Week 4 HW:Protein design PART I
Part A. Conceptual Questions
Answer any NINE of the following questions from Shuguang Zhang: (i.e. you can select two to skip)
- Why do humans eat beef but do not become a cow, eat fish but do not become fish?
Humans do not transform into fish or cws because dietary components are not assimilated as intact complex structures. When we ingest food, it is processed through a mechanism called digestion, which involves the enzymatic breakdown of macromolecules into their most basic units. These molecules are then used as raw materials to synthesize biomolecules acording to the instructions encoded in human DNA.
- Why are there only 20 natural amino acids?
The 20 amino acids constitute an almost ideal chemical spectrum that maximizes structural diversity with minimal redundancy. The ibosomal translation machinery efficiently harnesses this chemical diversity, nabling the formation of secondary and tertiary structures, hydrophobic interactions, disulfide bridges, and enzymatic catalysis.
- Can you make other non-natural amino acids? Design some new amino acids.
Currently, amino acids can be engineered through two main approaches. Chemical synthesis allows modification of the R side chain,he carboxyl group, and even the chirality (L or D configuration). In contrast, genetic code expansion techniques allow the reassignment of a stop codon, coupled with the introduction of an orthogonal tRNA and an engineered aminoacyl-tRNA synthetase, thereby enabling the site-specific incorporation of noncanonical amino acids into proteins.
- Where did amino acids come from before enzymes that make them, and before life started?
Amino acids originated through prebiotic chemical processes on early Earth or possibly even in outer space.The Miller–Urey experiment showed that lightning-like electrical discharges in a reducing atmosphere can produce amino acids.Furthermore, the detection of amino acids in meteorites supports the hypothesis of a partially exogenous origin for these biomolecules.
- If you make an α-helix using D-amino acids, what handedness (right or left) would you expect?
The α-helix formed by L-amino acids is right-handed due to the conformational constraints imposed by the chirality of the α-carbon. In contrast, a polypeptide consisting solely of D-amino acids inverts these geometric constraints, leading to the formation of a left-handed α-helix.
Can you discover additional helices in proteins? Helical conformations such as the 3₁₀ helix and the π-helix are defined by distinct hydrogen-bonding patterns and periodicities.The planar nature of the peptide bond, together with backbone dihedral angle restrictions defined by the Ramachandran plot, limits the range of energetically accessible helical conformations.However, through protein engineering and the incorporation of non-natural amino acids, it is possible to explore novel helical conformations.
Why are most molecular helices right-handed?
The predominance of right-handed helices in biological systems arises from their construction from L-amino acids and D-sugars.The chirality of these molecules creates spatial restrictions that make right-handed helices more stable and energetically favorable.
Why do β-sheets tend to aggregate?
- What is the driving force for β-sheet aggregation? β-sheets tend to aggregate because their extended structure enables the formation of extensive intermolecular hydrogen-bonding networks and altered exposure of hydrophobic residues, which promotes hydrophobic interactions between polypeptide chains.
Why do many amyloid diseases form β-sheets?
- Can you use amyloid β-sheets as materials? Amyloid diseases involve the conversion of partially denatured proteins into an extended β-sheet conformation, which enables the formation of highly stable cross-β structures through cooperative hydrogen-bonding networks and hydrophobic interactions.s. Remarkably, this fibrillar organization imparts outstanding mechanical rigidity and thermal stability, driving its investigation as a self-assembling biomaterial platform for nanotechnology and tissue engineering.
Part B: Protein Analysis and Visualization
In this part of the homework, you will be using online resources and 3D visualization software to answer questions about proteins. Pick any protein (from any organism) of your interest that has a 3D structure and answer the following questions:
Briefly describe the protein you selected and why you selected it.
