Week 2 HW: DNA Read, Write, & Edit
Part 0: Basics of Gel Electrophoresis
I reviewed the recorded class recitation.
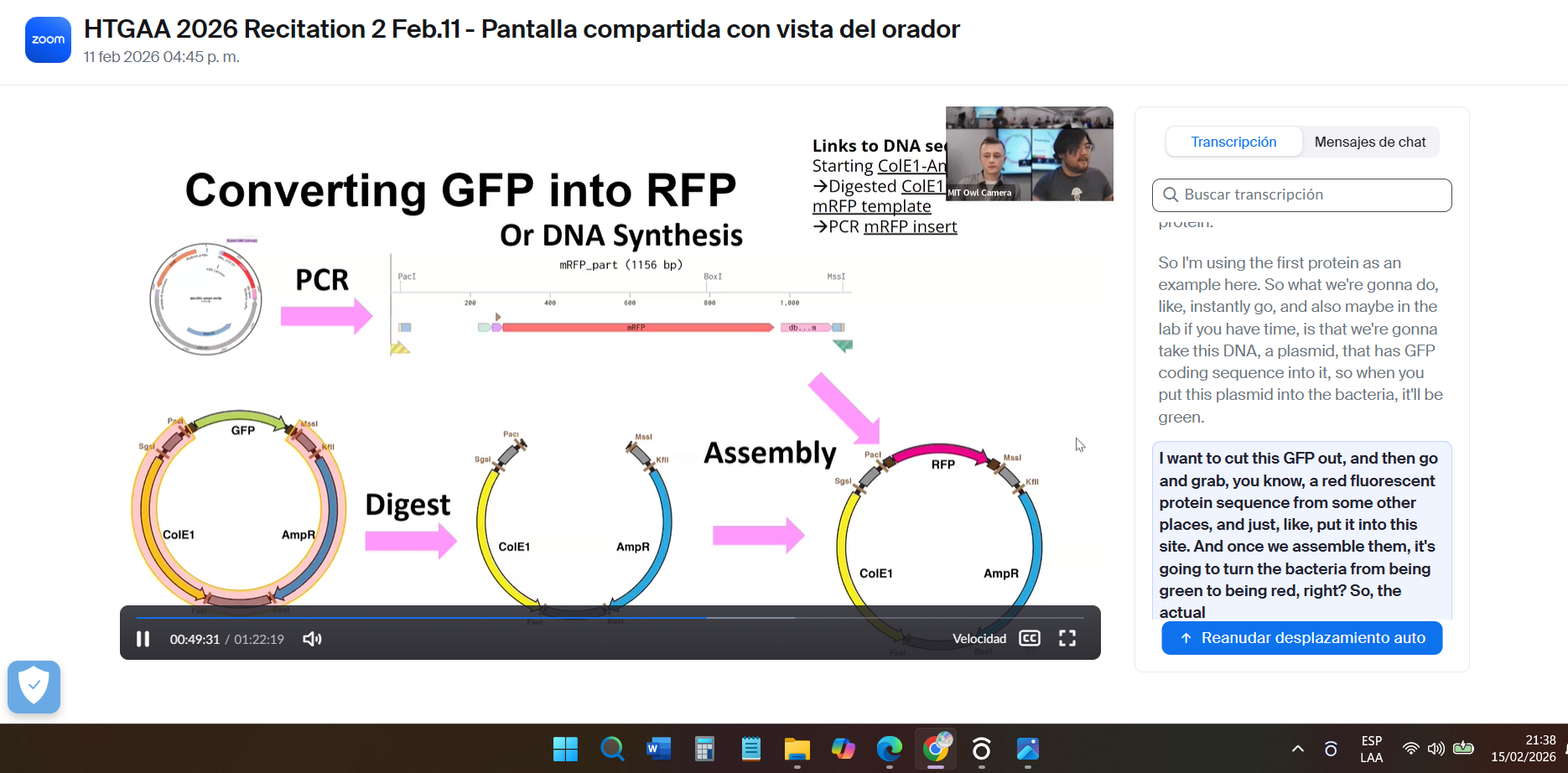
Part 1: Benchling & In-silico Gel Art
See the Gel Art: Restriction Digests and Gel Electrophoresis protocol for details. Overview:
Make a free account at benchling.com Import the Lambda DNA. Simulate Restriction Enzyme Digestion with the following Enzymes: EcoRI HindIII BamHI KpnI EcoRV SacI SalI Create a pattern/image in the style of Paul Vanouse’s Latent Figure Protocol artworks. You might find Ronan’s website a helpful tool for quickly iterating on designs!
