. Competition for replication material components within bacteria are a source of incompatibility.
Now for the previously chosen promoter and gene what will be the best origin or replication?
15A might be a reasonable candidate.
(Mandatory for Global listeners, Optional MIT/Harvard) Elaborate further on other bioparts like RBS, terminators, operators you would use for a correct design and further bioproduction?
For these, I would hunt parts found on IGEM given prior reserch, standardization, and testing. I would like an inducible promoter. For the RBS, terminators, and operators, I might choose the following noting initial steps for protoyping:
For the RBS: BBa_B0034
Terminator: BBa_B0015 (Double terminator)
OPerator: BBa_R0010 (lac regulated and inducible)
These would be emphasized for the prior reasons plus tigter regulation.
(Hot! Extra points) What are aptamers and riboswitches and how can they be used for metabolic tuning or engineering in prokaryotes?
A) Aptamers refer to short genomic sequences that bind to specific molecules.
B) Riboswitches are regulatory genomic elements that have a sensing region that acts like an aptamer and an expression-control region that can be used to modify either transcription or translation in the presence of a metabolite.
C) These can be used to tune protein expression in protkaryotic systems.
(Extra points) Now what approach can be used to join all these parts together? Make a quick analysis of their sequence in search of possibilities (search for restriction sites, etc)
Gibson and Golden Gate could be used. For a quick analysis, the availibility of compatible restriction sites may lean one towards Gibson whereas a desire to use multiple RNA regulatory parts could lean someone moreso towards Golden Gate Assembly.
(Extra Hot!!! Extra Points) Try to elaborate further on a biosynthetic pathway you would want to engineer in E. coli for production of a metabolite or product. What use could this bio-product have? Imagine dream applications!!!
In a very high-level way, a biosynethic pathway that is responsive to changing macro-host conditions could be neat to engineer. This bio-product could have medications.
(Extra points) For S. cerevisiae create an integration cassette for homologous recombination.
I’m not sure of the level that is desired here. At a high level: a cassette can include a left homology armm, promoter, Kozak sequence, our gene, terminator, a marker like KanMX (dominantn antibiotic selection), and a right homology arm.
First let’s check some concepts of yeast engineering and homologous recombination this in this notes
As well as for prokaryotes, eukaryotic DNA designs need bioparts used for construction of a function design and further expresion. Now search for a biosynthetic pathway if interested and describe one of the genes of the pathway.
I’m happy to use Carotoid biosynthesis via S. cerevisiae and use crtYB which encodes for phytoene synthase.
“Phytoene synthase (PSY) catalyzes the first committed step in the carotenoid biosynthesis pathway and is a major rate-limiting enzyme of carotenogenesis”
See: Ledetzky N, Osawa A, Iki K, Pollmann H, Gassel S, Breitenbach J, Shindo K, Sandmann G. Multiple transformation with the crtYB gene of the limiting enzyme increased carotenoid synthesis and generated novel derivatives in Xanthophyllomyces dendrorhous. Arch Biochem Biophys. 2014 Mar 1;545:141-7. doi: 10.1016/j.abb.2014.01.014. Epub 2014 Jan 30. PMID: 24486200.
Phytoene synthase has been described above.
Now, remember that for making a functional construct there are a variety of biological parts needed for this, like ribosome binding sites or Kozak sequences, terminators, and promoters. List the ones you could use for DNA design.
Again, using IGEM parts, some of my hypothethical parts might be:
In yeast engineering we use DNA construction designs for making genome integration. What chromosome site could you use for integration of these and why?
I’d consider the HO locus given research pointing at it being a relatively safe site.
See: Voth WP, Richards JD, Shaw JM, Stillman DJ. Yeast vectors for integration at the HO locus. Nucleic Acids Res. 2001 Jun 15;29(12):E59-9. doi: 10.1093/nar/29.12.e59. PMID: 11410682; PMCID: PMC55758.
Voth, W. P., Richards, J. D., Shaw, J. M., & Stillman, D. J. (2001). Yeast vectors for integration at the HO locus. Nucleic acids research, 29(12), E59–e59. https://doi.org/10.1093/nar/29.12.e59
(Hot! Extra points) Following the next chart of how a DNA integration cassette should be designed and with the previously chosen parts elaborate the DNA sequence you could use to synthesize with Twist.
At a high-level the following parts would be helpful to design: Homology arms (flanking the casette), Promoter, Kozak Sequence, our gene of interest (crtYB), terminator, and our selectible marker, to be eventually inserted into the HO locus. I’d be minfdul that the sequence does not contain incompatible restriction sites and minimize undesirable repeats and other sub-optimal sequence features.