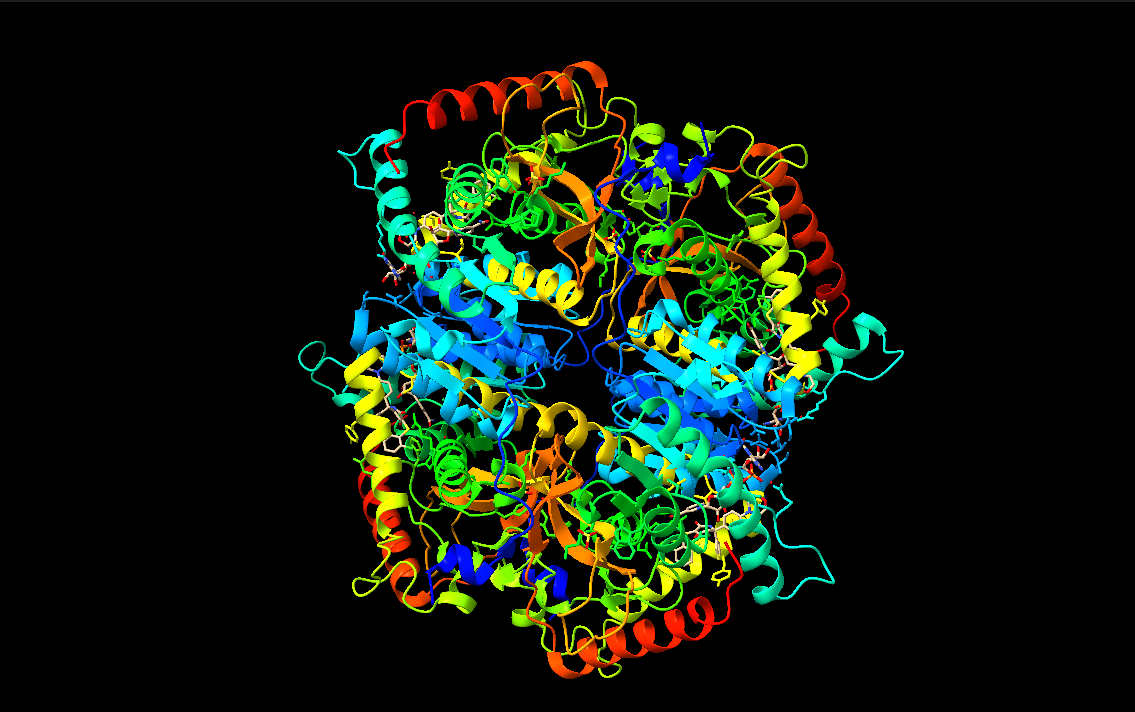
I chose lactate dehydrogenase because it is a fundamental enzyme in energy metabolism, particularly under anaerobic conditions. Its role in the conversion of pyruvate to lactate makes it biologically significant, and its tetrameric structure with a well-defined active site allows for a clear analysis of the relationship between three-dimensional structure and catalytic function. It is an excellent model for studying key concepts in biochemistry and enzymatic catalysis.
Identify the amino acid sequence of your protein.
sp|P00338|LDHA_HUMAN L-lactate dehydrogenase A chain OS=Homo sapiens OX=9606 GN=LDHA PE=1 SV=2 MATLKDQLIYNLLKEEQTPQNKITVVGVGAVGMACAISILMKDLADELALVDVIEDKLKG EMMDLQHGSLFLRTPKIVSGKDYNVTANSKLVIITAGARQQEGESRLNLVQRNVNIFKFI IPNVVKYSPNCKLLIVSNPVDILTYVAWKISGFPKNRVIGSGCNLDSARFRYLMGERLGV HPLSCHGWVLGEHGDSSVPVWSGMNVAGVSLKTLHPDLGTDKDKEQWKEVHKQVVESAYE VIKLKGYTSWAIGLSVADLAESIMKNLRRVHPVSTMIKGLYGIKDDVFLSVPCILGQNGI SDLVKVTLTSEEEARLKKSADTLWGIQKELQF
How long is it? What is the most frequent amino acid? You can use this Colab notebook to count the frequency of amino acids.