After experimenting with the simulator, I successfully generated three figures using different restriction enzymes.
HEART

CIRCLE

FLOWER

Part 3: DNA Design Challenge
3.1. Choose your protein.
In recitation, we discussed that you will pick a protein for your homework that you find interesting. Which protein have you chosen and why? Using one of the tools described in recitation (NCBI, UniProt, google), obtain the protein sequence for the protein you chose.
P0A7X3
I choose the Small Ribosomal Subunit Protein uS9 (P0A7X3) from Escherichia coli K-12 because it is a well-characterized and fundamental biological role in bacterial translation. Studying this protein is relevant for studies in ribosome engineering structural biology, and synthetic biology applications.
Ribosomal engineering is the targeted redesign of ribosomes to control how genetic information is translated into proteins. It has direct implications for biotechnology, pharmaceutical innovation and industrial bioproduction.
I used Uniprot:
sp|P0A7X3|RS9_ECOLI Small ribosomal subunit protein uS9 OS=Escherichia coli (strain K12) OX=83333 GN=rpsI PE=1 SV=2 MAENQYYGTGRRKSSAARVFIKPGNGKIVINQRSLEQYFGRETARMVVRQPLELVDMVEK LDLYITVKGGGISGQAGAIRHGITRALMEYDESLRSELRKAGFVTRDARQVERKKVGLRK ARRRPQFSKR
d’Aquino AE, Kim DS, Jewett MC. Engineered Ribosomes for Basic Science and Synthetic Biology. Annu Rev Chem Biomol Eng. 2018 Jun 7;9:311-340. doi: 10.1146/annurev-chembioeng-060817-084129. Epub 2018 Mar 28. PMID: 29589973.
3.2. Reverse Translate: Protein (amino acid) sequence to DNA (nucleotide) sequence.
The Central Dogma discussed in class and recitation describes the process in which DNA sequence becomes transcribed and translated into protein. The Central Dogma gives us the framework to work backwards from a given protein sequence and infer the DNA sequence that the protein is derived from. Using one of the tools discussed in class, NCBI or online tools (google “reverse translation tools”), determine the nucleotide sequence that corresponds to the protein sequence you chose above.
Central Dogma
The Central Dogma of molecular biology describes the flow of genetic information from DNA to ARNA and ultimately to protein. Given a known protein sequence, once can infer a plausible nucleotide sequence by considering the degeneracy of the genetic code.Because the genetic code is redundant, several diffirent codons can encode the same amino acid.
My sequence
reverse translation of sp|P0A7X3|RS9_ECOLI Small ribosomal subunit protein uS9 OS=Escherichia coli (strain K12) OX=83333 GN=rpsI PE=1 SV=2 to a 390 base sequence of most likely codons. atggcggaaaaccagtattatggcaccggccgccgcaaaagcagcgcggcgcgcgtgttt attaaaccgggcaacggcaaaattgtgattaaccagcgcagcctggaacagtattttggc cgcgaaaccgcgcgcatggtggtgcgccagccgctggaactggtggatatggtggaaaaa ctggatctgtatattaccgtgaaaggcggcggcattagcggccaggcgggcgcgattcgc catggcattacccgcgcgctgatggaatatgatgaaagcctgcgcagcgaactgcgcaaa gcgggctttgtgacccgcgatgcgcgccaggtggaacgcaaaaaagtgggcctgcgcaaa gcgcgccgccgcccgcagtttagcaaacgc
I used this website: https://www.bioinformatics.org/sms2/rev_trans.html , I generated a 390- base pair DNA sequence corresponding to the amino acid sequence.