Project Title: Project Z-Freeze, HTGAA 2026: Individual Final Project Documentation Section 1: Abstract Project Z-Freeze explores whether engineered variants of the Ice Nucleation Protein (InaZ) can be computationally designed and optimized to improve controlled freezing behavior for potential use in biohybrid thermal management systems. Data centers require large amounts of energy for cooling, and many existing cooling systems rely on resource-intensive or environmentally challenging approaches. Biological phase-change systems may provide an alternative strategy for improving thermal energy management. This project focuses on InaZ, an ice nucleation protein naturally produced by Pseudomonas syringae, which promotes freezing at relatively high sub-zero temperatures by organizing water molecules into ice-forming structures.
Project Title: Project Z-Freeze, HTGAA 2026: Individual Final Project Documentation
Section 1: Abstract
Project Z-Freeze explores whether engineered variants of the Ice Nucleation Protein (InaZ) can be computationally designed and optimized to improve controlled freezing behavior for potential use in biohybrid thermal management systems. Data centers require large amounts of energy for cooling, and many existing cooling systems rely on resource-intensive or environmentally challenging approaches. Biological phase-change systems may provide an alternative strategy for improving thermal energy management. This project focuses on InaZ, an ice nucleation protein naturally produced by Pseudomonas syringae, which promotes freezing at relatively high sub-zero temperatures by organizing water molecules into ice-forming structures.
The broad objective of this project is to establish a computationally guided synthetic biology workflow for designing and ranking engineered InaZ variants capable of initiating freezing at warmer temperatures than wild-type InaZ. The hypothesis is that engineered mutations to InaZ may alter nucleation efficiency, structural feasibility, and expression compatibility in ways that shift freezing onset temperatures upward relative to a control InaZ construct.
To test this hypothesis, InaZ variant constructs and controls will be computationally designed and evaluated in silico prior to experimental validation in Escherichia coli K-12 using modular synthetic biology methods. Constructs will include a promoter, ribosome binding site, mutated InaZ coding sequence, terminator, and plasmid backbone. Candidate constructs will be assembled conceptually using Golden Gate assembly within a pSB1C3 high-copy plasmid backbone and verified using PCR, gel electrophoresis, and DNA sequencing.
Functional testing will be performed using droplet-based freezing assays in which samples are gradually cooled under controlled conditions while thermal and optical imaging monitor freezing events in real time. The primary measurement collected will be freezing onset temperature. Comparisons between negative controls, wild-type InaZ, and engineered InaZ variants will be used to evaluate whether the mutations improve ice nucleation efficiency.
SECTION 2: PROJECT AIMS
Aim 1: Experimental Aim
The first aim of my final project is to design and validate a DNA construct encoding an InaZ-derived ice nucleation protein optimized for heterologous expression and functional feasibility in E. coli by utilizing codon optimization tools, modular plasmid design, computational sequence analysis, Golden Gate assembly, PCR verification, and controlled freezing assays.
Aim 2: Development Aim
The second aim is to develop a computational framework for evaluating InaZ variants based on predicted expression efficiency, sequence stability, and structural feasibility, enabling informed selection of constructs for experimental implementation.
Aim 3: Visionary Aim
The third aim is to extend this framework into a generalized multi-parameter optimization platform capable of designing and ranking biological systems for controlled phase-transition applications and related engineering contexts.
SECTION 3: BACKGROUND
Background and Literature Context
Current Knowledge and Research Context
Ice nucleation proteins produced by Pseudomonas syringae are among the most efficient known biological nucleators of ice formation. These proteins promote freezing by organizing interfacial water molecules into ordered structures that facilitate phase transition at relatively high sub-zero temperatures (Roeters et al., 2021). Early characterization of bacterial ice nucleation proteins demonstrated that InaZ proteins possess strong nucleation activity and can significantly alter freezing behavior in aqueous systems (Wolber et al., 1986).
Research into InaZ proteins has also demonstrated that these systems can be functionally expressed in engineered bacterial platforms, supporting their compatibility with synthetic biology workflows (Kassmannhuber et al., 2017). Additional studies have explored the use of ice nucleation genes as engineered biological tools in alternative bacterial systems, further supporting the feasibility of heterologous deployment strategies (Arvanitis et al., 1995). These findings suggest that engineered InaZ systems may be adaptable for synthetic biological applications beyond their native environmental role.