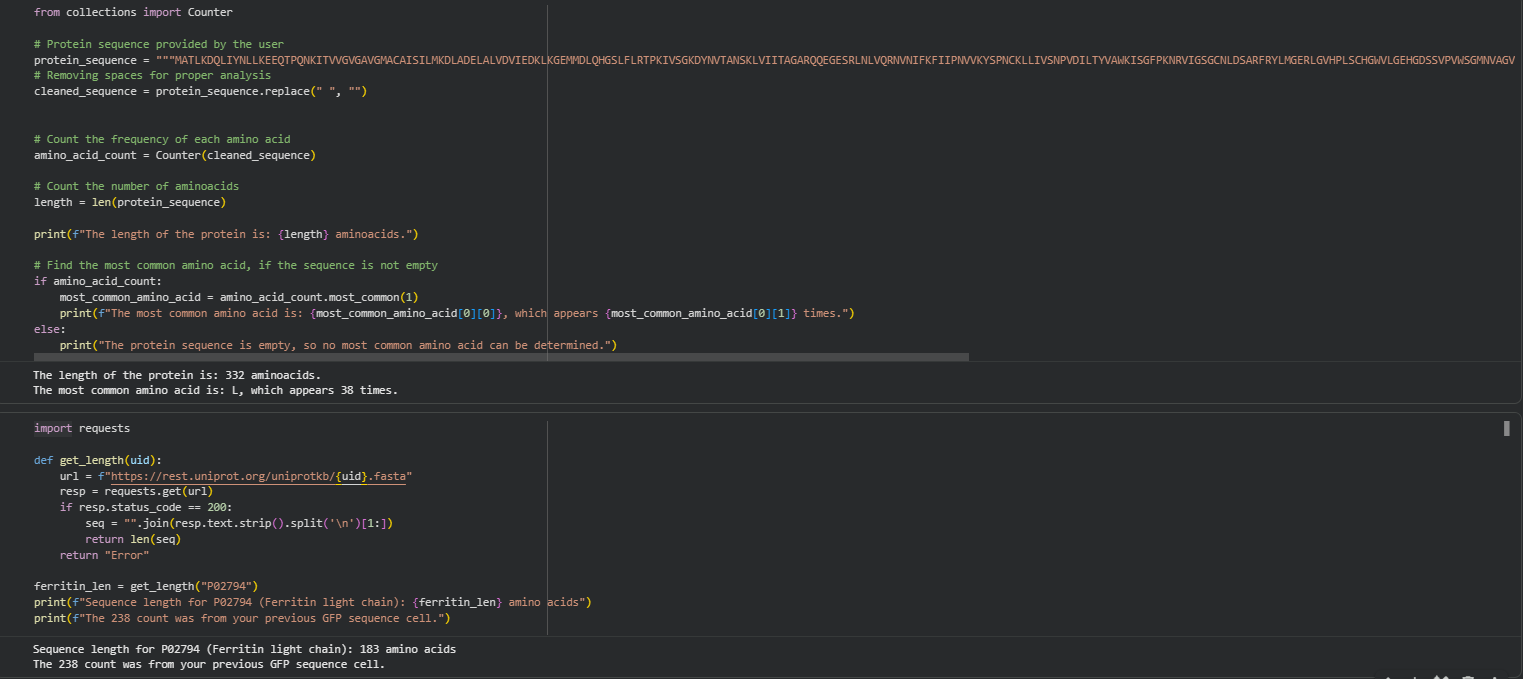
How many protein sequence homologs are there for your protein? Hint: Use Uniprot’s BLAST tool to search for homologs.
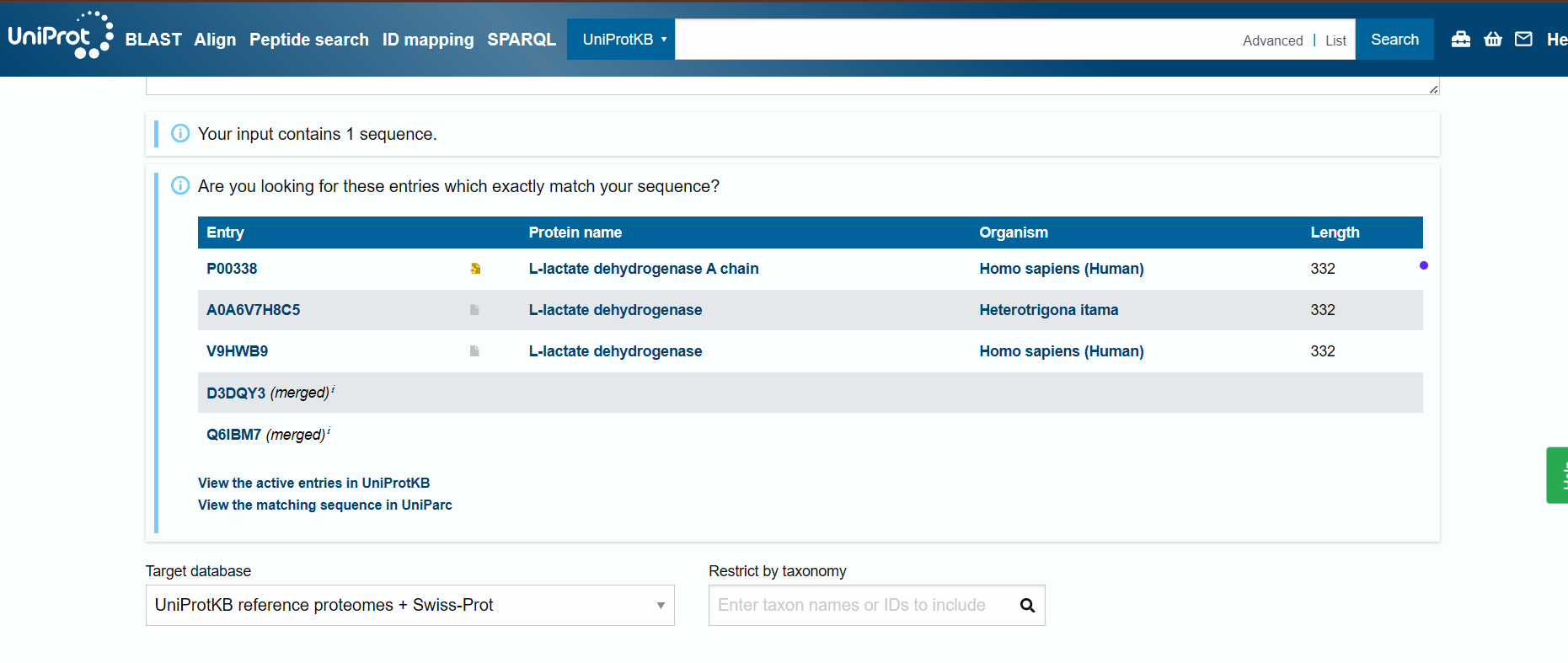
Does your protein belong to any protein family?
- Identify the structure page of your protein in RCSB https://www.rcsb.org/structure/4ZVV#entity-1