3.3. Codon optimization.
Once a nucleotide sequence of your protein is determined, you need to codon optimize your sequence. You may, once again, utilize google for a “codon optimization tool”. In your own words, describe why you need to optimize codon usage. Which organism have you chosen to optimize the codon sequence for and why?
ATG GCC GAA AAC CAG TAT TAT GGC ACC GGA CGG AGG AAA AGC TCT GCC GCA CGC GTG TTC ATT AAA CCA GGC AAT GGG AAG ATT GTG ATC AAT CAG AGA TCC TTG GAA CAG TAT TTT GGG CGG GAG ACT GCT AGG ATG GTG GTC AGA CAG CCT CTG GAA CTG GTG GAC ATG GTT GAG AAG CTG GAT CTG TAT ATT ACC GTG AAG GGA GGG GGC ATC TCC GGG CAG GCC GGC GCA ATC CGG CAT GGA ATT ACT CGA GCC CTT ATG GAG TAC GAC GAG TCC CTC CGC AGC GAG CTG AGA AAG GCG GGC TTC GTG ACC AGA GAT GCT CGA CAG GTG GAG AGG AAA AAG GTG GGA CTT CGC AAA GCT AGA AGA AGG CCA CAG TTT AGT AAA CGC
The following restriction enzyme sites have been found in the selected reading frame: BclI (TGATCA) MluI (ACGCGT) MlyI (GAGTC) NaeI (GCCGGC) XhoI (CTCGAG)
I used this website: https://www.idtdna.com/CodonOpt
3.4. You have a sequence! Now what?
What technologies could be used to produce this protein from your DNA? Describe in your words the DNA sequence can be transcribed and translated into your protein. You may describe either cell-dependent or cell-free methods, or both.
- Describe how a single gene codes for multiple proteins at the transcriptional level.
- Try aligning the DNA sequence, the transcribed RNA, and also the resulting translated Protein!!! See example below
a) Cell-free systems to produce RS9
Chemical synthesis The protein is small (80aa), so it could be chemically synthesized in a specialized laboratory. In chemical synthesis , Solid -Phase Peptide Synthesis (SPPS) is employed, in which the first amino acid is anchored to resin, subsequent amino acids are added one at a time to create sequential peptide bonds,and the synthesized protein is later detached and purified. This technique is particulary advantage because it grants full control over the peptide sequence and facilitates the integration of artificial molecular modifications
Cell free transcription system Conversely, the cell-free transcription strategy requires the prior design of a plasmid harboring a strong promoter, an optimized RBS and rpsl gene, which is subsequently added to the cell-free system to anable mRNA production.
Althought this system allows for fast protein production and rapid experimental turnaround,its implementation can be economically demanding.
Whittaker J. W. (2013). Cell-free protein synthesis: the state of the art. Biotechnology letters, 35(2), 143–152. https://doi.org/10.1007/s10529-012-1075-4
Part 4: Prepare a Twist DNA Synthesis Order
During the class, I learned how to design the plasmid to complete the exercise. After completing the tutorial, I understood the full process required to place a DNA synthesis order at Twist Bioscience.
