Yajaira Kelita Mellado Villanueva — HTGAA Spring 2026
About me
I am Yajaira Kelita, a Biotechnology Engineering student from Peru with a strong interest in understanding living systems ans developing technologies that coexist with nature.I am passionate about synthetic biology, tissue engineering, and medical, animal and agricultural biotechnology.
First, describe a biological engineering application or tool you want to develop and why. This could be inspired by an idea for your HTGAA class project and/or something for which you are already doing in your research, or something you are just curious about. Biosensors for Animal Health
I aim to develop molecular diagnostic biosensors for veterinary medice. Specifically, I am interested in creating biofluorescent biosensor kits capable of detecting animal pathogens in rural and remote areas. This application is particularly relevant in countries such as Peru, where many communities lacated far from urban centers depend on livestock for their livelihood but face limited access to laboratory diagnostic services. This often leads to delayed diagnoses and significant economic losses due to infectius diseases.
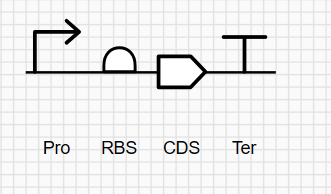
Part 0: Basics of Gel Electrophoresis I reviewed the recorded class recitation. Part 1: Benchling & In-silico Gel Art See the Gel Art: Restriction Digests and Gel Electrophoresis protocol for details. Overview:
Make a free account at benchling.com Import the Lambda DNA. Simulate Restriction Enzyme Digestion with the following Enzymes: EcoRI HindIII BamHI KpnI EcoRV SacI SalI Create a pattern/image in the style of Paul Vanouse’s Latent Figure Protocol artworks. You might find Ronan’s website a helpful tool for quickly iterating on designs!
Assignment: Python Script for Opentrons Artwork — DUE BY YOUR LAB TIME! Your task this week is to Create a Python file to run on an Opentrons liquid handling robot.
Post-Lab Questions — DUE BY START OF FEB 24 LECTURE To create the design, I employed several tests:
Part 1. I initially performed some tests by modifying the color in the first example provided in Google Colab.
Part A. Conceptual Questions Answer any NINE of the following questions from Shuguang Zhang: (i.e. you can select two to skip)
Why do humans eat beef but do not become a cow, eat fish but do not become fish? Humans do not transform into fish or cws because dietary components are not assimilated as intact complex structures. When we ingest food, it is processed through a mechanism called digestion, which involves the enzymatic breakdown of macromolecules into their most basic units. These molecules are then used as raw materials to synthesize biomolecules acording to the instructions encoded in human DNA.
Homework — DUE BY START OF MAR 10 LECTURE Part A: SOD1 Binder Peptide Design (From Pranam) Superoxide dismutase 1 (SOD1) is a cytosolic antioxidant enzyme that converts superoxide radicals into hydrogen peroxide and oxygen. In its native state, it forms a stable homodimer and binds copper and zinc.
Homework — DUE BY START OF MAR 17 LECTURE Assignment: DNA Assembly Answer these questions about the protocol in this week’s lab: 1. What are some components in the Phusion High-Fidelity PCR Master Mix and what is their purpose? The Phusion High-Fidelity master mix contains:
Homework — DUE BY Mar 31 2PM ET Assignment Part 1: Intracellular Artificial Neural Networks (IANNs)
What advantages do IANNs have over traditional genetic circuits, whose input/output behaviors are Boolean functions? Tradicional genetic rely on Boolean logic (AND, OR, NOT), producing discrete outputs. In contrast, IAANS are inspired by the perceptron by the perceptron model, where inputs correspond to : Activation is the gene expression threshold, Inputs are molecular concentrations,Weights is regulatory strength. Some advanges are: the continuous signal precessing, wich allows graded rather than binary responses, scalability through multilayer architectures, improved classification of complex cellular states, as multiple biological signals can be integrated and weighted simultaneously.
General homework questions 1. Explain the main advantages of cell-free protein synthesis over traditional in vivo methods, specifically in terms of flexibility and control over experimental variables. Name at least two cases where cell-free expression is more beneficial than cell production. 2. Describe the main components of a cell-free expression system and explain the role of each component. 3. Why is energy provision regeneration critical in cell-free systems? Describe a method you could use to ensure continuous ATP supply in your cell-free experiment. 4. Compare prokaryotic versus eukaryotic cell-free expression systems. Choose a protein to produce in each system and explain why. 5. How would you design a cell-free experiment to optimize the expression of a membrane protein? Discuss the challenges and how you would address them in your setup. 6. Imagine you observe a low yield of your target protein in a cell-free system. Describe three possible reasons for this and suggest a troubleshooting strategy for each. Homework question from Kate Adamala Design an example of a useful synthetic minimal cell as follows:
Homework — DUE BY START OF Apr 14 LECTURE Homework is partly based on data that will be generated in the Waters Immerse Lab in Cambridge, MA. Students will characterize green fluorescent protein (eGFP, a recombinant protein standard) structure (primary, secondary/tertiary) in the lab using liquid chromatography and mass spectrometry, as well as Keyhole Limpet Hemocyanin (KLH) oligomeric states using charge detection mass spectrometry (CDMS). Data generated in the lab needed to do the homework is included both within this document and in the Appendix of the laboratory protocol.
Part A: The 1,536 Pixel Artwork Canvas | Collective Artwork Contribute at least one pixel to this global artwork experiment before the editing ends on Sunday 4/19 at 11:59 PM EST. A personalized URL was sent to the email address associated with your Discourse account, and you can discuss the artwork on the Discourse. If you did not have a chance to contribute, it’s okay, just make sure you become a TA this fall! 😉
Subsections of Homework
Week 1 HW: Principles and Practices
First, describe a biological engineering application or tool you want to develop and why. This could be inspired by an idea for your HTGAA class project and/or something for which you are already doing in your research, or something you are just curious about.
Biosensors for Animal Health
I aim to develop molecular diagnostic biosensors for veterinary medice.
Specifically, I am interested in creating biofluorescent biosensor kits capable of detecting animal pathogens in rural and remote areas.
This application is particularly relevant in countries such as Peru, where many communities lacated far from urban centers depend on livestock for their livelihood but face limited access to laboratory diagnostic services. This often leads to delayed diagnoses and significant economic losses due to infectius diseases.
The biosensor would integrate a specific biological recognition element with a luciferase-based reporter system. When the target pathogen or its genetic material is present in the sample, it specifically interacts with the biological component of the biosensor. This interaction actives the luciferase enzyme, which catalizes a reaction that produces light.
The chemical reaction underlying the proposed biosensor is based on a bioluminescence process in whitch an enzyme catalyzes the transformation of substrate, releasing energy in the form of light. To generate this luminescent signal, the luciferase enzyme is activated in the presence of a pathogen or biomolecule. This mechanism offers several advantages, including high sensitivity and rapid results compared to other molecular techniques. Furthermore, it enable non-invasive observation of biological processes and the detection of pathogens in animals.
Figure 1. AI-generated image created using Ideogram
Next, describe one or more governance/policy goals related to ensuring that this application or tool contributes to an “ethical” future, like ensuring non-malfeasance (preventing harm). Break big goals down into two or more specific sub-goals. Below is one example framework (developed in the context of synthetic genomics) you can choose to use or adapt, or you can develop your own. The example was developed to consider policy goals of ensuring safety and security, alongside other goals, like promoting constructive uses, but you could propose other goals for example, those relating to equity or autonomy.
Objective 1: Minimizing Harm Throught Safe Biosensor Design and Deployment
This objective is essential to ensuring the principle of non-maleficience, meaning that the design and implementation of the biosensor must prioritize the prevention of harm to animals and the environment.
Specific objectives
- Establish biosafety guidelines for the use of pathogenic biological components and containment strategies.
- Promote responsible use and ensure that biosensor is use for veterinary diagnostic purposes through user guidelines and target training,
Objective 2: Promoting fair and Inclusive Access
Policies should promote the affordability and accessibility of the technology for small-scale livestock farmers.
Next, describe at least three different potential governance “actions” by considering the four aspects below (Purpose, Design, Assumptions, Risks of Failure & “Success”). Try to outline a mix of actions (e.g. a new requirement/rule, incentive, or technical strategy) pursued by different “actors” (e.g. academic researchers, companies, federal regulators, law enforcement, etc). Draw upon your existing knowledge and a little additional digging, and feel free to use analogies to other domains (e.g. 3D printing, drones, financial systems, etc.).
Purpose: What is done now and what changes are you proposing?
Design: What is needed to make it “work”? (including the actor(s) involved - who must opt-in, fund, approve, or implement, etc)
Assumptions: What could you have wrong (incorrect assumptions, uncertainties)?
Risks of Failure & “Success”: How might this fail, including any unintended consequences of the “success” of your proposed actions?
🐮
ACTION 1 : Establishment of biosafety and use protocols
Purpose
To implement biosafety measures for the use of biological components such as luciferase, genetically modified organisms, or active enzymatic extracts
Design
In context of Peru, With the support of authorities such as the Ministry of Agrarian Development and Rick Management, research groups, and international organizations, protocols and regulations canbe developed to ensure safety.
Assumptions
This proposal assumes that standardized testing protocols can be adapted to diverse rural contexts and financial incentives will effectively motivate to focus on equity and that reduced-cost technologies will still meet quality and safety standards.
Risks
Improper use due to insufficent user training and lack of adequate capacity-building initiatives.
🐮
ACTION 2 : Accessibility-oriented economic and logistical desing
Purpose
To reduce access barriers related to cost, equipment requirements and supply chains
Design
selection of low cost materials and the pursuit of funding from government agencies and external organizations.
Assumptions
Reduced complexity does not significantly compromise analytical sensitivity.
Risks
Reduced analytical performance
🐮
ACTION 3: Participatory validation with rural veterinarians and livestock keepers
Purpose
Currently, molecular detección methods such as PCR, ELISA, require sample pre-treatment in centralized laboratories and longer turnaround times to obtain results.In contrast, lusiferase-based biosensors enable rapid, accurate, and real-time data acquisition.Therefore,this action proposes a basic training and certication program for veterinary biosensors in rural areas.
Design
This accion require collaboration between governments,funding agencies would provide grants, subsidies and research groups that prioritize affordability and rural deployment.
Assumptions
This proposal assumes that standardized testing protocols can be adapted to diverse rural contexts and financial incentives will effectively motivate to focus on equity and that reduced-cost technologies will still meet quality and safety standars.
Risks
The policy could fail if incentives are insufficient or poorly target. Also, limited stakeholder engagement or resistance to adopting new technologies could hinder effective implementation.
Next, score (from 1-3 with, 1 as the best, or n/a) each of your governance actions against your rubric of policy goals. The following is one framework but feel free to make your own:
Does the option:
Establishment of biosafety and use protocols
Accessibility-oriented economic and logistical desing
Participatory validation with rural veterinarians and livestock keepers
Enhance Biosecurity
• By preventing incidents
2
3
1
• By helping respond
3
2
2
Foster Lab Safety
• By preventing incident
2
3
2
• By helping respond
2
2
1
Protect the environment
• By preventing incidents
2
2
1
• By helping respond
2
1
n/a
Other considerations
• Minimizing costs and burdens to stakeholders
3
2
n/a
• Feasibility?
3
2
2
• Not impede research
1
2
3
• Promote constructive applications
2
2
1
Last, drawing upon this scoring, describe which governance option, or combination of options, you would prioritize, and why. Outline any trade-offs you considered as well as assumptions and uncertainties. For this, you can choose one or more relevant audiences for your recommendation, which could range from the very local (e.g. to MIT leadership or Cambridge Mayoral Office) to the national (e.g. to President Biden or the head of a Federal Agency) to the international (e.g. to the United Nations Office of the Secretary-General, or the leadership of a multinational firm or industry consortia). These could also be one of the “actor” groups in your matrix.
Reflecting on what you learned and did in class this week, outline any ethical concerns that arose, especially any that were new to you. Then propose any governance actions you think might be appropriate to address those issues. This should be included on your class page for this week.
🌿 My Reflection
Completing this task allowed me to become more aware of the ethical considerations involved in implementing a technology within a specific context. I came to understand that such implementing a technology within a specific context. I come to understand that such implementation requires close collaboration between researchers and government institutions to establish safaty protocols that ensure animal health.In addition, effective public policies are necessary to promote appropriate and equitable use in order to fulfill the mission of applicability in rural settings.
Identification of the most common diseases in domestic animals in Peru that require molecular diagnosis and present high clinical demand.
Exploration of current global solutions for these diseases within the fields of synthetic biology, biotechnology, and biological engineering.
Detailed explanation of the molecular and biological mechanisms underlying these technological solutions.
Comparative evaluation of isothermal molecular diagnostics (RPA and LAMP) and biosensor-based diagnostic approaches.
Identification of relevant scientific literature supporting isothermal amplification methods and biosensor technologies.
All interpretations, critical analysis, and conclusions derived from these prompts are the author’s own.
Assignment (Final Project) – Due as part of your Final Project presentation (not Feb 10)
Assignees for the following sections
MIT/Harvard students
Required
Committed Listeners
Required
As part of your final project, design one or more strategies to ensure that your project, and what it enables, contributes to growing an ethical biological future.
Assignment (Lab Preparation) — DUE BY START OF FEB 10 LECTURE
Assignees for the following sections
MIT/Harvard students
Required
Committed Listeners
(Not Applicable)
Lab Training (failure to complete this will jeopardize your acceptance into the course)
Complete Lab Specific Training in Person.
Complete Safety Training in Atlas
Navigate to atlas.mit.edu and on the right-hand side, click “Learning Center”
Head to the Course Catalog and find the following two courses:
General Biosafety for Researchers (EHS00260w)
Managing Hazardous Waste (EHS00501w)
Assignment (Week 2 Lecture Prep) — DUE BY START OF FEB 10 LECTURE
Assignees for the following sections
MIT/Harvard students
Required
Committed Listeners
Required
In preparation for Week 2’s lecture on “DNA Read, Write, and Edit," please review these materials:
Lecture 2 slides as posted below.
The associated papers that are referenced in those slides.
In addition, answer these questions in each faculty member’s section:
Nature’s machinery for copying DNA is called polymerase. What is the error rate of polymerase? How does this compare to the
length of the human genome. How does biology deal with that discrepancy?
The error rate of natural polymerase is aproximatly 1 in 10^6 nucleotides. When this is compared to the human genome (3 billion base pairs) the discrepancy is significativa. It is important to emphasize the mechanisms used by cells, particularly enzymes such as exonuclease 3-5 and exonuclease 5-3
How many different ways are there to code (DNA nucleotide code) for an average human protein? In practice what are some of the
reasons that all of these different codes don’t work to code for the protein of interest?
There are 20 amino acids, which can generate a large number of comabinations. Nevertheless, some factors limit functionality, such as extreme GC content homopolymers, and free energy.
What’s the most commonly used method for oligo synthesis currently?
Currently, the phosphoramidite method is the most commonlty used approach for oligonucleotide synthesis. This technique was introduced by Caarathers in 1981. This chemestry process involves the sequential addition of nucleotides onto a polymer-supported chain.
Why is it difficult to make oligos longer than 200nt via direct synthesis?
The first limitation is yield. In some cases, the error rate can reach approximately 1 in 100. As a result, the yield decreases exponentially with each additional base.