When was the structure solved? Is it a good quality structure? Good quality structure is the one with good resolution. Smaller the better (Resolution: 2.70 Å)
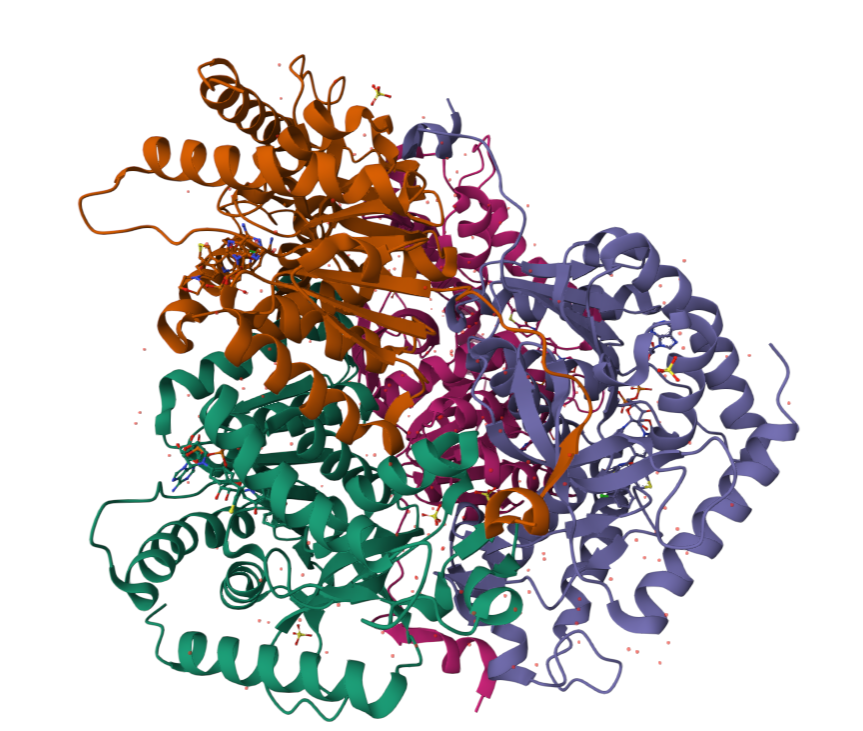
Are there any other molecules in the solved structure apart from protein?