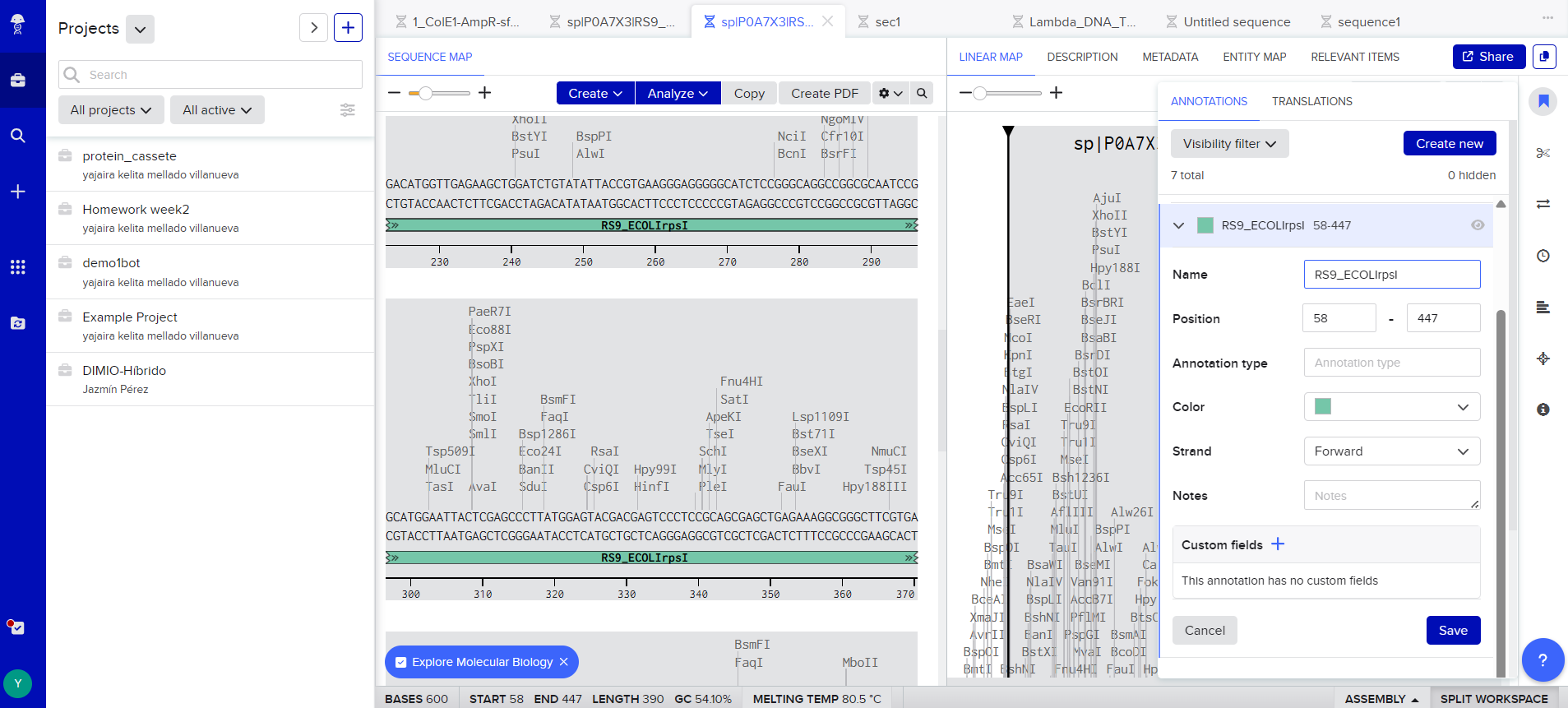
As it was my first time making a vector, with the help of ChatGPT and DeepSeek I was able to create these pieces.
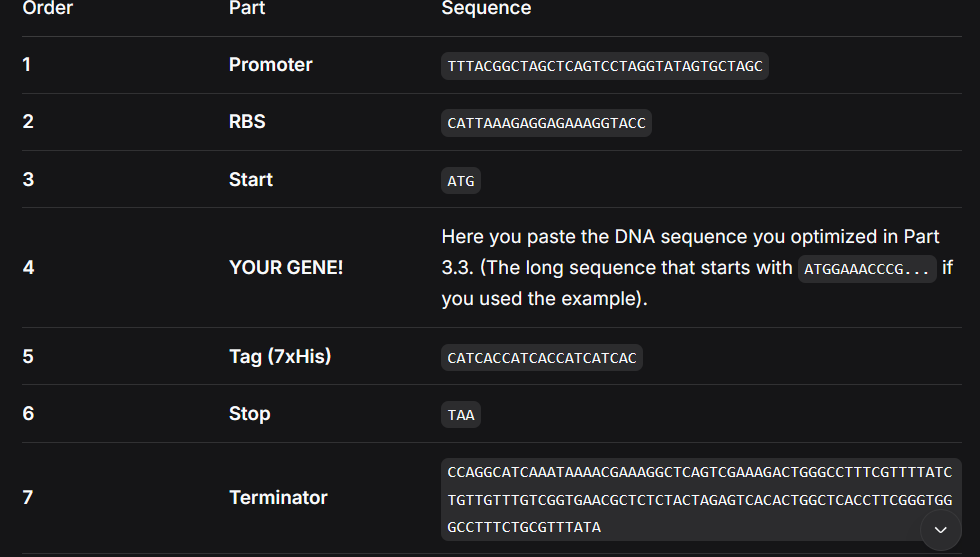
I used the same basic structure to represent my synthetic circuit with the Rs9 protein.