At the same time, increasing computational demand has intensified interest in alternative thermal management technologies for data centers and advanced computing infrastructure. Existing cooling systems often consume substantial amounts of energy and water, motivating interest in phase-change cooling systems and bio-inspired thermal management strategies. Recent reviews of bio-inspired phase-change materials and thermal energy systems suggest that biologically informed approaches may contribute to future thermal engineering solutions (An et al., 2023; Pielichowska et al., 2025).
This project addresses the gap between naturally occurring ice nucleation proteins and engineered synthetic biology systems capable of computational optimization and functional ranking for programmable phase-transition applications. Rather than studying InaZ only as a natural biological phenomenon, Project Z-Freeze explores whether computational design, sequence optimization, and structural evaluation may be combined into a predictive framework for engineering improved nucleation behavior.
Peer-Reviewed Research Citation Summary 1
Roeters et al. (2021) investigated how ice-nucleating proteins influence the structure of interfacial water at low temperatures. Their work demonstrated that ice nucleation proteins become activated under colder conditions and organize surrounding water molecules into ordered arrangements that promote freezing. This study provided mechanistic insight into how biological nucleators initiate phase transitions more efficiently than abiotic controls. These findings support the hypothesis that engineered modifications to InaZ may alter nucleation efficiency and freezing onset behavior.
Peer-Reviewed Research Citation Summary 2
Kassmannhuber et al. (2017) demonstrated successful functional display of the InaZ ice nucleation protein on engineered bacterial ghost systems. Their work showed that InaZ proteins can remain functionally active in heterologous biological platforms and may be incorporated into engineered systems outside their native bacterial context. This study also supports the feasibility of modular synthetic biology approaches for expressing and testing engineered InaZ constructs. The research provides a relevant foundation for the construct design and expression framework proposed in Project Z-Freeze.
Novelty and Innovation
This project is novel because it combines computational protein design, synthetic biology construct engineering, and functional freezing assays to investigate whether InaZ proteins can be rationally modified to improve ice nucleation efficiency. While naturally occurring InaZ proteins have been extensively studied in environmental and biological systems, fewer studies have explored computationally guided optimization workflows for engineering InaZ variants within modular synthetic biology frameworks. Project Z-Freeze also extends beyond simple expression studies by proposing a predictive ranking system capable of evaluating variant feasibility prior to experimental implementation.
The project further expands the boundaries of synthetic biology by applying biological phase-transition systems to problems traditionally addressed through mechanical or industrial engineering solutions. Rather than viewing biological ice nucleation solely as an atmospheric or agricultural phenomenon, this project explores whether engineered nucleation systems may contribute to programmable thermal management technologies.
Why the Project Matters
Thermal management remains a major challenge for data centers and other computational infrastructure systems. Existing cooling technologies can require substantial energy consumption and water usage, creating economic and environmental concerns. Improving the efficiency of phase-change cooling systems may contribute to more sustainable approaches for controlling thermal energy transfer in high-performance computing environments.
Project Z-Freeze matters because it explores whether synthetic biology can contribute to programmable thermal management systems through engineered biological phase-transition control. If successful, engineered InaZ variants may help expand the temperature range at which controlled freezing can occur, potentially improving the flexibility of future biohybrid cooling systems. Even if the engineered variants do not outperform wild-type InaZ, the project may still contribute useful information regarding the relationship between protein structure, expression feasibility, and ice nucleation behavior.
The project may also contribute more broadly to synthetic biology by demonstrating how computational sequence optimization, modular construct engineering, and functional phenotypic assays can be integrated into a unified experimental workflow. Additionally, the predictive framework proposed in this project may eventually support the design and ranking of other biological systems intended for engineered phase-transition applications.
Ethical Implications
One ethical consideration associated with this project involves the use of genetically engineered organisms and synthetic biological systems. Although the proposed work utilizes a non-pathogenic E. coli K-12 laboratory chassis, responsible handling of engineered biological materials remains important. Ethical principles such as non-maleficence and responsibility are especially relevant because unintended environmental release or misuse of engineered organisms could create ecological or biosafety concerns (Hodgeson et al, 2025). The project must therefore prioritize proper laboratory containment, safe handling procedures, and responsible communication of results.