Another important limitation is error accumulation. Chemical synthesis has a higher error rate than synthesis performed by biological polymerases.
Why can’t you make a 2000bp gene via direct oligo synthesis?
Las principales razones se deben a la complejidad de la secuencia de 2000 pb que las regiones poco visibles tienen contenido GC muy alto o bajo. Otro aspecto es el rendimiento ya que el metodo de fosforamidita no funcionaria con eficiencia.
Choose ONE of the following three questions to answer; and please cite AI prompts or paper citations used, if any.
[Using Google & Prof. Church’s slide #4] What are the 10 essential amino acids in all animals
and how does this affect your view of the “Lysine Contingency”?
The 10 essential amino acids are Lysine(K),Leucine(L),Arginine (R), Histidine (H), Methionine(M), Isoleucine (I),Tryptophan (W), Threonine (T), Valine (V), Phenylalanine (F).
The lysine contingency is Lysine Contingency functions as a genetic safaty mechanism. In my view, this idea presents interesting implications. First, desingning an organism that lacks an essential amino acid, as it would be considered biologically fragile from the outset.That is, it creates the illusion of a biological switch; howeve, even in the hypothetical case that it works,the organism would be too fragile under normal conditions, as it would depend entirely on dietary availability, leading to imbalance and massive metabolic constraints.
From a synthetic biology perspective, it is important to incorporate additional safety features such as dependence on non-standard amino acids for engineered organisms.
Assignment (Your HTGAA Website) — DUE BY START OF FEB 10 LECTURE
Assignees for the following sections
MIT/Harvard students
Required
Committed Listeners
Required
Begin personalizing your HTGAA website in in https://edit.htgaa.org/, starting with your homepage —
fill in the template with information about yourself, or remove what’s there and make it your own. Be creative!
As with all assignments in HTGAA, be sure to write up every part of this Homework on your HTGAA website in order to receive credit.
Important
For this week only, once your homework is complete and written up on your HTGAA website (and you’ve checked your published
website at pages.htgaa.org and are happy with it), fill out the Homework 1 Completion form which David emailed out just after
Lecture 1. This Google form expresses your interest in continuing with the course; without it you will not be accepted in HTGAA!
Week 2 HW: DNA Read, Write, & Edit
Part 0: Basics of Gel Electrophoresis
I reviewed the recorded class recitation.
Part 1: Benchling & In-silico Gel Art
See the Gel Art: Restriction Digests and Gel Electrophoresis protocol for details. Overview:
Make a free account at benchling.com
Import the Lambda DNA.
Simulate Restriction Enzyme Digestion with the following Enzymes:
EcoRI
HindIII
BamHI
KpnI
EcoRV
SacI
SalI
Create a pattern/image in the style of Paul Vanouse’s Latent Figure Protocol artworks.
You might find Ronan’s website a helpful tool for quickly iterating on designs!
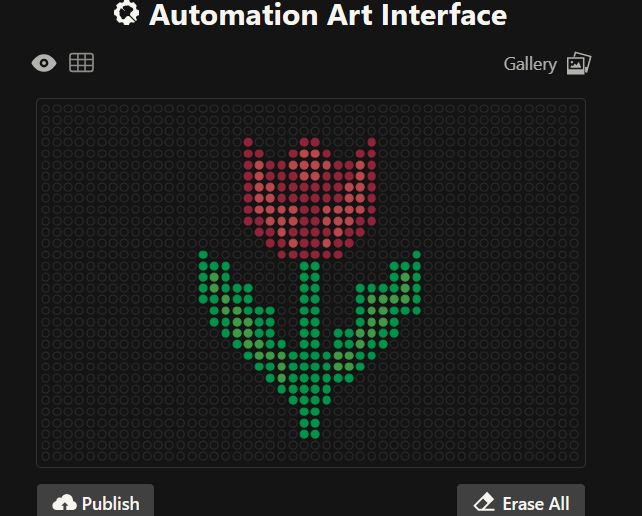
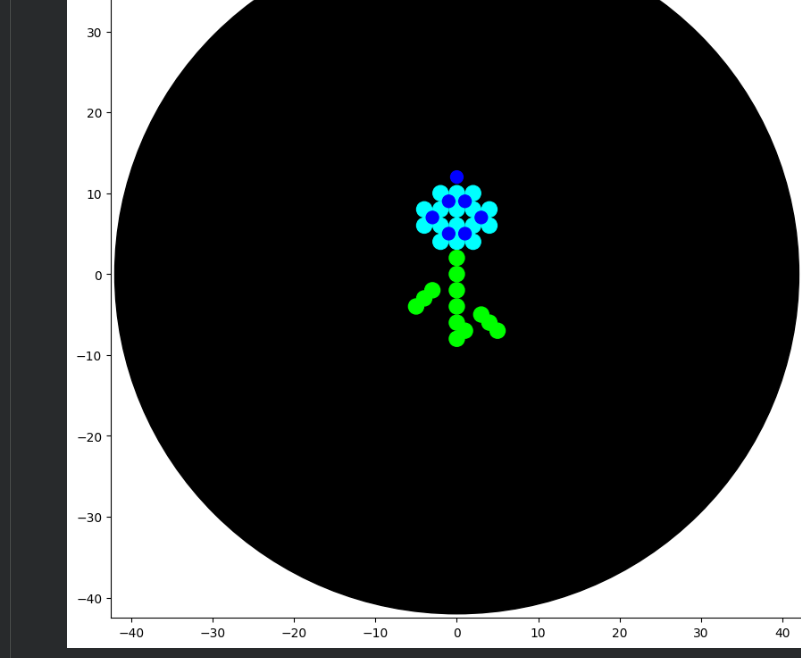
After experimenting with the simulator, I successfully generated three figures using different restriction enzymes.
HEART
CIRCLE
FLOWER
Part 3: DNA Design Challenge
3.1. Choose your protein.
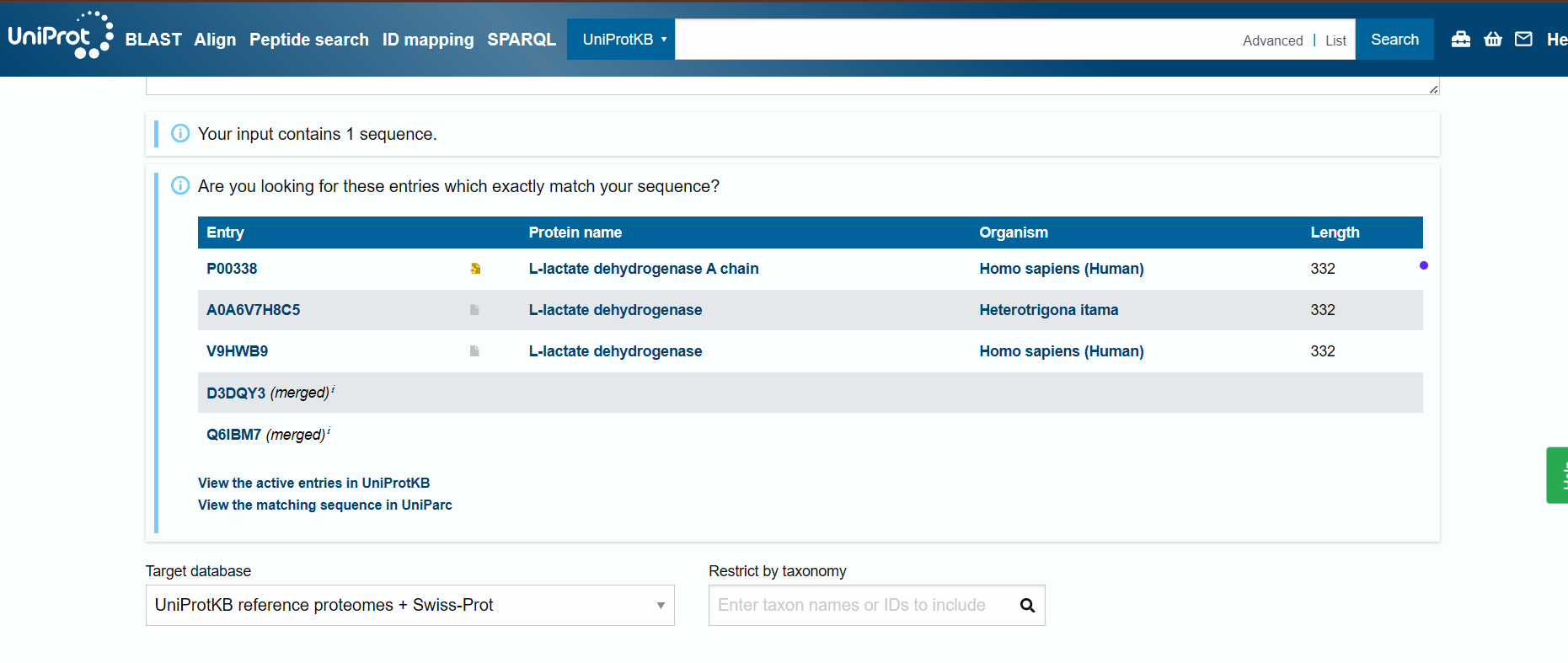
In recitation, we discussed that you will pick a protein for your homework that you find interesting. Which protein have you chosen and why? Using one of the tools described in recitation (NCBI, UniProt, google), obtain the protein sequence for the protein you chose.
P0A7X3
I choose the Small Ribosomal Subunit Protein uS9 (P0A7X3) from Escherichia coli K-12 because it is a well-characterized and fundamental biological role in bacterial translation. Studying this protein is relevant for studies in ribosome engineering structural biology, and synthetic biology applications.
Ribosomal engineering is the targeted redesign of ribosomes to control how genetic information is translated into proteins. It has direct implications for biotechnology, pharmaceutical innovation and industrial bioproduction.
I used Uniprot:
sp|P0A7X3|RS9_ECOLI Small ribosomal subunit protein uS9 OS=Escherichia coli (strain K12) OX=83333 GN=rpsI PE=1 SV=2
MAENQYYGTGRRKSSAARVFIKPGNGKIVINQRSLEQYFGRETARMVVRQPLELVDMVEK
LDLYITVKGGGISGQAGAIRHGITRALMEYDESLRSELRKAGFVTRDARQVERKKVGLRK
ARRRPQFSKR
d’Aquino AE, Kim DS, Jewett MC. Engineered Ribosomes for Basic Science and Synthetic Biology. Annu Rev Chem Biomol Eng. 2018 Jun 7;9:311-340. doi: 10.1146/annurev-chembioeng-060817-084129. Epub 2018 Mar 28. PMID: 29589973.
3.2. Reverse Translate: Protein (amino acid) sequence to DNA (nucleotide) sequence.
The Central Dogma discussed in class and recitation describes the process in which DNA sequence becomes transcribed and translated into protein. The Central Dogma gives us the framework to work backwards from a given protein sequence and infer the DNA sequence that the protein is derived from. Using one of the tools discussed in class, NCBI or online tools (google “reverse translation tools”), determine the nucleotide sequence that corresponds to the protein sequence you chose above.
Central Dogma
The Central Dogma of molecular biology describes the flow of genetic information from DNA to ARNA and ultimately to protein. Given a known protein sequence, once can infer a plausible nucleotide sequence by considering the degeneracy of the genetic code.Because the genetic code is redundant, several diffirent codons can encode the same amino acid.
My sequence
reverse translation of sp|P0A7X3|RS9_ECOLI Small ribosomal subunit protein uS9 OS=Escherichia coli (strain K12) OX=83333 GN=rpsI PE=1 SV=2 to a 390 base sequence of most likely codons.
atggcggaaaaccagtattatggcaccggccgccgcaaaagcagcgcggcgcgcgtgttt
attaaaccgggcaacggcaaaattgtgattaaccagcgcagcctggaacagtattttggc
cgcgaaaccgcgcgcatggtggtgcgccagccgctggaactggtggatatggtggaaaaa
ctggatctgtatattaccgtgaaaggcggcggcattagcggccaggcgggcgcgattcgc
catggcattacccgcgcgctgatggaatatgatgaaagcctgcgcagcgaactgcgcaaa
gcgggctttgtgacccgcgatgcgcgccaggtggaacgcaaaaaagtgggcctgcgcaaa
gcgcgccgccgcccgcagtttagcaaacgc
Once a nucleotide sequence of your protein is determined, you need to codon optimize your sequence. You may, once again, utilize google for a “codon optimization tool”. In your own words, describe why you need to optimize codon usage. Which organism have you chosen to optimize the codon sequence for and why?
ATG GCC GAA AAC CAG TAT TAT GGC ACC GGA CGG AGG AAA AGC TCT GCC GCA CGC GTG TTC ATT AAA CCA GGC AAT GGG AAG ATT GTG ATC AAT CAG AGA TCC TTG GAA CAG TAT TTT GGG CGG GAG ACT GCT AGG ATG GTG GTC AGA CAG CCT CTG GAA CTG GTG GAC ATG GTT GAG AAG CTG GAT CTG TAT ATT ACC GTG AAG GGA GGG GGC ATC TCC GGG CAG GCC GGC GCA ATC CGG CAT GGA ATT ACT CGA GCC CTT ATG GAG TAC GAC GAG TCC CTC CGC AGC GAG CTG AGA AAG GCG GGC TTC GTG ACC AGA GAT GCT CGA CAG GTG GAG AGG AAA AAG GTG GGA CTT CGC AAA GCT AGA AGA AGG CCA CAG TTT AGT AAA CGC
The following restriction enzyme sites have been found in the selected reading frame:
BclI (TGATCA)
MluI (ACGCGT)
MlyI (GAGTC)
NaeI (GCCGGC)
XhoI (CTCGAG)
What technologies could be used to produce this protein from your DNA? Describe in your words the DNA sequence can be transcribed and translated into your protein. You may describe either cell-dependent or cell-free methods, or both.
Describe how a single gene codes for multiple proteins at the transcriptional level.
Try aligning the DNA sequence, the transcribed RNA, and also the resulting translated Protein!!! See example below
a) Cell-free systems to produce RS9
Chemical synthesis
The protein is small (80aa), so it could be chemically synthesized in a specialized laboratory.
In chemical synthesis , Solid -Phase Peptide Synthesis (SPPS) is employed, in which the first amino acid is anchored to resin, subsequent amino acids are added one at a time to create sequential peptide bonds,and the synthesized protein is later detached and purified.
This technique is particulary advantage because it grants full control over the peptide sequence and facilitates the integration of artificial molecular modifications
Cell free transcription system
Conversely, the cell-free transcription strategy requires the prior design of a plasmid harboring a strong promoter, an optimized RBS and rpsl gene, which is subsequently added to the cell-free system to anable mRNA production.
Althought this system allows for fast protein production and rapid experimental turnaround,its implementation can be economically demanding.