The most complicated part was fitting my sequence with the vector.
but in the end I chose this vector: pTwist Amp High Copy - (2221bp) and I got to this part:
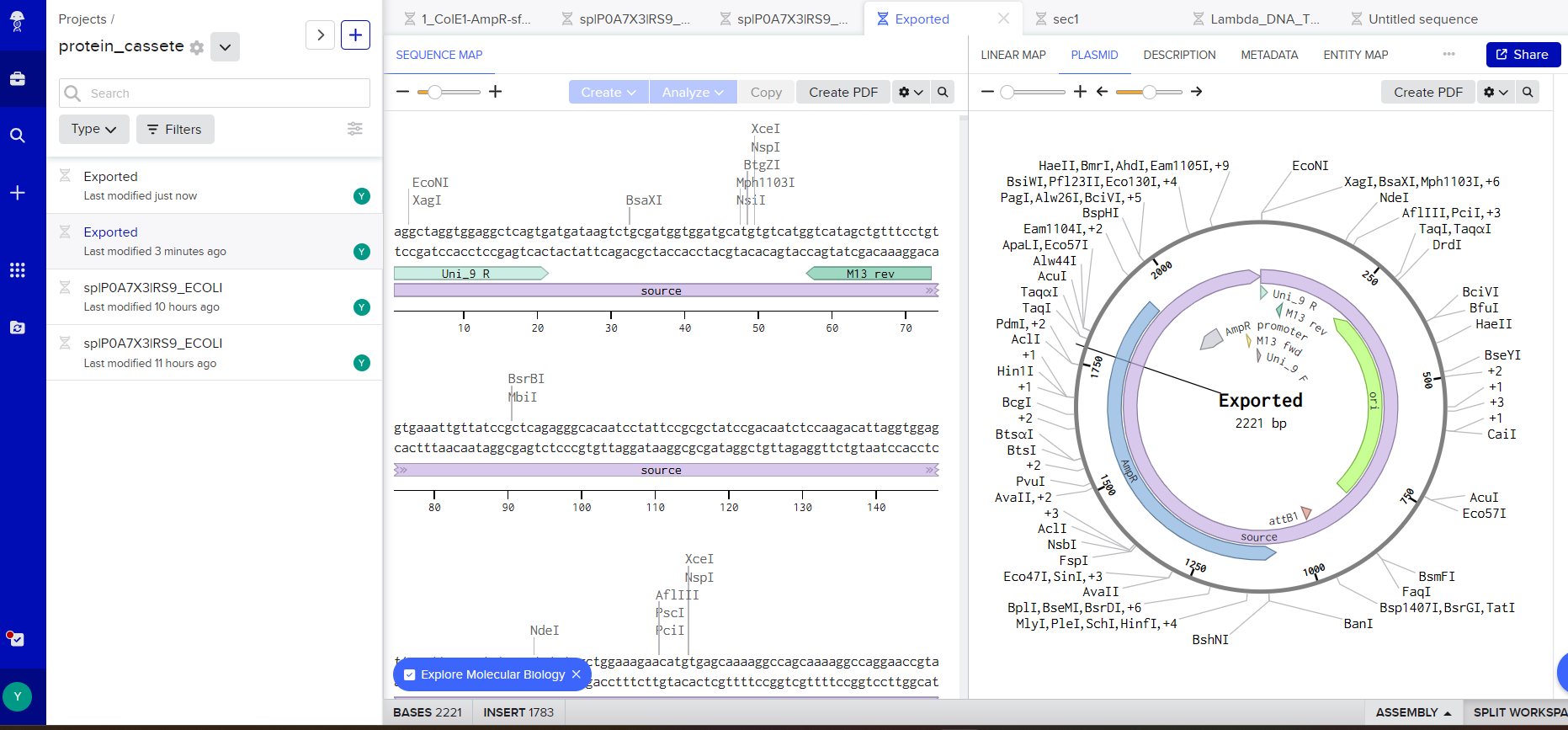
Part 5: DNA Read/Write/Edit
5.1 DNA Read
(i) What DNA would you want to sequence (e.g., read) and why? This could be DNA related to human health (e.g. genes related to disease research), environmental monitoring (e.g., sewage waste water, biodiversity analysis), and beyond (e.g. DNA data storage, biobank).