Another ethical consideration involves the broader implications of applying engineered biological systems to industrial infrastructure and thermal management technologies. Although biologically inspired cooling systems may provide sustainability benefits, uncertainties remain regarding scalability, ecological impact, and long-term implementation. Measures to maintain ethical responsibility include following biosafety guidelines, limiting experimental work to controlled laboratory environments, and clearly communicating uncertainties and limitations in both experimental findings and predictive computational models. Alternative approaches, including non-biological thermal technologies, should also be considered when evaluating the broader implications of the project.
References for Section 3
An, S., Shi, B., Jiang, M., Fu, B., Song, C., Tao, P., Shang, W. and Deng, T. (2023) ‘Bio-inspired phase-change and thermal management systems’, Chemical Reviews, 123(11), pp. 7081–7118. Available at: https://doi.org/10.1021/acs.chemrev.3c00136.
Arvanitis, N., Vargas, C., Tegos, G., Perysinakis, A., Nieto, J.J., Ventosa, A. and Drainas, C. (1995) ‘Development of a gene reporter system in moderately halophilic bacteria by employing the ice nucleation gene of Pseudomonas syringae’, Applied and Environmental Microbiology, 61(11), pp. 3821–3825. Available at: https://doi.org/10.1128/aem.61.11.3821-3825.1995.
Kassmannhuber, J., Rauscher, M., Schöner, L., Witte, A. and Lubitz, W. (2017) ‘Functional display of ice nucleation protein InaZ on the surface of bacterial ghosts’, Bioengineered, 8(5), pp. 488–500. Available at: https://doi.org/10.1080/21655979.2017.1284712.
Pielichowska, K., Szatkowska, M. and Pielichowski, K. (2025) ‘Thermal energy storage in bio-inspired PCM-based systems’, Energies, 18(13), pp. 1–28.
Roeters, S.J., Golbek, T.W., Bregnhøj, M. et al. (2021) ‘Ice-nucleating proteins are activated by low temperatures to control the structure of interfacial water’, Nature Communications, 12, p. 1183. Available at: https://doi.org/10.1038/s41467-021-21349-3.
Valeriani, C. (2022) ‘Deep learning for unravelling features of heterogeneous ice nucleation’, Proceedings of the National Academy of Sciences of the United States of America, 119(35), e2211295119. Available at: https://doi.org/10.1073/pnas.2211295119.
Wolber, P.K., Deininger, C.A., Southworth, M.W., Vandekerckhove, J., van Montagu, M. and Warren, G.J. (1986) ‘Identification and purification of a bacterial ice-nucleation protein’, Proceedings of the National Academy of Sciences of the United States of America, 83(19), pp. 7256–7260. Available at: https://doi.org/10.1073/pnas.83.19.7256.
SECTION 4: EXPERIMENTAL DESIGN, TECHNIQUES, TOOLS, AND TECHNOLOGY
Review literature on InaZ structure, ice nucleation mechanisms, protein engineering, and synthetic biology expression systems.
Identify candidate InaZ regions for potential mutation, codon optimization, or structural engineering.
Design wild-type and mutated InaZ coding sequences using computational sequence analysis and predictive evaluation methods.
Select modular construct components including promoter, RBS, terminator, and plasmid backbone.
Use pSB1C3 as the high-copy plasmid backbone.
Design expression constructs using Benchling or similar DNA design software.
Simulate construct organization, compatibility, and predicted expression feasibility using in silico tools.
Conceptually assemble constructs using Golden Gate assembly.
Transform constructs into E. coli K-12 or related laboratory strains.
Screen colonies using PCR and gel electrophoresis.
Verify construct identity and mutation accuracy through DNA sequencing.
Culture engineered strains under controlled laboratory conditions.
Prepare samples containing engineered InaZ constructs and controls.
Perform droplet-based freezing assays under gradual cooling conditions.
Use thermal and optical imaging to monitor freezing events.
Record freezing onset temperatures for individual droplets.
Compare freezing behavior between controls and mutated InaZ variants.
Analyze whether engineered variants shift freezing onset temperatures upward relative to wild-type InaZ and compare experimental results against computational predictions.
Expected Results
The negative control is expected to freeze at the lowest temperatures. Wild-type InaZ constructs are expected to demonstrate higher ice nucleation activity than negative controls. Successful engineered variants may demonstrate freezing onset temperatures higher than wild-type InaZ.