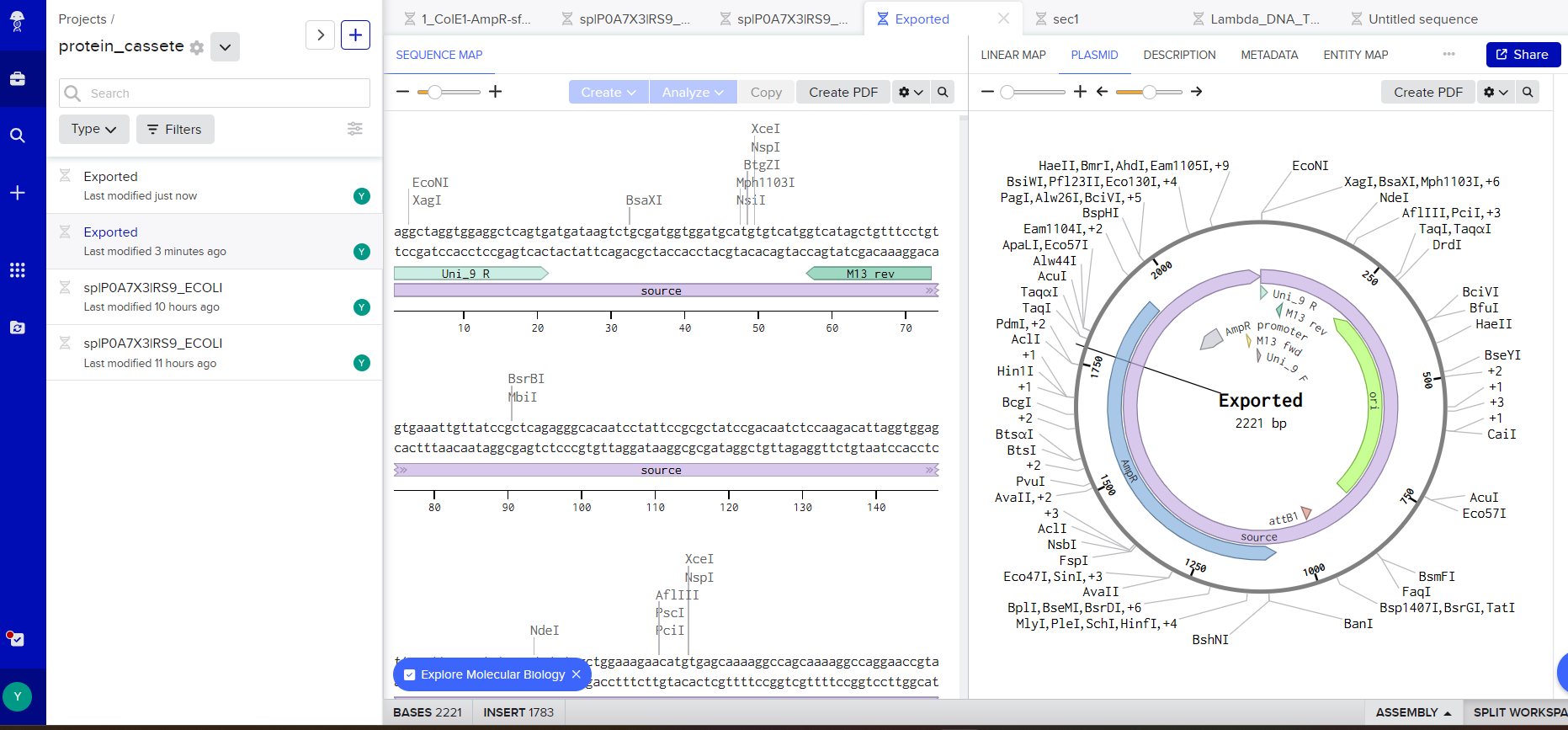
During the class, I learned how to design the plasmid to complete the exercise.
After completing the tutorial, I understood the full process required to place a DNA synthesis order at Twist Bioscience.
As it was my first time making a vector, with the help of ChatGPT and DeepSeek I was able to create these pieces.
I used the same basic structure to represent my synthetic circuit with the Rs9 protein.
The most complicated part was fitting my sequence with the vector.
but in the end I chose this vector: pTwist Amp High Copy - (2221bp) and I got to this part:
Part 5: DNA Read/Write/Edit
5.1 DNA Read
(i) What DNA would you want to sequence (e.g., read) and why? This could be DNA related to human health (e.g. genes related to disease research), environmental monitoring (e.g., sewage waste water, biodiversity analysis), and beyond (e.g. DNA data storage, biobank).
I want to sequence the DNA of South American camelids in Peru, such as alpacas, vicuñas, huanacos and llamas with the aim of identiying genes that confer resistance to extreme cold events (known as friajes).
Knowledge of these sequences, it would be possible to identify high-value biactive molecules, such as nanobodies or inmune-related proteins, antimicrobial peptides, for development of therapeutic strateigies and vaccine design, especifically for diseases affecting the respiratory system.
(ii) In lecture, a variety of sequencing technologies were mentioned. What technology or technologies would you use to perform sequencing on your DNA and why?
Also answer the following questions:
Is your method first-, second- or third-generation or other? How so?
What is your input? How do you prepare your input (e.g. fragmentation, adapter ligation, PCR)? List the essential steps.
What are the essential steps of your chosen sequencing technology, how does it decode the bases of your DNA sample (base calling)?
What is the output of your chosen sequencing technology?
To sequence the DNA of South American camelid and study genes involved in cold resistance, I would use a hybrid sequencing strategy is employed because cold-adaptation genes include regulatory regions and gene duplications and mammalian genomes are large and repetitive.
Oxford Nanopore
PacBio
Ilumina
*third-generation
*third-generation
*second-generation
Nanopore sequencing reads DNA molecules in real time
Sequencing reads individual DNA molecules without PCR
Technology based on DNA amplification and synthesis
function: In Oxford Nanopore sequencing, DNA passes through a pore and detecting electrical signals
function: Sequencing uses a polymerase that copies DNA while emitting fluorescent signals.
function: Uses sequential incorporation of fluorescent nucleotides, generating images in cycles with high accuracy.
ChatGPT was used with the following prompts to assist in data organization and text refinement varuety of sequencing technologies such as Oxford Nanopore, PacBio, and Ilumina.
SPPS adicional: Peptide synthesis: a review of classical and emerging methods (Zhangping Cai et al., 2025).
Cell-free adicional: Cell-Free Protein Synthesis: Pros and Cons of Prokaryotic and Eukaryotic Systems (Anne Zemella et al., 2015).
5.2 DNA Write
(i) What DNA would you want to synthesize (e.g., write) and why? These could be individual genes, clusters of genes or genetic circuits, whole genomes, and beyond. As described in class thus far, applications could range from therapeutics and drug discovery (e.g., mRNA vaccines and therapies) to novel biomaterials (e.g. structural proteins), to sensors (e.g., genetic circuits for sensing and responding to inflammation, environmental stimuli, etc.), to art (DNA origamis). If possible, include the specific genetic sequence(s) of what you would like to synthesize! You will have the opportunity to actually have Twist synthesize these DNA constructs! :)
I want to synthesize a modular synthetic cold-adaptation operon optimized for E.coli. The construct would include: The cold-shock gene cspA, an antifreeze protein (AFP) coding sequence and the trehalose biosynthesis genes otsA and otsB enhance osmoprotection, a strong double terminator
These genes would be assembled under a cold-inducible promoter to create a synthetic operon capable of enhancing cellular survival and function at low temperatures. Cold environments impose multiple molecular stresses, including RNA secondary structure stabilization, reduced enzymatic kinetics, membrane rigity.
(ii) What technology or technologies would you use to perform this DNA synthesis and why?
Also answer the following questions:
What are the essential steps of your chosen sequencing methods?
What are the limitations of your sequencing method (if any) in terms of speed, accuracy, scalability?
a) Phosphoramidite solid-phase DNA synthesis for oligonucleotides
This standard method for producing short oligonucleotides (20-200 nucleotides). DNA is synthesized base by base on a solid support through repeatec cycles of deprotection,coupling, oxidation and washing. While this technique provides precise control over sequence composition and enables chemical modifications.
b) Gibson Assembly to assemble fragments
Gibson Assembly is an efficient DNA assembly method that allowas multiple fragments ti joined simultaneously in a single-tube, isothermal reaction. It uses a combination of exonuclease, DNA polymerase, and DNA ligase to join fragments that share overlapping homologous regios.
c) Commercial gene synthesis services in Twist Bioscience
These synthesis services, such as those offered by Twist Bioscience provide end-to-end solutios for gene construcction, and sequencing based validation. Also provide sequence optimization options, such as codon optimization and restriction site removal, reducing experimental workload while increasing reliabity
Limitations
Detection sensitivity threshold: There is an inherent limit to the sensitivity of error deteccion during queality control.
False positives due:Errors introduced during PCR amplification, sequencing, or DNA assembly can be incorrectly interpreted as true mutations.
Lenght-dependent error rates : Making long sequences more prone to insertions, deletions, and substitutions and necessitating assembly from shorter fragments.
GC-rich sequence limitations: GC-rich regions reduce synthesis ans assembly efficency due to the formation of stable secondary structures, often requiring sequence redesing.
Cost constraints: Gene synthesis costs increase with sequence length, GC content, and desing complexity, as longer and more complex genes require additional synthesis, assembly, and quality steps.
5.3 DNA Edit
(i) What DNA would you want to edit and why? In class, George shared a variety of ways to edit the genes and genomes of humans and other organisms. Such DNA editing technologies have profound implications for human health, development, and even human longevity and human augmentation. DNA editing is also already commonly leveraged for flora and fauna, for example in nature conservation efforts, (animal/plant restoration, de-extinction), or in agriculture (e.g. plant breeding, nitrogen fixation). What kinds of edits might you want to make to DNA (e.g., human genomes and beyond) and why?
I propose editing genes implicated in maize abiotic stress responses, with a focus on drought and elevanted temperature tolerance. These genetic modifications would be direted toward regulatory genes and molecular pathways responsible for efficient water use, osmotic regulation, and oxidative strees defense. It represents a potential response to climate change, which is increasing the frequency of extreme conditions that reduce agricultural yields in high-Andean regions.
(ii) What technology or technologies would you use to perform these DNA edits and why?
Also answer the following questions:
How does your technology of choice edit DNA? What are the essential steps?
What preparation do you need to do (e.g. design steps) and what is the input (e.g. DNA template, enzymes, plasmids, primers, guides, cells) for the editing?
What are the limitations of your editing methods (if any) in terms of efficiency or precision?
I would employ CRISPR-CAS9 in combination with base editing or prime editing to achieve precise genome modifications without the insertion of exogenous DNA. The editing process include candidate gene identification, the desing of CRISPR guides, delivery of editing system into maize cells, and the regeneration of edited plants.
The main limitations of this approach are varible editing efficiency and the genetic complex genetic architecture underlying these traits.
REFERENCES:
Molla, K. A., Sretenovic, S., Bansal, K. C., & Qi, Y. (2021). Precise plant genome editing using base editors and prime editors. Nature Plants, 7, 1166–1187. https://doi.org/10.1021/acsagscitech.2c00090
The following prompts were employed in ChatGPT to organize and refine the information:
“Explain Gibson Assembly step by step, highlighting the roles of exonuclease, DNA polymerase, and ligase, as well as common sources of assembly errors.”
“Compare Gibson Assembly with restriction enzyme cloning for synthetic gene construction, including advantages, limitations, and typical use cases.
Explain how CRISPR-Cas9 combined with base editing or prime editing enables precise genome modifications without introducing exogenous DNA.”
Discuss the main technical limitations of CRISPR-based genome editing in plants, including delivery, efficiency, and polygenic traits.
“Identify and explain categories of genes involved in abiotic stress tolerance in maize, including water-use efficiency, osmotic balance, and oxidative stress pathways.
Week 3 HW: Lab Automation
Assignment: Python Script for Opentrons Artwork — DUE BY YOUR LAB TIME!
Your task this week is to Create a Python file to run on an Opentrons liquid handling robot.
Post-Lab Questions — DUE BY START OF FEB 24 LECTURE
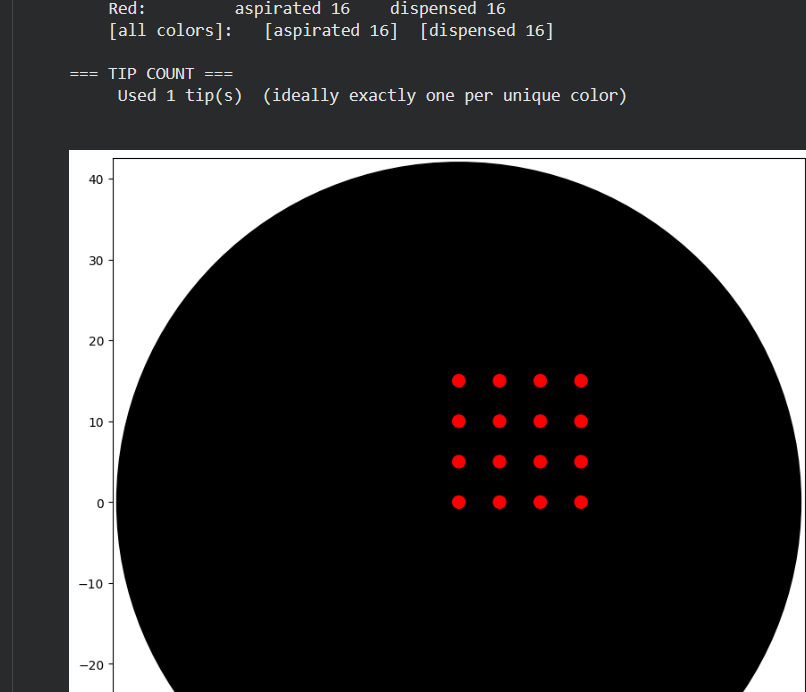
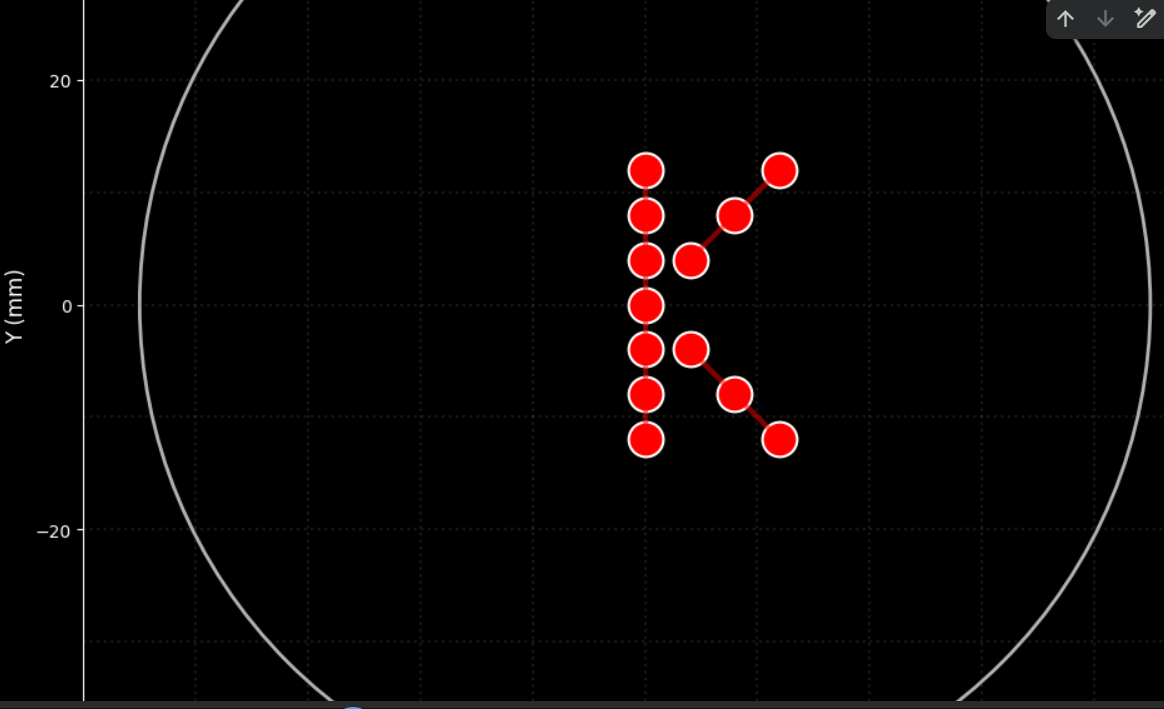
To create the design, I employed several tests:
Part 1.