I want to sequence the DNA of South American camelids in Peru, such as alpacas, vicuñas, huanacos and llamas with the aim of identiying genes that confer resistance to extreme cold events (known as friajes). Knowledge of these sequences, it would be possible to identify high-value biactive molecules, such as nanobodies or inmune-related proteins, antimicrobial peptides, for development of therapeutic strateigies and vaccine design, especifically for diseases affecting the respiratory system.
(ii) In lecture, a variety of sequencing technologies were mentioned. What technology or technologies would you use to perform sequencing on your DNA and why?
Also answer the following questions: Is your method first-, second- or third-generation or other? How so? What is your input? How do you prepare your input (e.g. fragmentation, adapter ligation, PCR)? List the essential steps. What are the essential steps of your chosen sequencing technology, how does it decode the bases of your DNA sample (base calling)? What is the output of your chosen sequencing technology?
To sequence the DNA of South American camelid and study genes involved in cold resistance, I would use a hybrid sequencing strategy is employed because cold-adaptation genes include regulatory regions and gene duplications and mammalian genomes are large and repetitive.
| Oxford Nanopore | PacBio | Ilumina |
|---|---|---|
| *third-generation | *third-generation | *second-generation |
| Nanopore sequencing reads DNA molecules in real time | Sequencing reads individual DNA molecules without PCR | Technology based on DNA amplification and synthesis |
| function: In Oxford Nanopore sequencing, DNA passes through a pore and detecting electrical signals | function: Sequencing uses a polymerase that copies DNA while emitting fluorescent signals. | function: Uses sequential incorporation of fluorescent nucleotides, generating images in cycles with high accuracy. |
- ChatGPT was used with the following prompts to assist in data organization and text refinement varuety of sequencing technologies such as Oxford Nanopore, PacBio, and Ilumina.
- SPPS adicional: Peptide synthesis: a review of classical and emerging methods (Zhangping Cai et al., 2025).
- Cell-free adicional: Cell-Free Protein Synthesis: Pros and Cons of Prokaryotic and Eukaryotic Systems (Anne Zemella et al., 2015).
5.2 DNA Write
(i) What DNA would you want to synthesize (e.g., write) and why? These could be individual genes, clusters of genes or genetic circuits, whole genomes, and beyond. As described in class thus far, applications could range from therapeutics and drug discovery (e.g., mRNA vaccines and therapies) to novel biomaterials (e.g. structural proteins), to sensors (e.g., genetic circuits for sensing and responding to inflammation, environmental stimuli, etc.), to art (DNA origamis). If possible, include the specific genetic sequence(s) of what you would like to synthesize! You will have the opportunity to actually have Twist synthesize these DNA constructs! :)
I want to synthesize a modular synthetic cold-adaptation operon optimized for E.coli. The construct would include: The cold-shock gene cspA, an antifreeze protein (AFP) coding sequence and the trehalose biosynthesis genes otsA and otsB enhance osmoprotection, a strong double terminator These genes would be assembled under a cold-inducible promoter to create a synthetic operon capable of enhancing cellular survival and function at low temperatures. Cold environments impose multiple molecular stresses, including RNA secondary structure stabilization, reduced enzymatic kinetics, membrane rigity.
(ii) What technology or technologies would you use to perform this DNA synthesis and why?