Relevant Techniques
Checked Techniques
Pipetting
Lab Safety
Bioethical Considerations
DNA Sequencing
DNA Construct Design
Gel Electrophoresis
Databases (GenBank, NCBI)
Use of Benchling
Chassis Selection
Registry of Standard Biological Parts
Plasmid Preparation
Bacterial Culturing
Bacterial Processing
Primer Design
PCR Reactions
Golden Gate or Gibson Assembly
Protein Design
Expanded Technique 1: DNA Construct Design
DNA construct design will be used to generate modular InaZ expression systems containing a promoter, ribosome binding site, mutated InaZ coding sequence, and terminator within a pSB1C3 plasmid backbone. Benchling and Registry of Standard Biological Parts resources will be used to organize and simulate constructs prior to assembly. Construct design will also include consideration of promoter strength, translation efficiency, and plasmid compatibility. Comparative construct design will allow systematic testing of different InaZ variants while keeping other expression components constant.
Expanded Technique 2: PCR and Gel Electrophoresis
PCR and gel electrophoresis will be used to verify the presence and approximate size of engineered InaZ constructs following assembly. PCR primers will be designed to amplify regions containing the mutated InaZ sequence. Amplified DNA fragments will be visualized through agarose gel electrophoresis to confirm expected fragment sizes. These methods will provide an initial quality-control step prior to DNA sequencing and functional freezing assays.
Industry Council Companies Associated with Project
The aspect of the project selected for validation is the design and verification of mutated InaZ DNA constructs intended for comparative freezing assays. This validation focuses on confirming that engineered constructs contain the expected promoter, RBS, InaZ variant, and terminator arrangement prior to functional testing. As this project was dry and without lab access, Digital constructs have been designed but not fully tested. The validation protocol below reflects an ideal case provided lab access is made possible.
Validation Protocol
Design mutated InaZ construct sequences using Asimov (Kernel).
Select promoter, RBS, terminator, and pSB1C3 plasmid backbone.
Design primers for PCR amplification of the construct region.
Simulate Golden Gate assembly compatibility.
Assemble constructs conceptually or experimentally.
Transform constructs into E. coli.
Select colonies using antibiotic resistance.
Perform colony PCR.
Run PCR products on agarose gels.
Compare observed DNA fragment sizes to expected construct sizes.
Submit successful constructs for DNA sequencing.
Confirm mutation presence and sequence accuracy.
Synthetic Biology Techniques Used
Hypothetical validation incorporates DNA construct design, plasmid assembly, PCR, gel electrophoresis, and DNA sequencing. DNA construct design allows modular organization of promoter, RBS, InaZ coding sequence, and terminator elements. PCR and gel electrophoresis provide rapid screening methods to evaluate whether constructs contain inserts of the expected size. DNA sequencing would be used to confirm that the mutated InaZ sequence was assembled correctly and does not contain unintended mutations.
Data and Analysis
Data generated during validation may include agarose gel images, sequencing alignments, construct maps, and simulated freezing assay data. Quantitative analysis may involve comparing freezing onset temperatures across negative controls, wild-type InaZ constructs, and engineered variants.
Challenges and Limitations
One potential challenge is that InaZ is a large and structurally complex membrane-associated protein that may not express efficiently in E. coli. High-copy plasmid expression could also increase cellular stress or affect protein folding. Another limitation is that freezing behavior may depend on protein localization and aggregation state rather than sequence changes alone. Alternative strategies could include modifying promoter strength, testing different chassis strains, or evaluating cell-free expression systems.
SECTION 6: ADDITIONAL INFORMATION
References
Alsante, A.N., Thornton, D.C.O. and Brooks, S.D. (2024) ‘[Article title not provided]’, Environmental Science & Technology, 58(10), pp. 4594–4605. Available at: https://doi.org/10.1021/acs.est.3c06835.
Arvanitis, N., Vargas, C., Tegos, G., Perysinakis, A., Nieto, J.J., Ventosa, A. and Drainas, C. (1995) ‘Development of a gene reporter system in moderately halophilic bacteria by employing the ice nucleation gene of Pseudomonas syringae’, Applied and Environmental Microbiology, 61(11), pp. 3821–3825. Available at: https://doi.org/10.1128/aem.61.11.3821-3825.1995.
Deininger, C.A., Mueller, G.M. and Wolber, P.K. (1988) ‘Immunological characterization of ice nucleation proteins from Pseudomonas syringae, Pseudomonas fluorescens, and Erwinia herbicola’, Journal of Bacteriology, 170(2), pp. 669–675. Available at: https://doi.org/10.1128/jb.170.2.669-675.1988.