I initially performed some tests by modifying the color in the first example provided in Google Colab.
class OpentronsMock:
def init(self, well_colors):
self.well_colors = well_colors
self.dots = [] # List to store drawn points
self.current_position = (0, 0, 0)
self.tip_attached = False
self.labware = {} # Dictionary to store labware
self.modules = {} # Dictionary to store modules
self.instruments = {} # Dictionary to store instruments
def load_labware(self, name, slot, label=None):
labware = MockLabware(name, slot, label, self)
self.labware[slot] = labware
return labware
def load_instrument(self, name, mount, tip_racks):
instrument = MockPipette(name, mount, tip_racks, self)
self.instruments[mount] = instrument
return instrument
def load_module(self, name, slot):
module = MockModule(name, slot, self)
self.modules[slot] = module
return module
def visualize(self):
"""Visualize the points drawn on the agar plate with black background"""
# Configure style with black background
plt.style.use('dark_background')
fig, ax = plt.subplots(figsize=(10, 10), facecolor='black')
ax.set_facecolor('black')
# Draw the agar plate outline (90mm diameter circle)
circle = plt.Circle((0, 0), 45, fill=False, color='white', linewidth=2, alpha=0.7)
ax.add_patch(circle)
# Draw a faint grid for reference
for i in range(-40, 41, 10):
ax.axhline(y=i, color='gray', linestyle=':', alpha=0.2)
ax.axvline(x=i, color='gray', linestyle=':', alpha=0.2)
# Draw the plate center
ax.plot(0, 0, 'w+', markersize=10, linewidth=2, label='Center', alpha=0.7)
# Draw the points
if self.dots:
x_coords = [dot[0] for dot in self.dots]
y_coords = [dot[1] for dot in self.dots]
colors = [dot[2] for dot in self.dots]
# Color mapping (adjusted for black background)
color_map = {'Red': 'red', 'Yellow': 'yellow', 'Green': 'lime',
'Cyan': 'cyan', 'Blue': 'dodgerblue'}
for x, y, color in zip(x_coords, y_coords, colors):
ax.plot(x, y, 'o', color=color_map.get(color, 'white'),
markersize=20, markeredgecolor='white', markeredgewidth=1.5)
# Connect the points to show the K shape
if len(self.dots) >= 13: # If we have all points
# Sort points to draw the K lines
# Vertical points (sorted by y)
vertical = sorted([(x, y) for x, y, _ in self.dots if x == 0], key=lambda p: p[1])
if len(vertical) >= 7:
v_x, v_y = zip(*vertical)
ax.plot(v_x, v_y, 'red', alpha=0.5, linewidth=3, linestyle='-')
# Lower diagonal points
diagonal_lower = [(x, y) for x, y, _ in self.dots if x > 0 and y < 0]
if diagonal_lower:
d_x, d_y = zip(*sorted(diagonal_lower, key=lambda p: p[0]))
ax.plot(d_x, d_y, 'red', alpha=0.5, linewidth=3, linestyle='-')
# Upper diagonal points
diagonal_upper = [(x, y) for x, y, _ in self.dots if x > 0 and y > 0]
if diagonal_upper:
d_x, d_y = zip(*sorted(diagonal_upper, key=lambda p: p[0]))
ax.plot(d_x, d_y, 'red', alpha=0.5, linewidth=3, linestyle='-')
# Configure the plot
ax.set_xlim(-50, 50)
ax.set_ylim(-50, 50)
ax.set_aspect('equal')
# Customize axes for black background
ax.spines['bottom'].set_color('white')
ax.spines['top'].set_color('white')
ax.spines['left'].set_color('white')
ax.spines['right'].set_color('white')
ax.tick_params(colors='white')
ax.xaxis.label.set_color('white')
ax.yaxis.label.set_color('white')
ax.title.set_color('white')
ax.set_title('Agar Plate Visualization - Letter K in Red',
fontsize=16, fontweight='bold', color='white')
ax.set_xlabel('X (mm)', fontsize=12, color='white')
ax.set_ylabel('Y (mm)', fontsize=12, color='white')
# Add legend with light colors
from matplotlib.patches import Patch
legend_elements = [
Patch(facecolor='red', edgecolor='white', label='Red drops'),
plt.Line2D([0], [0], color='red', alpha=0.5, linewidth=3, label='K connections'),
plt.Line2D([0], [0], marker='+', color='white', linestyle='None',
markersize=10, markeredgewidth=2, label='Center')
]
legend = ax.legend(handles=legend_elements, loc='upper right', facecolor='black',
edgecolor='white', labelcolor='white')
for text in legend.get_texts():
text.set_color('white')
plt.tight_layout()
plt.show()
# Print information
print(f"\n{'='*50}")
print("📊 EXPERIMENT SUMMARY")
print(f"{'='*50}")
print(f"🎨 Color used: Red")
print(f"🔴 Total drops deposited: {len(self.dots)}")
print(f"📏 Shape: Letter K")
print(f"📐 Drop spacing: 4 mm")
print(f"📏 Dimensions: 24 mm high x 12 mm wide")
# Show point coordinates
print(f"\n📍 POINT COORDINATES (x, y) in mm:")
print(f"{'-'*40}")
for i, (x, y, color) in enumerate(self.dots, 1):
print(f" Point {i:2d}: ({x:+5.1f}, {y:+5.1f})")
# Create simulated wells
if 'agar_plate' in name:
# For agar plate, we only need A1
self.wells_dict['A1'] = MockWell('A1', slot, mock_env)
else:
# For other plates, create some wells
for well_name in ['A1', 'B1', 'C1', 'D1', 'E1']:
self.wells_dict[well_name] = MockWell(well_name, slot, mock_env)
def wells(self):
return list(self.wells_dict.values())
def __getitem__(self, well_name):
"""Make the object subscriptable"""
if well_name in self.wells_dict:
return self.wells_dict[well_name]
else:
# If it doesn't exist, create a new one
self.wells_dict[well_name] = MockWell(well_name, self.slot, self.mock_env)
return self.wells_dict[well_name]
class MockWell:
def init(self, name, slot, mock_env):
self.name = name
self.slot = slot
self.mock_env = mock_env
class MockPipette:
def init(self, name, mount, tip_racks, mock_env):
self.name = name
self.mount = mount
self.tip_racks = tip_racks
self.mock_env = mock_env
self.current_volume = 0
def pick_up_tip(self):
self.mock_env.tip_attached = True
print("🔄 Tip picked up")
def drop_tip(self):
self.mock_env.tip_attached = False
print("🔄 Tip discarded")
def aspirate(self, volume, location):
self.current_volume = volume
if hasattr(location, 'name'):
print(f"💧 Aspirating {volume}uL from {location.name}")
else:
print(f"💧 Aspirating {volume}uL")
def dispense(self, volume, location):
self.current_volume -= volume
# Register the point for visualization
if hasattr(location, 'point'):
self.mock_env.dots.append((location.point.x, location.point.y, 'Red'))
print(f"💧 Dispensing {volume}uL at ({location.point.x:+5.1f}, {location.point.y:+5.1f})")
def move_to(self, location):
if hasattr(location, 'point'):
self.mock_env.current_position = (location.point.x, location.point.y, location.point.z)
def run(protocol):
##############################################################################
### Load labware, modules and pipettes
##############################################################################
# Tips
tips_20ul = protocol.load_labware('opentrons_96_tiprack_20ul', TIP_RACK_DECK_SLOT, 'Opentrons 20uL Tips')
# Pipettes
pipette_20ul = protocol.load_instrument("p20_single_gen2", "right", [tips_20ul])
# Modules
temperature_module = protocol.load_module('temperature module gen2', COLORS_DECK_SLOT)
# Temperature Module Plate
temperature_plate = temperature_module.load_labware('opentrons_96_aluminumblock_generic_pcr_strip_200ul',
'Cold Plate')
# Choose where to take the colors from
color_plate = temperature_plate
# Agar Plate
agar_plate = protocol.load_labware('htgaa_agar_plate', AGAR_DECK_SLOT, 'Agar Plate')
# Get the top-center of the plate
center_location = agar_plate['A1'].top()
##############################################################################
### Patterning
##############################################################################
###
### Helper functions for this lab
###
def location_of_color(color_string):
for well, color in well_colors.items():
if color.lower() == color_string.lower():
return color_plate[well].top()
raise ValueError(f"No well found with color {color_string}")
def dispense_and_detach(pipette, volume, location):
"""
Move laterally 5mm above the plate; then drop down to the plate,
dispense, move back up 5mm to detach drop.
"""
assert(isinstance(volume, (int, float)))
if hasattr(location, 'point'):
above_location = location.move(types.Point(z=location.point.z + 5))
pipette.move_to(above_location)
pipette.dispense(volume, location)
pipette.move_to(above_location)
###
### YOUR CODE HERE to create your design
###
###
### Code to create the letter K in red
###
print("\n" + "="*50)
print("🚀 STARTING PROTOCOL: Letter K in Red")
print("="*50 + "\n")
# Aspirate red color
pipette_20ul.pick_up_tip()
# Calculate required volume (13 points x 1uL = 13uL)
pipette_20ul.aspirate(13, location_of_color('Red'))
# Define points to form the letter K
# Letter size (spacing between points in mm)
spacing = 4
# Points for the vertical line (7 points)
vertical_points = []
for i in range(-3, 4):
vertical_points.append((0, i * spacing))
# Points for the upper diagonal (3 points)
diagonal_upper = []
for i in range(1, 4):
diagonal_upper.append((i * spacing, i * spacing))
# Points for the lower diagonal (3 points)
diagonal_lower = []
for i in range(1, 4):
diagonal_lower.append((i * spacing, -i * spacing))
print("🎨 Drawing letter K...")
# Draw the vertical line
print("\n📏 Vertical line (7 points):")
print(" " + "─" * 35)
for i, (x, y) in enumerate(vertical_points):
adjusted_location = center_location.move(types.Point(x=x, y=y))
dispense_and_detach(pipette_20ul, 1, adjusted_location)
print(f" Point {i+1:2d}: ({x:+5.1f}, {y:+5.1f})")
# Draw the lower diagonal
print("\n📐 Lower diagonal (3 points):")
print(" " + "─" * 35)
for i, (x, y) in enumerate(diagonal_lower):
adjusted_location = center_location.move(types.Point(x=x, y=y))
dispense_and_detach(pipette_20ul, 1, adjusted_location)
print(f" Point {i+1:2d}: ({x:+5.1f}, {y:+5.1f})")
# Draw the upper diagonal
print("\n📐 Upper diagonal (3 points):")
print(" " + "─" * 35)
for i, (x, y) in enumerate(diagonal_upper):
adjusted_location = center_location.move(types.Point(x=x, y=y))
dispense_and_detach(pipette_20ul, 1, adjusted_location)
print(f" Point {i+1:2d}: ({x:+5.1f}, {y:+5.1f})")
# Clean up
pipette_20ul.drop_tip()
print("\n" + "="*50)
print("✅ PROTOCOL COMPLETED!")
print("="*50)
print(f"🎯 Total drops: {len(vertical_points) + len(diagonal_lower) + len(diagonal_upper)}")
print(f"🎨 Color: Red")
print(f"📏 Shape: Letter 'K'")
Post-Lab Questions — DUE BY START OF FEB 24 LECTURE
One of the great parts about having an automated robot is being able to precisely mix, deposit, and run reactions without much intervention, and design and deploy experiments remotely.
For this week, we’d like for you to do the following:
Find and describe a published paper that utilizes the Opentrons or an automation tool to achieve novel biological applications.
Write a description about what you intend to do with automation tools for your final project. You may include example pseudocode, Python scripts, 3D printed holders, a plan for how to use Ginkgo Nebula, and more. You may reference this week’s recitation slide deck for lab automation details.
The article “Automated High-Throughtput Flow Cytometry for High-Content”
Describe the development of a fully integrated automated designed to enhance antibody discovery thorugh high-throughput cytometric analysis.
In this study, the researchers integrated robotic liquid handling with automated flow cytometry. The platform automated sample preparation, dilution steps, incubation procedures, and data acquisition.
Automated Tasks
Plate Initialization
Dispense 100 µL of cell suspension into each well of a 96-well plate.
Ensure uniform cell density (e.g., 1 × 10⁵ cells per well).
Serial Dilution of Antibodies
Perform 1:3 or 1:5 serial dilutions across designated wells.
Transfer precise microliter volumes (e.g., 20 µL) to generate standardized concentration gradients.
Antibody Incubation Setup
Add diluted antibody solutions to corresponding wells.
Mix gently using programmed pipette mixing cycles.
Incubate for a defined period (e.g., 30 minutes at room temperature).
Wash Steps
Aspirate supernatant without disturbing the cell pellet.
This application focuses on the high-throughput screening of single-domain antibodies (VHH nanobodies) derived from South American camelids, such as llamas and alpacas. These antibodies are smaller and structurally simpler than conventional IgG molecules, making them highly suitable for recombinant expression and screening workflows. Using Opentrons automation, the robot can perform serial dilutions of VHH candidates, dispense them into 96-well plates containing target cells expressing the antigen of interest, and execute standardized wash and incubation steps prior to flow cytometry analysis. Because nanobody screening requires precise concentration gradients to evaluate binding affinity, automated liquid handling significantly improves reproducibility and quantitative reliability.
REFERENCES:
The table was generated with the assistance of ChatGPT (OpenAI) to summarize the key advantages described in the referenced article.
Wang Y, Yoshihara T, King S, Le T, Leroy P, Zhao X, Chan CK, Yan ZH, Menon S. Automated High-Throughput Flow Cytometry for High-Content Screening in Antibody Development. SLAS Discov. 2018 Aug;23(7):656-666. doi: 10.1177/2472555218776607. Epub 2018 Jun 13. PMID: 29898633.
Week 4 HW:Protein design PART I
Part A. Conceptual Questions
Answer any NINE of the following questions from Shuguang Zhang: (i.e. you can select two to skip)
Why do humans eat beef but do not become a cow, eat fish but do not become fish?
Humans do not transform into fish or cws because dietary components are not assimilated as intact complex structures. When we ingest food, it is processed through a mechanism called digestion, which involves the enzymatic breakdown of macromolecules into their most basic units. These molecules are then used as raw materials to synthesize biomolecules acording to the instructions encoded in human DNA.
Why are there only 20 natural amino acids?