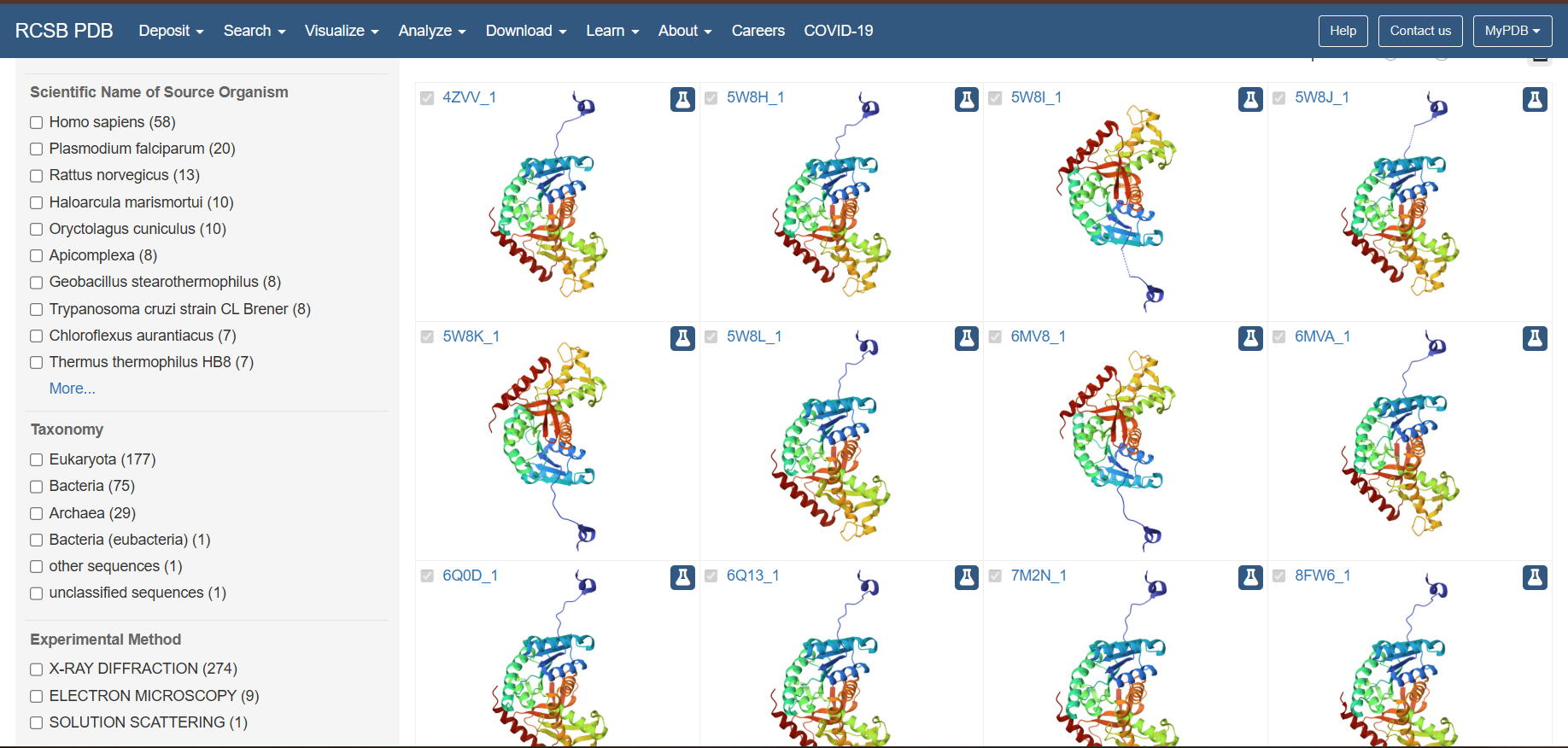
Does your protein belong to any structure classification family?