Hodgson, A., Frow, E., Palmer, X.-L., Rohwer, F., Elcock, L., Johnson, A., Zimmerman, E.H. and Voigt, C. (2025) ‘1.3 Public infrastructure for analyzing and assessing beyond biocontainment biotechnologies’, Rice University. Available at: https://doi.org/10.25611/Z7YJ-EA10
Kassmannhuber, J., Rauscher, M., Schöner, L., Witte, A. and Lubitz, W. (2017) ‘Functional display of ice nucleation protein InaZ on the surface of bacterial ghosts’, Bioengineered, 8(5), pp. 488–500. Available at: https://doi.org/10.1080/21655979.2017.1284712.
Melillo, J.H., Nikulina, E., Iriarte-Alonso, M.A., Cerveny, S. and Bittner, A.M. (2022) ‘Electron microscopy and calorimetry of proteins in supercooled water’, Scientific Reports, 12(1), p. 16512. Available at: https://doi.org/10.1038/s41598-022-20430-1.
National Center for Biotechnology Information (NCBI) (n.d.) Protein database entry: CBX54760.1 hypothetical protein. Available at: https://www.ncbi.nlm.nih.gov/protein/CBX54760.1 (Accessed: 7 April 2026).
Pielichowska, K., Szatkowska, M. and Pielichowski, K. (2025) ‘Thermal energy storage in bio-inspired PCM-based systems’, Energies, 18(13), pp. 1–28.
Roeters, S.J., Golbek, T.W., Bregnhøj, M. et al. (2021) ‘Ice-nucleating proteins are activated by low temperatures to control the structure of interfacial water’, Nature Communications, 12, p. 1183. Available at: https://doi.org/10.1038/s41467-021-21349-3.
Shun, A., Shi, B., Jiang, M., Fu, B., Song, C., Tao, P., Shang, W. and Deng, T. (2023) ‘[Article title not provided]’, Chemical Reviews, 123(11), pp. 7081–7118. Available at: https://doi.org/10.1021/acs.chemrev.3c00136.
Valeriani, C. (2022) ‘Deep learning for unravelling features of heterogeneous ice nucleation’, Proceedings of the National Academy of Sciences of the United States of America, 119(35), e2211295119. Available at: https://doi.org/10.1073/pnas.2211295119.
Wolber, P.K., Deininger, C.A., Southworth, M.W., Vandekerckhove, J., van Montagu, M. and Warren, G.J. (1986) ‘Identification and purification of a bacterial ice-nucleation protein’, Proceedings of the National Academy of Sciences of the United States of America, 83(19), pp. 7256–7260. Available at: https://doi.org/10.1073/pnas.83.19.7256
pSB1C3 plasmid backbone
Restriction enzymes / Golden Gate assembly reagents
PCR reagents and primers
Agarose gel electrophoresis materials
DNA sequencing services
E. coli K-12 competent cells
Growth media and antibiotics
Pipettes and consumables
Thermal imaging system
Optical imaging camera
Controlled cooling platform
Benchling or equivalent software
Estimated project budget: variable depending on sequencing and imaging equipment availability
Special Note: Organization of the above was facilited through a combination of pre-written material and organization through Claude, Gemini, and ChatGPT.
Below is the draft version, for posterity:
Final Project Name: Project Z Freeze
Final Project Description: Project Z Freeze aims to address limitations in current data center and general computing phase-change cooling strategies by developing an alternative system based on engineered variants of the ice-nucleating protein InaZ, designed to reliably initiate freezing at higher subzero temperatures. Through computational design, sequence optimization, and structural evaluation, this project establishes a predictive framework to rank variants for functional feasibility and heterologous expression. Ultimately, this work aims to enable programmable control of phase transitions in high-performance thermal management systems.
Aim 1: The first aim of my final project is to design and validate a DNA construct encoding an InaZ-derived ice nucleation protein optimized for expression in E. coli by utilizing codon optimization tools, modular plasmid design, and computational analysis of protein size and stability.
Aim 2: The second aim is to develop a computational evaluation framework to compare InaZ variants based on predicted expression efficiency, sequence stability, and structural feasibility, enabling informed selection of constructs for experimental implementation.
Aim 3: The third aim is to extend this framework into a generalized multi-parameter optimization platform capable of designing and ranking biological systems for controlled phase-transition applications and related engineering contexts.
Potential Companies: Asimov (Kernel), Gingko Bioworks, and Twist Biosciences
Below is an alternative presentation design that was offered via Gemini via Google Slides through enhance feature. It look neat, but I ultimately did not adopt it.
Here’s the prompt: “Redesign this slide to most effectively communicate the content. Prefer well structured visual layouts.”