The 20 amino acids constitute an almost ideal chemical spectrum that maximizes structural diversity with minimal redundancy. The ibosomal translation machinery efficiently harnesses this chemical diversity, nabling the formation of secondary and tertiary structures, hydrophobic interactions, disulfide bridges, and enzymatic catalysis.
Can you make other non-natural amino acids? Design some new amino acids.
Currently, amino acids can be engineered through two main approaches. Chemical synthesis allows modification of the R side chain,he carboxyl group, and even the chirality (L or D configuration).
In contrast, genetic code expansion techniques allow the reassignment of a stop codon, coupled with the introduction of an orthogonal tRNA and an engineered aminoacyl-tRNA synthetase, thereby enabling the site-specific incorporation of noncanonical amino acids into proteins.
Where did amino acids come from before enzymes that make them, and before life started?
Amino acids originated through prebiotic chemical processes on early Earth or possibly even in outer space.The Miller–Urey experiment showed that lightning-like electrical discharges in a reducing atmosphere can produce amino acids.Furthermore, the detection of amino acids in meteorites supports the hypothesis of a partially exogenous origin for these biomolecules.
If you make an α-helix using D-amino acids, what handedness (right or left) would you expect?
The α-helix formed by L-amino acids is right-handed due to the conformational constraints imposed by the chirality of the α-carbon. In contrast, a polypeptide consisting solely of D-amino acids inverts these geometric constraints, leading to the formation of a left-handed α-helix.
Can you discover additional helices in proteins?
Helical conformations such as the 3₁₀ helix and the π-helix are defined by distinct hydrogen-bonding patterns and periodicities.The planar nature of the peptide bond, together with backbone dihedral angle restrictions defined by the Ramachandran plot, limits the range of energetically accessible helical conformations.However, through protein engineering and the incorporation of non-natural amino acids, it is possible to explore novel helical conformations.
Why are most molecular helices right-handed?
The predominance of right-handed helices in biological systems arises from their construction from L-amino acids and D-sugars.The chirality of these molecules creates spatial restrictions that make right-handed helices more stable and energetically favorable.
Why do β-sheets tend to aggregate?
What is the driving force for β-sheet aggregation?
β-sheets tend to aggregate because their extended structure enables the formation of extensive intermolecular hydrogen-bonding networks and altered exposure of hydrophobic residues, which promotes hydrophobic interactions between polypeptide chains.
Why do many amyloid diseases form β-sheets?
Can you use amyloid β-sheets as materials?
Amyloid diseases involve the conversion of partially denatured proteins into an extended β-sheet conformation, which enables the formation of highly stable cross-β structures through cooperative hydrogen-bonding networks and hydrophobic interactions.s. Remarkably, this fibrillar organization imparts outstanding mechanical rigidity and thermal stability, driving its investigation as a self-assembling biomaterial platform for nanotechnology and tissue engineering.
Part B: Protein Analysis and Visualization
In this part of the homework, you will be using online resources and 3D visualization software to answer questions about proteins. Pick any protein (from any organism) of your interest that has a 3D structure and answer the following questions:
Briefly describe the protein you selected and why you selected it.
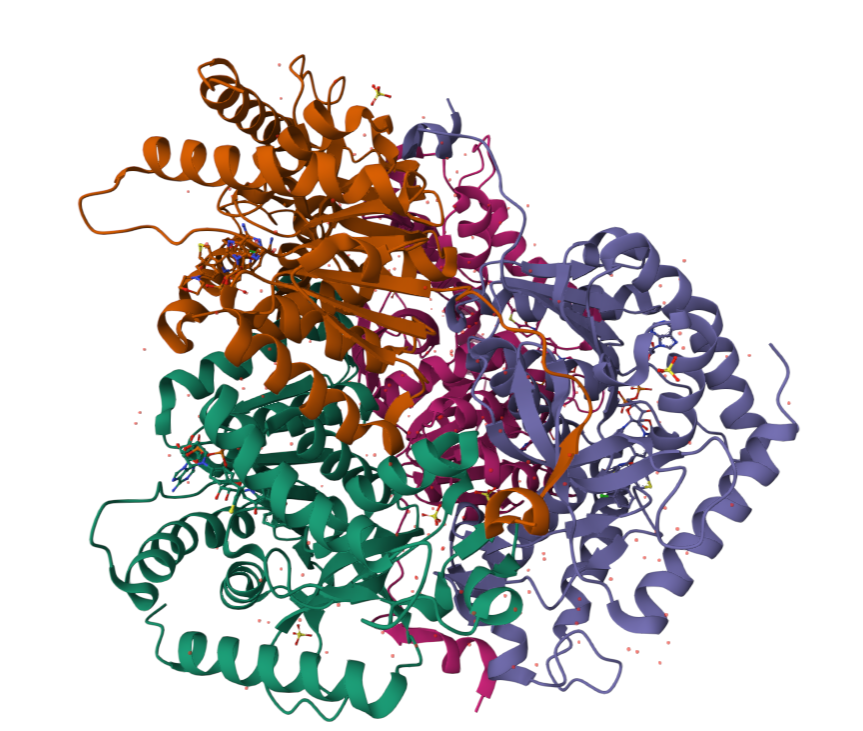
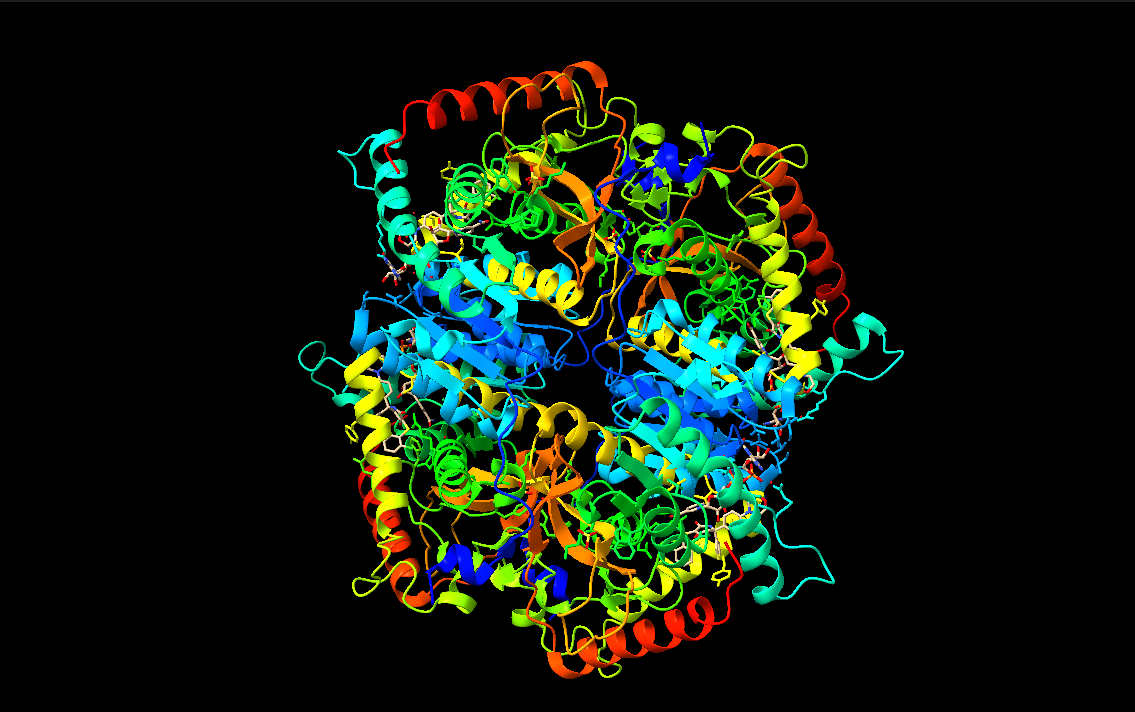
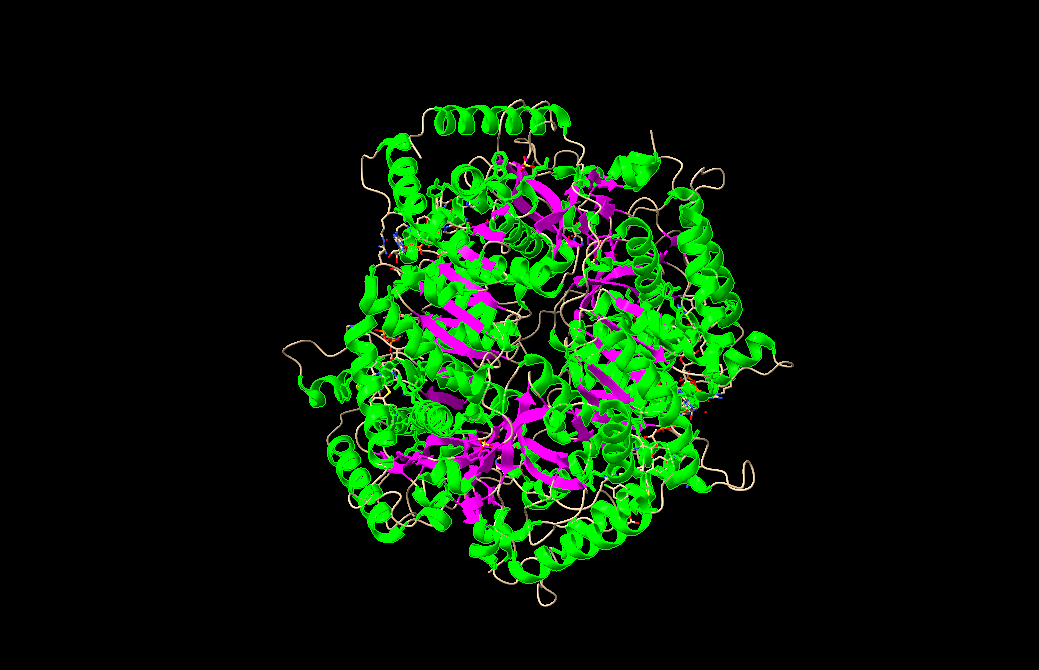
I chose lactate dehydrogenase because it is a fundamental enzyme in energy metabolism, particularly under anaerobic conditions. Its role in the conversion of pyruvate to lactate makes it biologically significant, and its tetrameric structure with a well-defined active site allows for a clear analysis of the relationship between three-dimensional structure and catalytic function. It is an excellent model for studying key concepts in biochemistry and enzymatic catalysis.
When was the structure solved? Is it a good quality structure? Good quality structure is the one with good resolution. Smaller the better (Resolution: 2.70 Å)
Are there any other molecules in the solved structure apart from protein?
Does your protein belong to any structure classification family?
Open the structure of your protein in any 3D molecule visualization software:
PyMol Tutorial Here (hint: ChatGPT is good at PyMol commands)
Visualize the protein as “cartoon”, “ribbon” and “ball and stick”.
Color the protein by secondary structure. Does it have more helices or sheets?
Color the protein by residue type. What can you tell about the distribution of hydrophobic vs hydrophilic residues?
Visualize the surface of the protein. Does it have any “holes” (aka binding pockets)?
Part C. Using ML-Based Protein Design Tools
In this section, we will learn about the capabilities of modern protein AI models and test some of them in your chosen protein.
Copy the HTGAA_ProteinDesign2026.ipynb notebook and set up a colab instance with GPU.
Choose your favorite protein from the PDB.
We will now try multiple things in the three sections below; report each of these results in your homework writeup on your HTGAA website:
PROTEIN SECUENCE:
MATLKDQLIYNLLKEEQTPQNKITVVGVGAVGMACAISILMKDLADELALVDVIEDKLKG
EMMDLQHGSLFLRTPKIVSGKDYNVTANSKLVIITAGARQQEGESRLNLVQRNVNIFKFI
IPNVVKYSPNCKLLIVSNPVDILTYVAWKISGFPKNRVIGSGCNLDSARFRYLMGERLGV
HPLSCHGWVLGEHGDSSVPVWSGMNVAGVSLKTLHPDLGTDKDKEQWKEVHKQVVESAYE
VIKLKGYTSWAIGLSVADLAESIMKNLRRVHPVSTMIKGLYGIKDDVFLSVPCILGQNGI
SDLVKVTLTSEEEARLKKSADTLWGIQKELQF
C1. Protein Language Modeling
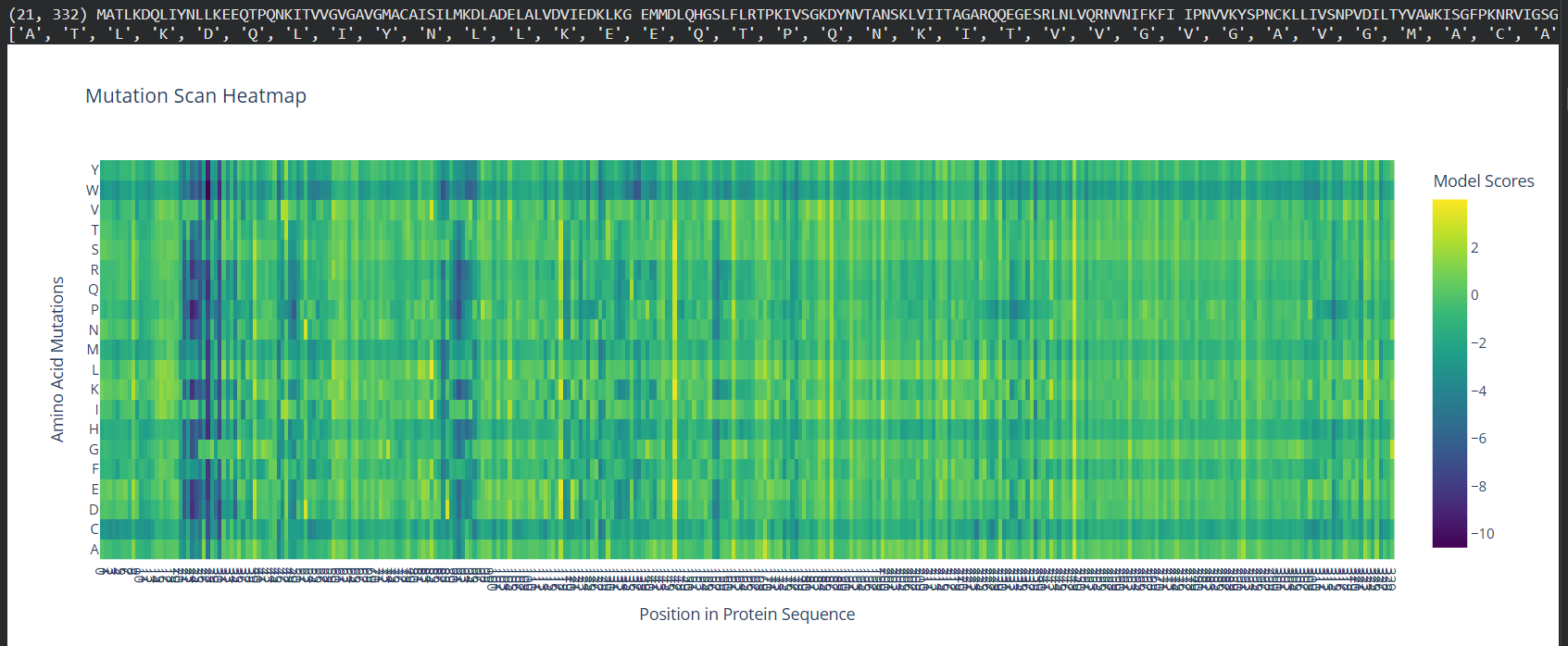
Deep Mutational Scans
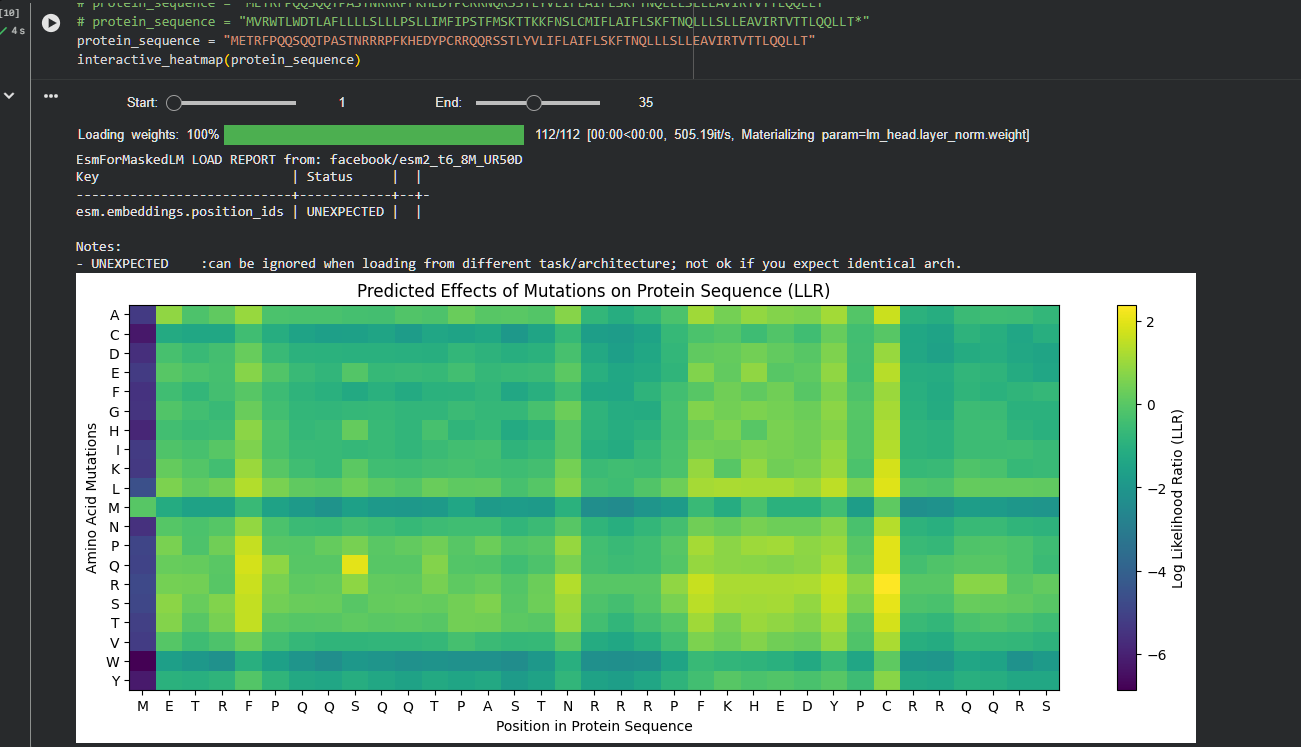
Use ESM2 to generate an unsupervised deep mutational scan of your protein based on language model likelihoods.