- Open the structure of your protein in any 3D molecule visualization software:
- PyMol Tutorial Here (hint: ChatGPT is good at PyMol commands)
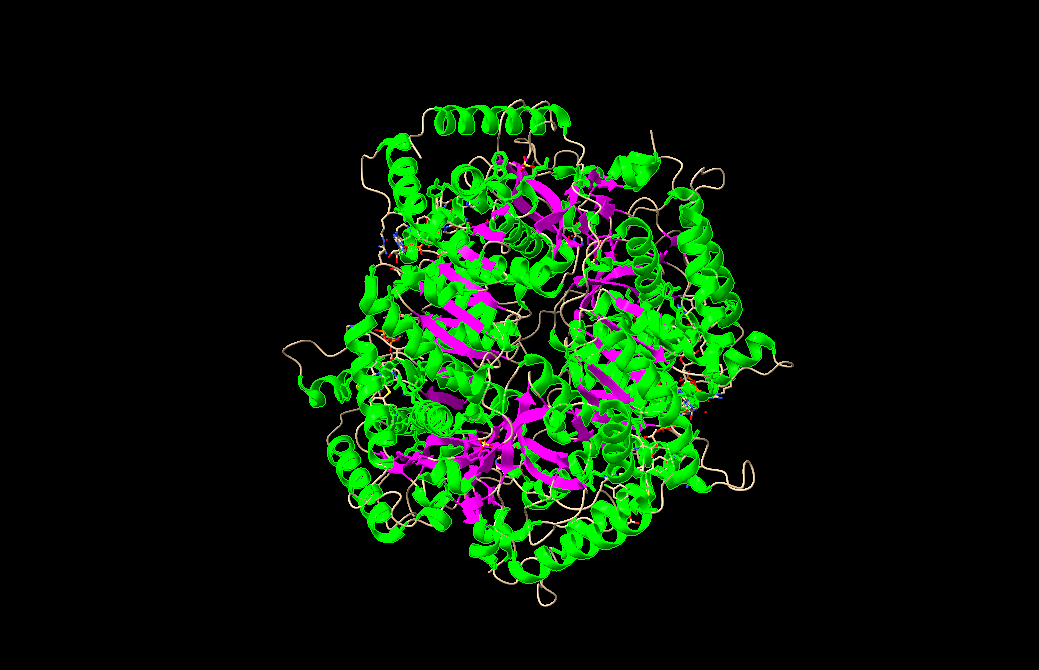
- Visualize the protein as “cartoon”, “ribbon” and “ball and stick”.
- Color the protein by secondary structure. Does it have more helices or sheets?
- Color the protein by residue type. What can you tell about the distribution of hydrophobic vs hydrophilic residues?
- Visualize the surface of the protein. Does it have any “holes” (aka binding pockets)?
Part C. Using ML-Based Protein Design Tools
In this section, we will learn about the capabilities of modern protein AI models and test some of them in your chosen protein.
Copy the HTGAA_ProteinDesign2026.ipynb notebook and set up a colab instance with GPU. Choose your favorite protein from the PDB. We will now try multiple things in the three sections below; report each of these results in your homework writeup on your HTGAA website:
PROTEIN SECUENCE: MATLKDQLIYNLLKEEQTPQNKITVVGVGAVGMACAISILMKDLADELALVDVIEDKLKG EMMDLQHGSLFLRTPKIVSGKDYNVTANSKLVIITAGARQQEGESRLNLVQRNVNIFKFI IPNVVKYSPNCKLLIVSNPVDILTYVAWKISGFPKNRVIGSGCNLDSARFRYLMGERLGV HPLSCHGWVLGEHGDSSVPVWSGMNVAGVSLKTLHPDLGTDKDKEQWKEVHKQVVESAYE VIKLKGYTSWAIGLSVADLAESIMKNLRRVHPVSTMIKGLYGIKDDVFLSVPCILGQNGI SDLVKVTLTSEEEARLKKSADTLWGIQKELQF
C1. Protein Language Modeling
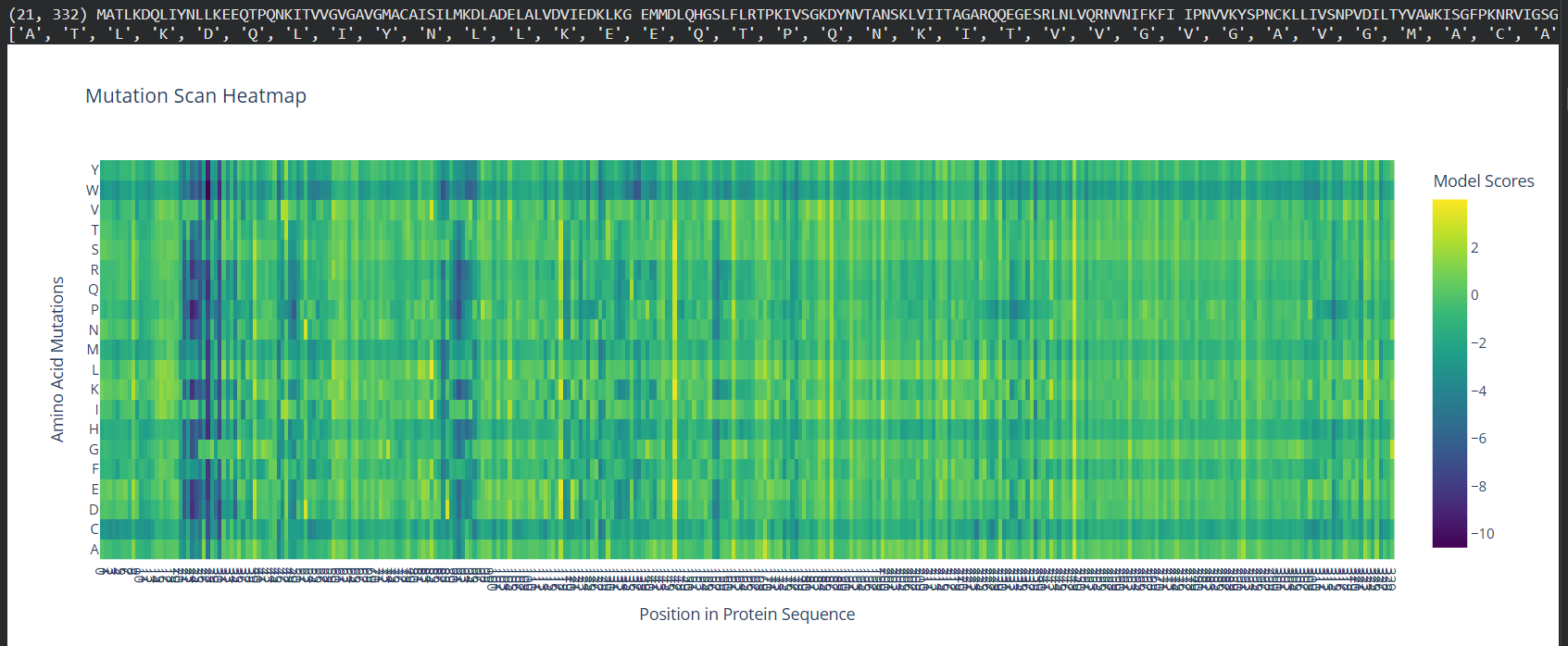
- Deep Mutational Scans
- Use ESM2 to generate an unsupervised deep mutational scan of your protein based on language model likelihoods.
- Can you explain any particular pattern? (choose a residue and a mutation that stands out)
(Bonus) Find sequences for which we have experimental scans, and compare the prediction of the language model to experiment.