Also answer the following questions: What are the essential steps of your chosen sequencing methods? What are the limitations of your sequencing method (if any) in terms of speed, accuracy, scalability?
a) Phosphoramidite solid-phase DNA synthesis for oligonucleotides This standard method for producing short oligonucleotides (20-200 nucleotides). DNA is synthesized base by base on a solid support through repeatec cycles of deprotection,coupling, oxidation and washing. While this technique provides precise control over sequence composition and enables chemical modifications.
b) Gibson Assembly to assemble fragments Gibson Assembly is an efficient DNA assembly method that allowas multiple fragments ti joined simultaneously in a single-tube, isothermal reaction. It uses a combination of exonuclease, DNA polymerase, and DNA ligase to join fragments that share overlapping homologous regios.
c) Commercial gene synthesis services in Twist Bioscience These synthesis services, such as those offered by Twist Bioscience provide end-to-end solutios for gene construcction, and sequencing based validation. Also provide sequence optimization options, such as codon optimization and restriction site removal, reducing experimental workload while increasing reliabity
Limitations
Detection sensitivity threshold: There is an inherent limit to the sensitivity of error deteccion during queality control.
False positives due:Errors introduced during PCR amplification, sequencing, or DNA assembly can be incorrectly interpreted as true mutations.
Lenght-dependent error rates : Making long sequences more prone to insertions, deletions, and substitutions and necessitating assembly from shorter fragments.
GC-rich sequence limitations: GC-rich regions reduce synthesis ans assembly efficency due to the formation of stable secondary structures, often requiring sequence redesing.
Cost constraints: Gene synthesis costs increase with sequence length, GC content, and desing complexity, as longer and more complex genes require additional synthesis, assembly, and quality steps.
5.3 DNA Edit
(i) What DNA would you want to edit and why? In class, George shared a variety of ways to edit the genes and genomes of humans and other organisms. Such DNA editing technologies have profound implications for human health, development, and even human longevity and human augmentation. DNA editing is also already commonly leveraged for flora and fauna, for example in nature conservation efforts, (animal/plant restoration, de-extinction), or in agriculture (e.g. plant breeding, nitrogen fixation). What kinds of edits might you want to make to DNA (e.g., human genomes and beyond) and why?
I propose editing genes implicated in maize abiotic stress responses, with a focus on drought and elevanted temperature tolerance. These genetic modifications would be direted toward regulatory genes and molecular pathways responsible for efficient water use, osmotic regulation, and oxidative strees defense. It represents a potential response to climate change, which is increasing the frequency of extreme conditions that reduce agricultural yields in high-Andean regions.
(ii) What technology or technologies would you use to perform these DNA edits and why?
Also answer the following questions: How does your technology of choice edit DNA? What are the essential steps? What preparation do you need to do (e.g. design steps) and what is the input (e.g. DNA template, enzymes, plasmids, primers, guides, cells) for the editing? What are the limitations of your editing methods (if any) in terms of efficiency or precision?
I would employ CRISPR-CAS9 in combination with base editing or prime editing to achieve precise genome modifications without the insertion of exogenous DNA. The editing process include candidate gene identification, the desing of CRISPR guides, delivery of editing system into maize cells, and the regeneration of edited plants.
The main limitations of this approach are varible editing efficiency and the genetic complex genetic architecture underlying these traits.
REFERENCES:
Molla, K. A., Sretenovic, S., Bansal, K. C., & Qi, Y. (2021). Precise plant genome editing using base editors and prime editors. Nature Plants, 7, 1166–1187. https://doi.org/10.1021/acsagscitech.2c00090
The following prompts were employed in ChatGPT to organize and refine the information:
“Explain Gibson Assembly step by step, highlighting the roles of exonuclease, DNA polymerase, and ligase, as well as common sources of assembly errors.”
“Compare Gibson Assembly with restriction enzyme cloning for synthetic gene construction, including advantages, limitations, and typical use cases.
Explain how CRISPR-Cas9 combined with base editing or prime editing enables precise genome modifications without introducing exogenous DNA.”
Discuss the main technical limitations of CRISPR-based genome editing in plants, including delivery, efficiency, and polygenic traits.
“Identify and explain categories of genes involved in abiotic stress tolerance in maize, including water-use efficiency, osmotic balance, and oxidative stress pathways.