Can you explain any particular pattern? (choose a residue and a mutation that stands out)
(Bonus) Find sequences for which we have experimental scans, and compare the prediction of the language model to experiment.
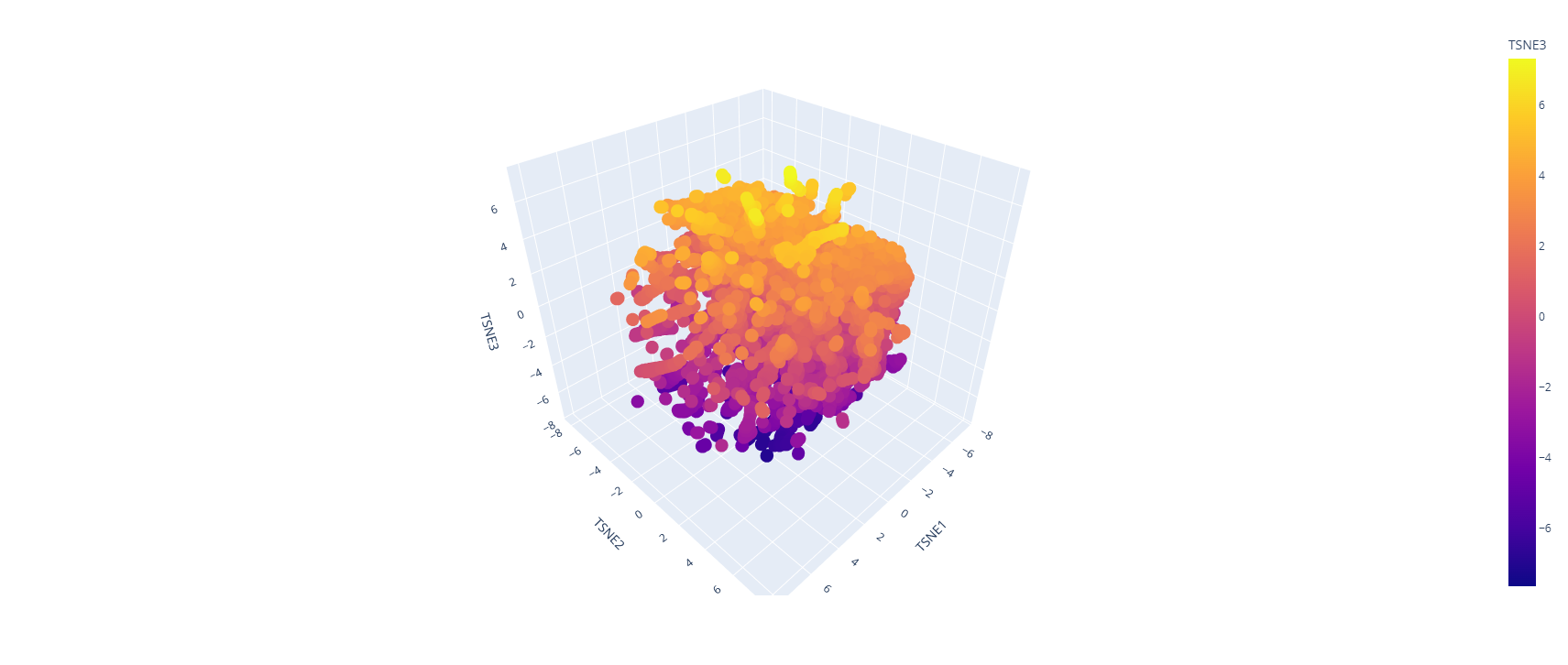
Latent Space Analysis
Use the provided sequence dataset to embed proteins in reduced dimensionality.
Analyze the different formed neighborhoods: do they approximate similar proteins?
Place your protein in the resulting map and explain its position and similarity to its neighbors.
C2. Protein Folding
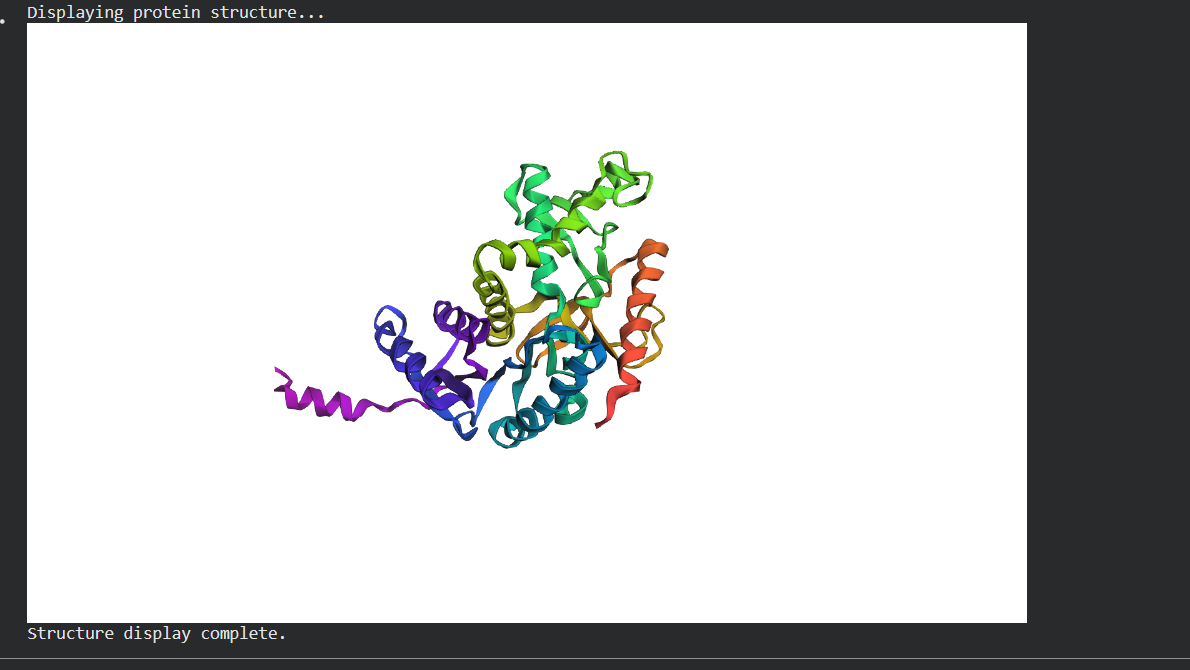
Folding a protein
Fold your protein with ESMFold. Do the predicted coordinates match your original structure?
Try changing the sequence, first try some mutations, then large segments. Is your protein structure resilient to mutations?
C3. Protein Generation
Inverse-Folding a protein: Let’s now use the backbone of your chosen PDB to propose sequence candidates via ProteinMPNN
Analyze the predicted sequence probabilities and compare the predicted sequence vs the original one.
Input this sequence into ESMFold and compare the predicted structure to your original.
Part D. Group Brainstorm on Bacteriophage Engineering
Find a group of ~3–4 students
Read through the Phage Reading material listed under “Reading & Resources” below.
Review the Bacteriophage Final Project Goals for engineering the L Protein:
Increased stability (easiest)
Higher titers (medium)
Higher toxicity of lysis protein (hard)
Brainstorm Session
Choose one or two main goals from the list that you think you can address computationally (e.g., “We’ll try to stabilize the lysis protein,” or “We’ll attempt to disrupt its interaction with E. coli DnaJ.”).
Write a 1-page proposal (bullet points or short paragraphs) describing:
Which tools/approaches from recitation you propose using (e.g., “Use Protein Language Models to do in silico mutagenesis, then AlphaFold-Multimer to check complexes.”).
Why do you think those tools might help solve your chosen sub-problem?
Name one or two potential pitfalls (e.g., “We lack enough training data on phage–bacteria interactions.”).
Include a schematic of your pipeline.
This resource may be useful: HTGAA Protein Engineering Tools
Each individually put your plan on your HTGAA website
Include your group’s short plan for engineering a bacteriophage
Superoxide dismutase 1 (SOD1) is a cytosolic antioxidant enzyme that converts superoxide radicals into hydrogen peroxide and oxygen. In its native state, it forms a stable homodimer and binds copper and zinc.
Mutations in SOD1 cause familial Amyotrophic Lateral Sclerosis (ALS). Among them, the A4V mutation (Alanine → Valine at residue 4) leads to one of the most aggressive forms of the disease. The mutation subtly destabilizes the N-terminus, perturbs folding energetics, and promotes toxic aggregation.
Your challenge:
Design short peptides that bind mutant SOD1.
Then decide which ones are worth advancing toward therapy.
You will use three models developed in our lab:
PepMLM: target sequence-conditioned peptide generation via masked language modeling
PeptiVerse: therapeutic property prediction
moPPIt: motif-specific multi-objective peptide design using Multi-Objective Guided Discrete Flow Matching (MOG-DFM)
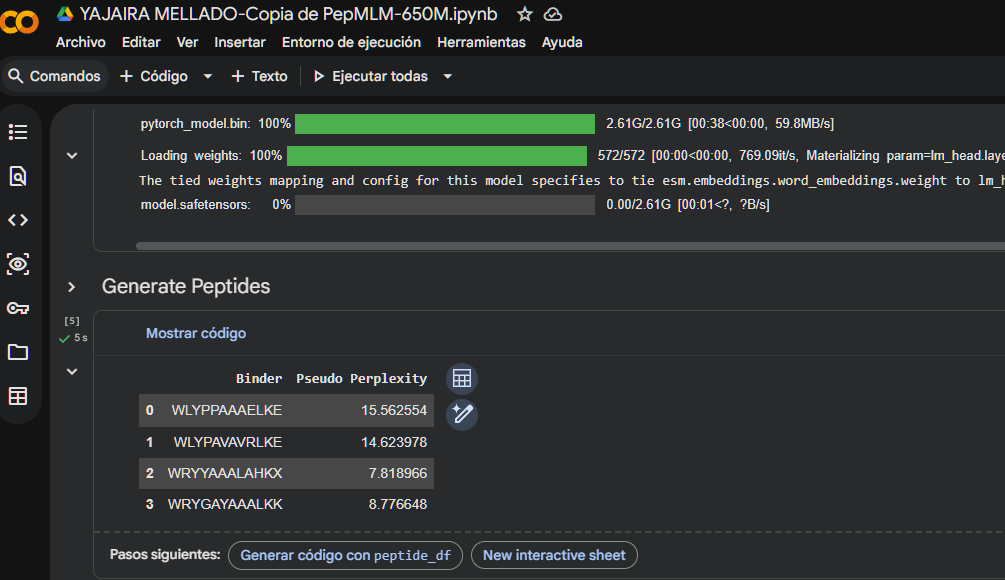
Part 1: Generate Binders with PepMLM
Begin by retrieving the human SOD1 sequence from UniProt (P00441) and introducing the A4V mutation.
Using the PepMLM Colab linked from the HuggingFace PepMLM-650M model card:
Generate four peptides of length 12 amino acids conditioned on the mutant SOD1 sequence.
To your generated list, add the known SOD1-binding peptide FLYRWLPSRRGG for comparison.
Record the perplexity scores that indicate PepMLM’s confidence in the binders.
I generated these four peptides using PepMLM-650M.ipynb
The perplexity scores were recorded to evaluate PepMLM’s confidence in the generated peptide binders. Lower perplexity values indicate higher confidence in the sequence. Among the designed peptides, P3 (7.82) showed the lowest perplexity score, followed by P4 (8.78), P2 (14.62), and P1 (15.56), suggesting that P3 and P4 are the most plausible sequences according to the model.