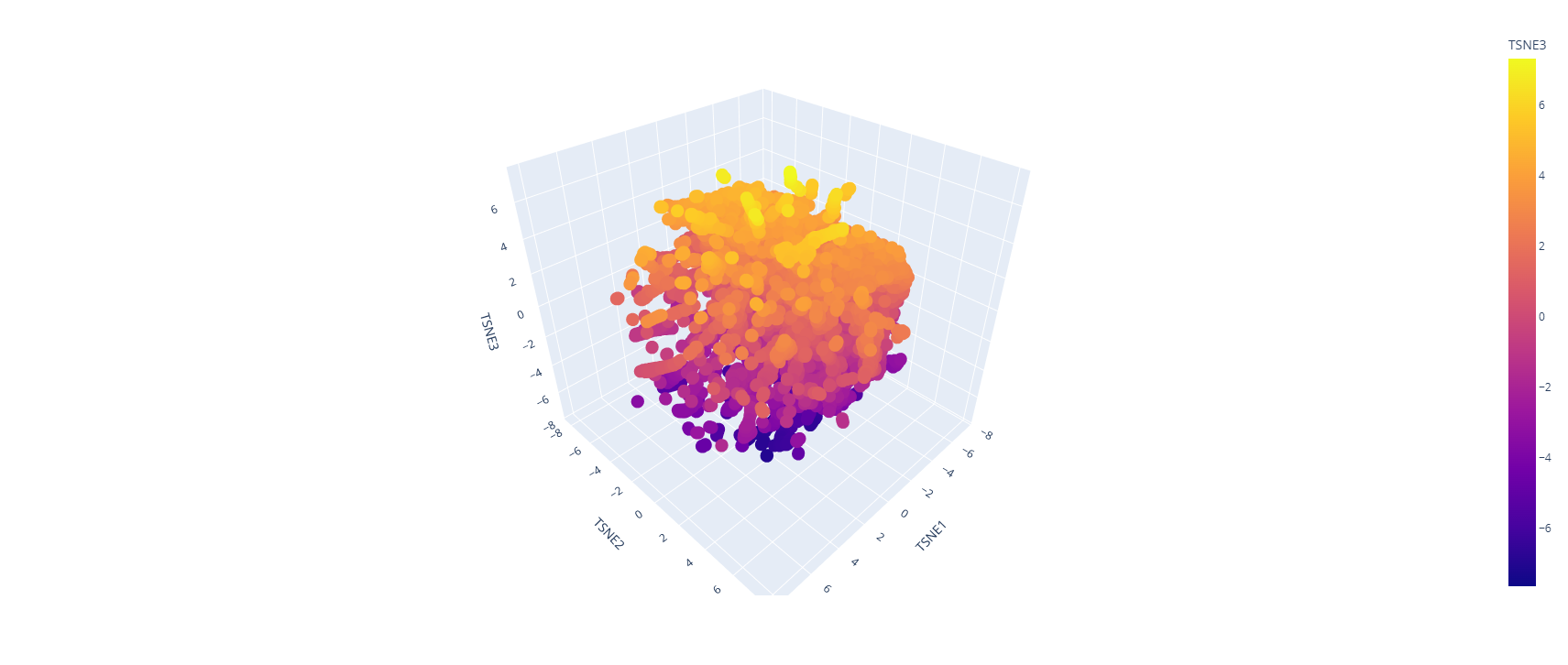
- Latent Space Analysis
Use the provided sequence dataset to embed proteins in reduced dimensionality.
Analyze the different formed neighborhoods: do they approximate similar proteins?
Place your protein in the resulting map and explain its position and similarity to its neighbors.
C2. Protein Folding Folding a protein
- Fold your protein with ESMFold. Do the predicted coordinates match your original structure?
Try changing the sequence, first try some mutations, then large segments. Is your protein structure resilient to mutations?
C3. Protein Generation Inverse-Folding a protein: Let’s now use the backbone of your chosen PDB to propose sequence candidates via ProteinMPNN Analyze the predicted sequence probabilities and compare the predicted sequence vs the original one. Input this sequence into ESMFold and compare the predicted structure to your original.
Part D. Group Brainstorm on Bacteriophage Engineering
Find a group of ~3–4 students
Read through the Phage Reading material listed under “Reading & Resources” below.
Review the Bacteriophage Final Project Goals for engineering the L Protein: Increased stability (easiest) Higher titers (medium) Higher toxicity of lysis protein (hard)
Brainstorm Session
- Choose one or two main goals from the list that you think you can address computationally (e.g., “We’ll try to stabilize the lysis protein,” or “We’ll attempt to disrupt its interaction with E. coli DnaJ.”).
- Write a 1-page proposal (bullet points or short paragraphs) describing:
- Which tools/approaches from recitation you propose using (e.g., “Use Protein Language Models to do in silico mutagenesis, then AlphaFold-Multimer to check complexes.”).
- Why do you think those tools might help solve your chosen sub-problem?
- Name one or two potential pitfalls (e.g., “We lack enough training data on phage–bacteria interactions.”).
- Include a schematic of your pipeline.
- This resource may be useful: HTGAA Protein Engineering Tools
- Each individually put your plan on your HTGAA website Include your group’s short plan for engineering a bacteriophage

PROYECT IDEA
https://docs.google.com/presentation/d/1yNann1oBmofVF4Cz4Ti-MHT5e2Iq76Sn2N9hpjaFEFA/edit?usp=sharing