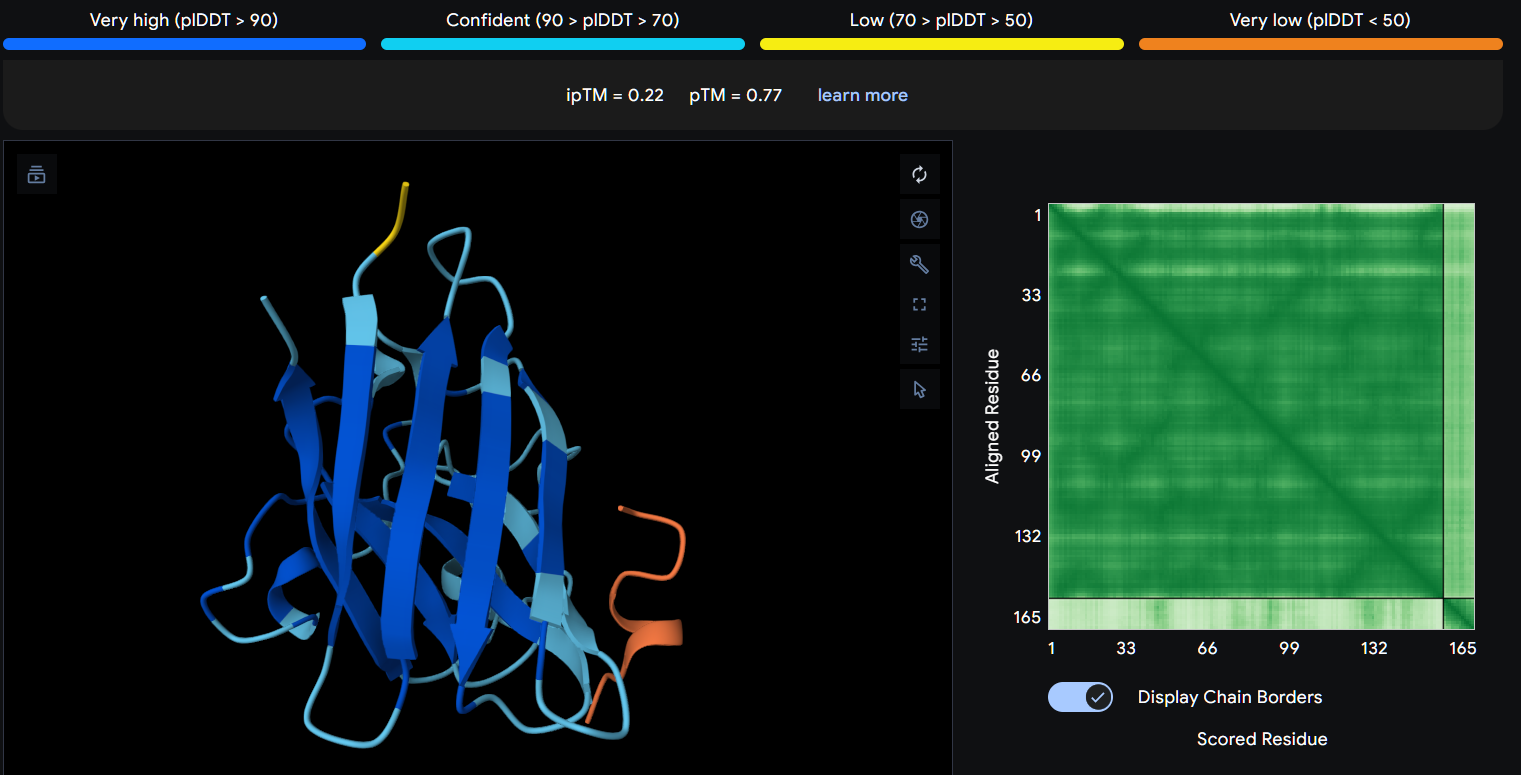
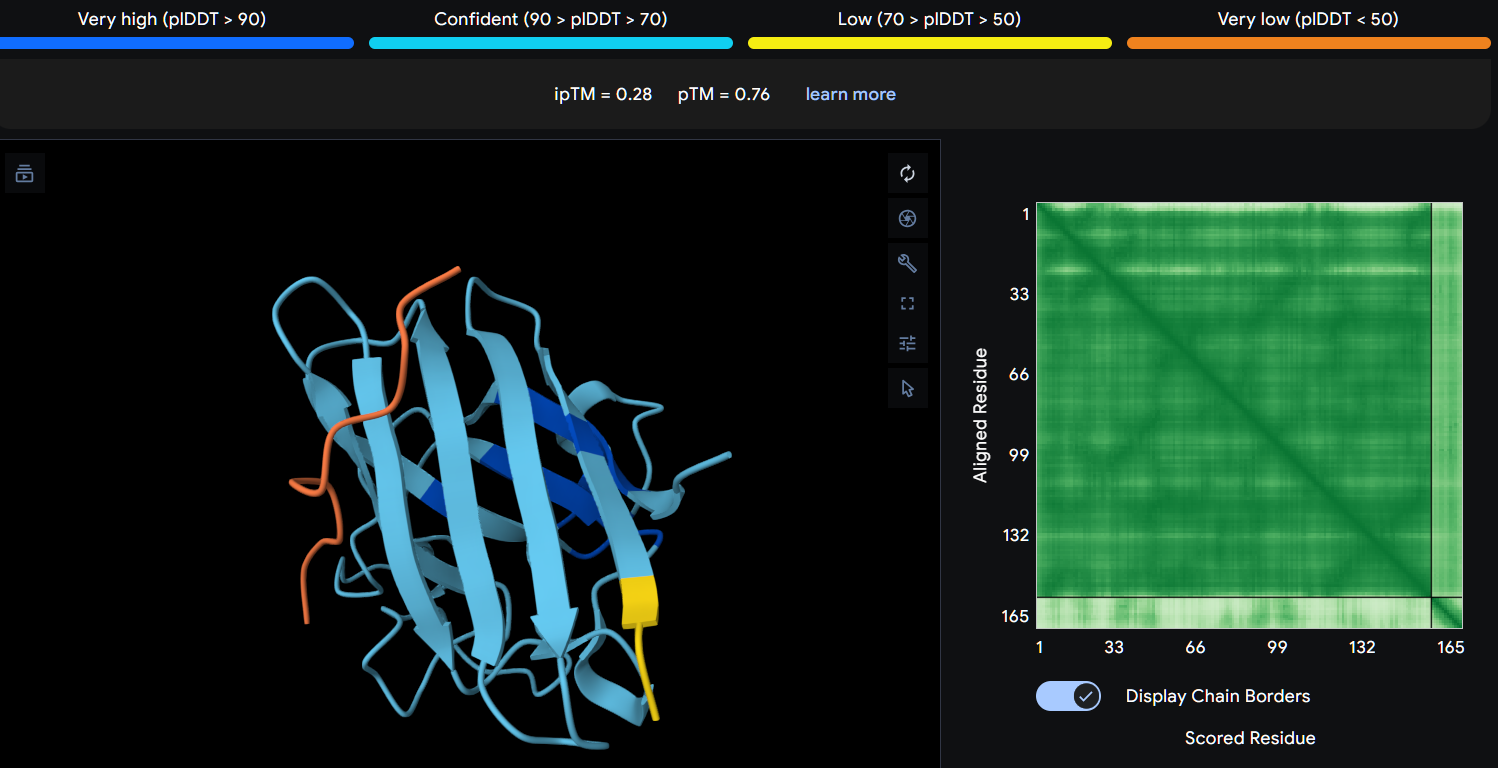
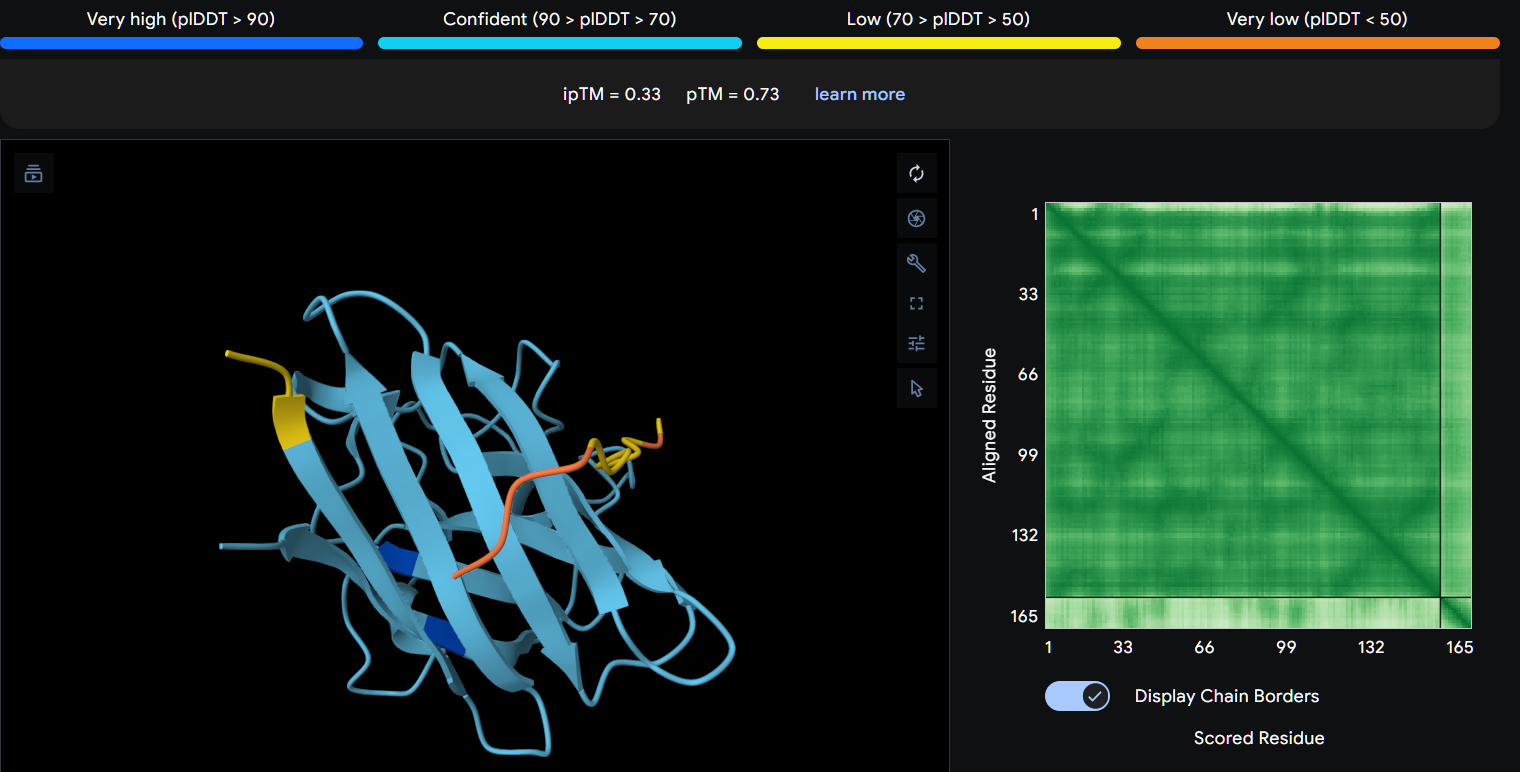
Part 2: Evaluate Binders with AlphaFold3
Navigate to the AlphaFold Server: alphafoldserver.com
For each peptide, submit the mutant SOD1 sequence followed by the peptide sequence as separate chains to model the protein-peptide complex.
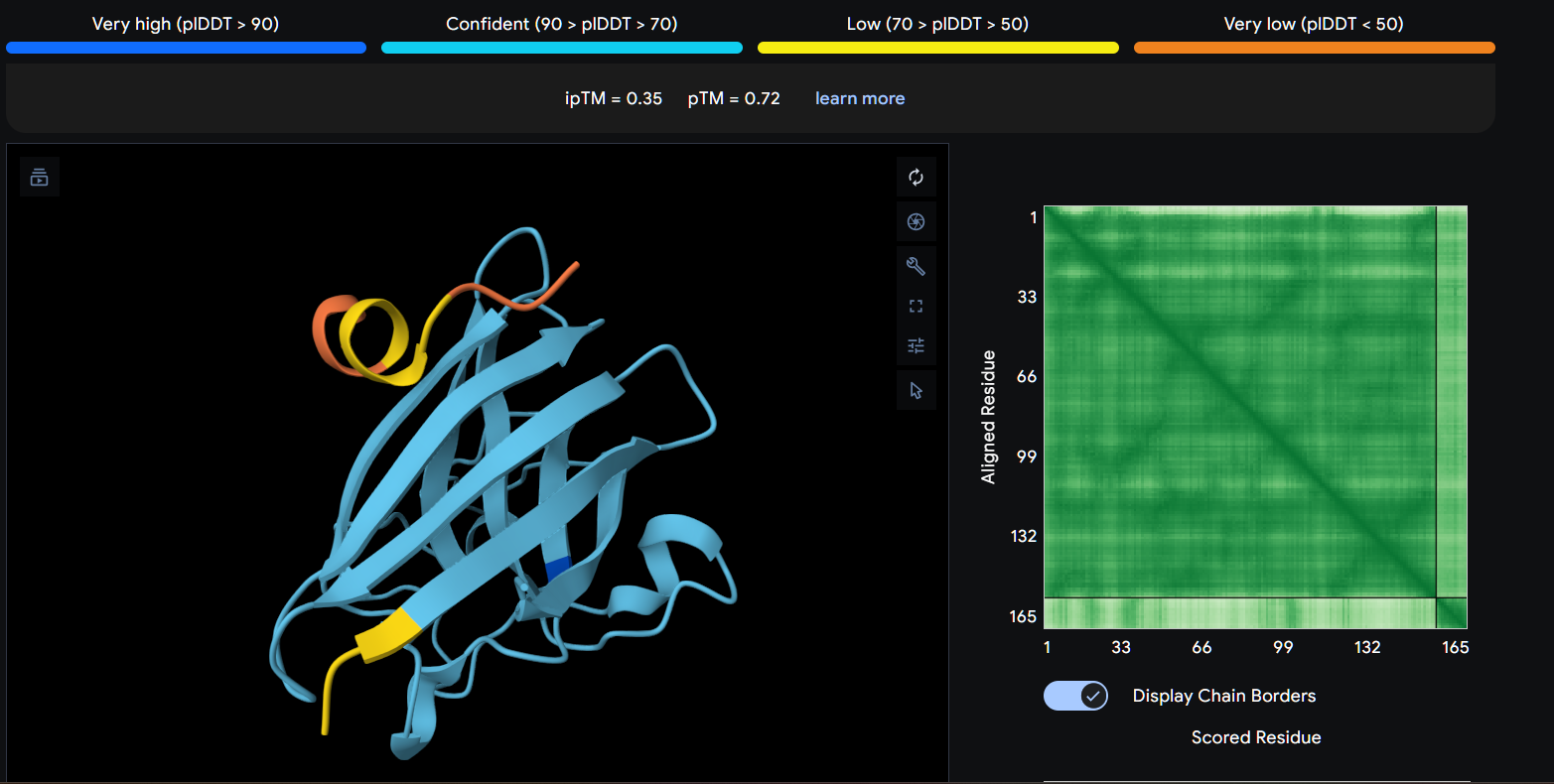
Record the ipTM score and briefly describe where the peptide appears to bind. Does it localize near the N-terminus where A4V sits? Does it engage the β-barrel region or approach the dimer interface? Does it appear surface-bound or partially buried?
In a short paragraph, describe the ipTM values you observe and whether any PepMLM-generated peptide matches or exceeds the known binder.
Peptide 1 (P1)
Peptide 2 (P2)
Peptide 3 (P3)
Peptide 4 (P4)
Using AlphaFold3, I determines that P2 and P1 appear to hav more stable structures, while P3 and P4 may exhibit greater interaction capability.
Also AlphaFold3 analysis showed differences in structural confidence and interaction potential among the peptides. Based on the predicted TM-score (pTM), the ranking was P1 ≈ P2 > P3 > P4, indicating that P1 and P2 have the most reliable predicted structures. In contrast, the interface TM-score (ipTM) followed the order P4 > P3 > P2 > P1, suggesting that P4 has the highest potential for molecular interactions.
Part 3: Evaluate Properties of Generated Peptides in the PeptiVerse
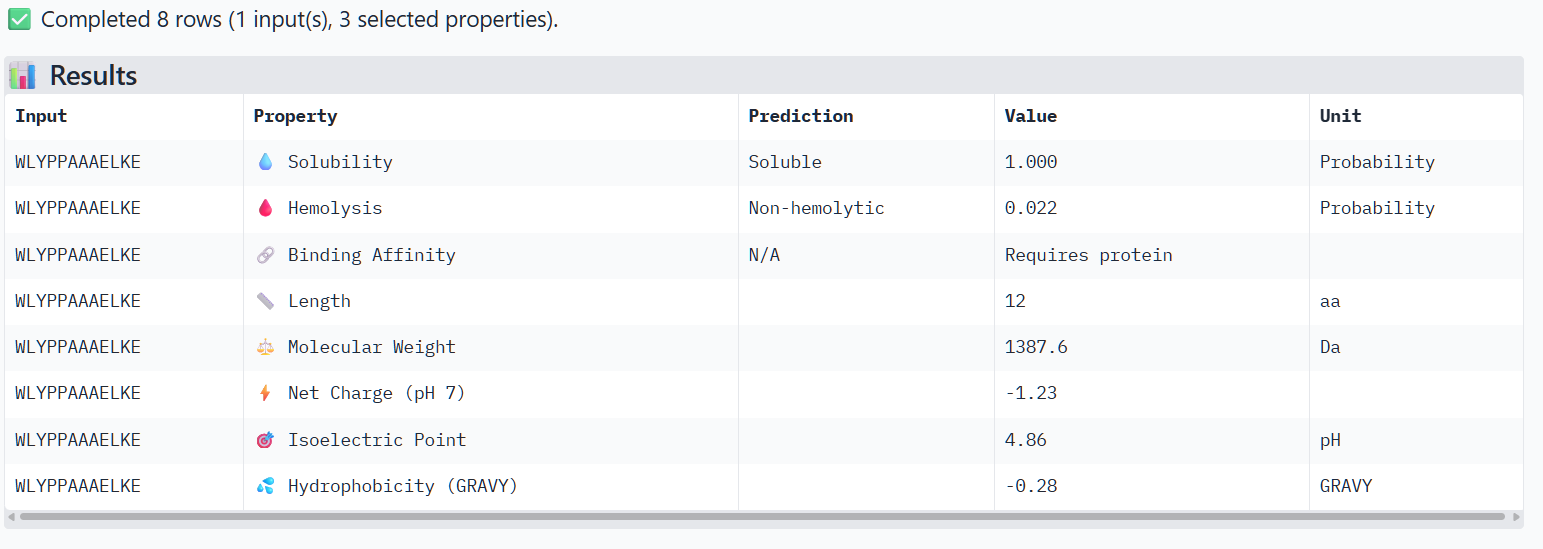
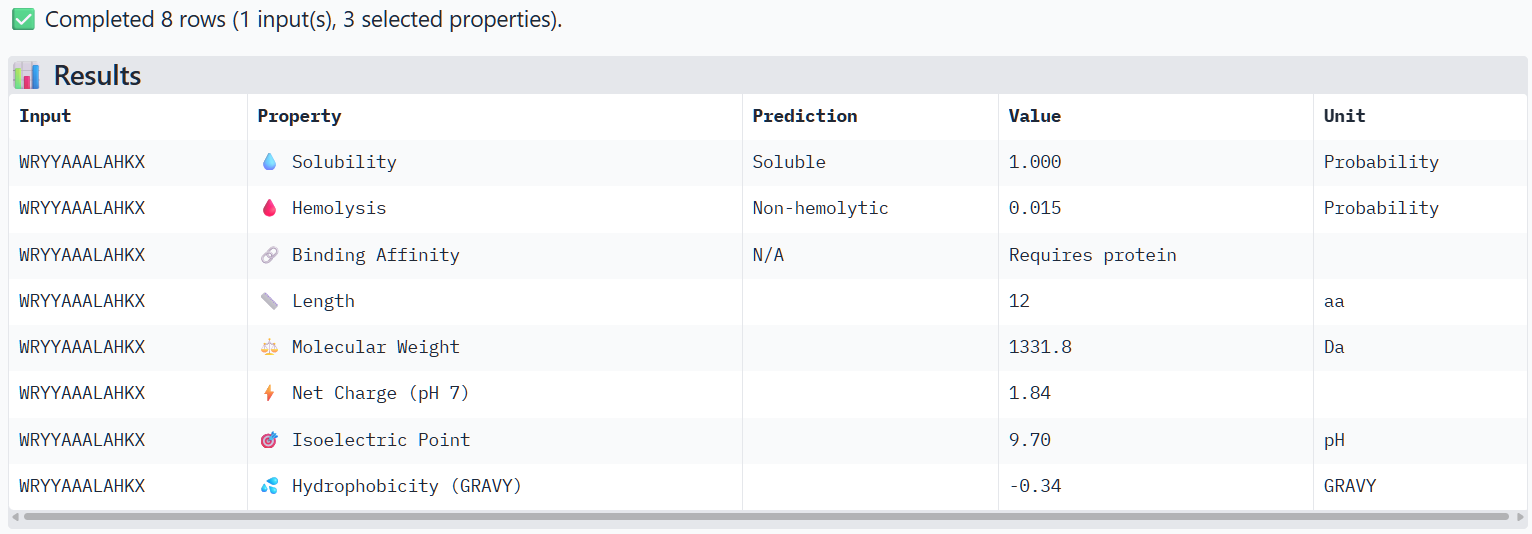
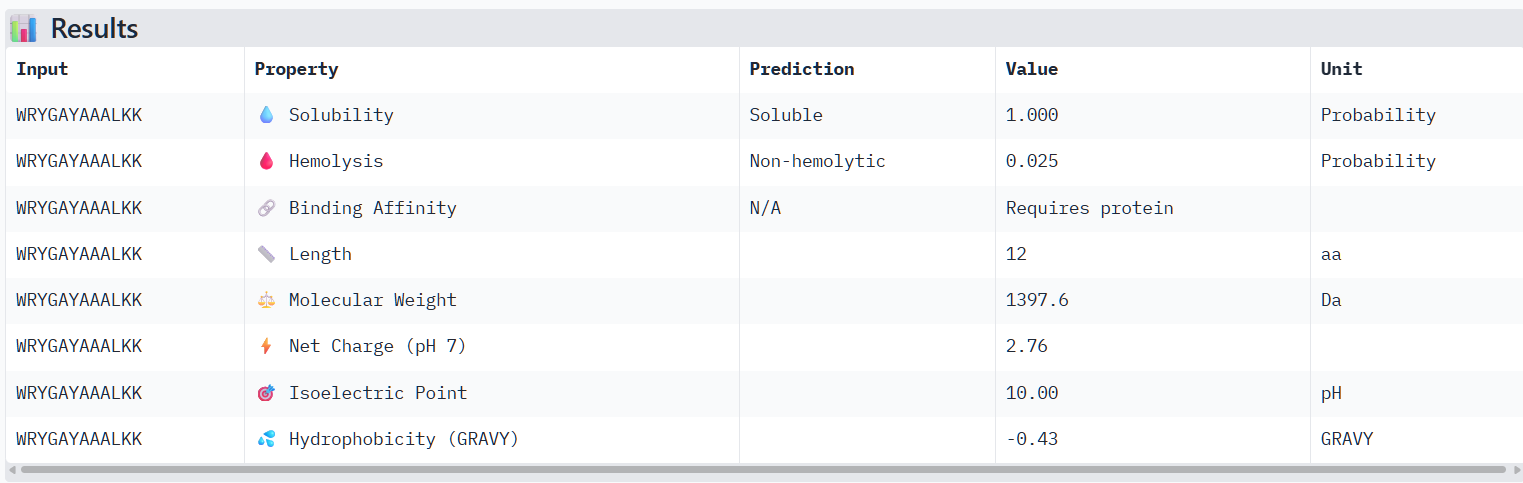
Structural confidence alone is insufficient for therapeutic development. Using PeptiVerse, let’s evaluate the therapeutic properties of your peptide! For each PepMLM-generated peptide:
Paste the peptide sequence.
Paste the A4V mutant SOD1 sequence in the target field.
Check the boxes
Predicted binding affinity
Solubility
Hemolysis probability
Net charge (pH 7)
Molecular weight
Compare these predictions to what you observed structurally with AlphaFold3. In a short paragraph, describe what you see. Do peptides with higher ipTM also show stronger predicted affinity? Are any strong binders predicted to be hemolytic or poorly soluble? Which peptide best balances predicted binding and therapeutic properties?
Choose one peptide you would advance and justify your decision briefly.
Peptide 1 (P1)
Peptide 2 (P2)
Peptide 3 (P3)
Peptide 4 (P3)
The structural predictions from AlphaFold3 were compared with the physicochemical properties predicted for the generated peptides. Peptides with higher ipTM scores, particularly P4 and P3, showed greater predicted interaction potential, suggesting stronger binding capability compared to P1 and P2. Importantly, all peptides were predicted to be non-hemolytic and soluble, indicating favorable therapeutic safety profiles. Considering both interaction potential and physicochemical properties, P3 appears to provide the best balance between predicted binding capability and therapeutic characteristics, while P4 also shows strong interaction potential but slightly lower structural confidence.
Part 4: Generate Optimized Peptides with moPPIt
Now, move from sampling to controlled design. moPPIt uses Multi-Objective Guided Discrete Flow Matching (MOG-DFM) to steer peptide generation toward specific residues and optimize binding and therapeutic properties simultaneously. Unlike PepMLM, which samples plausible binders conditioned on just the target sequence, moPPIt lets you choose where you want to bind and optimize multiple objectives at once.
Open the moPPit Colab linked from the HuggingFace moPPIt model card
Make a copy and switch to a GPU runtime.
In the notebook:
Paste your A4V mutant SOD1 sequence.
Choose specific residue indices on SOD1 that you want your peptide to bind (for example, residues near position 4, the dimer interface, or another surface patch).
Set peptide length to 12 amino acids.
Enable motif and affinity guidance (and solubility/hemolysis guidance if available). Generate peptides.
After generation, briefly describe how these moPPit peptides differ from your PepMLM peptides. How would you evaluate these peptides before advancing them to clinical studies?
I couldn’t run the Colab notebook because I don’t have access to the required environment.
Note: Lysis protein contains a soluble N-terminal domain followed by a transmembrane protein (blue/last 35 residues). Transmembrane protein affects the lysis activity. The soluble domain (green) is the domain responsible for interaction with DnaJ.
Designing these mutants with good computational confidence is hard. It will show you limitations of some of the structure based models. Ultimately, you can pick various combinations of mutations and get lab results and then decide to pick the next round of mutations, but this assay will not be easy to run at scale in this class.
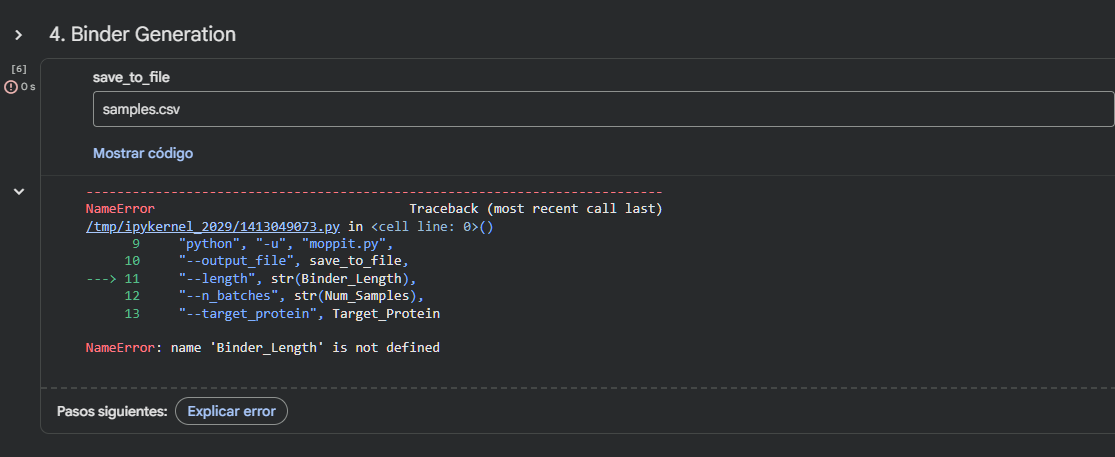
Run this notebook to generate for each position in the amino acid sequence, a “score” for what would happen to the protein if you mutated into another amino acid. It can be positive or negative for the protein. We want to identify possible mutations that are “positive” If you run this notebook - you will see a .csv file in the sidebar. You can download it and look at it in the google sheets if that’s easier
Use the experimental data here. This dataset contains information about mutants of the L-Protein and their effect on lysis in the lab - L-Protein Mutants
First check, does the experimental data correlate with the scores from the notebook in (b)? This should give you a clue on how well these language embeddings capture information about this protein sequence.
Using information about the effect of protein mutations at these sites - both the scores and the experimental data in the drive, come up with 5 mutations for each student along with how you came up with them and why you believe they would work. 2 of the variants you submit must have mutations in the transmembrane region (refer to notes above on what amino acid positions these are) and 2 of them must be in the soluble region . Remember that you can also use the pBLAST to see which residues are conserved and not mutate them if you want to.
One easy way to generate sequence mutations could be to look for residue positions and mutations that have a positive mutational effect either in the experimental or have a positive score from step 1. And pick a combination of those mutations.
You can utilize Af2_Multimer to generate a Multimeric Assembly; you can do this by making your query sequence as. We want to do this because - A running hypothesis for how this protein functions is that it assembles to make a perforation in the bacterial membrane.
Figure generated using the following multimeric assembly (where each chain is separated from the other with a :):
Answer these questions about the protocol in this week’s lab:
1. What are some components in the Phusion High-Fidelity PCR Master Mix and what is their purpose?
The Phusion High-Fidelity master mix contains:
Phusion DNA polymerase: An enzyme that synthesizes new DNA
dNTPs (deoxynucleotide triphotes): The blocks that the polymerase incorporates to build the new DNA strand.
Reaction buffer: Provides optimal pH and salt conditions for the polymerase.
Stabilizers and additives: Help maintain enzyme activity during temperature cycling.
2. What are some factors that determine primer annealing temperature during PCR?
The annealing temperature (Tm) is determinaned mainly by:
1. Base composition: G-C pairs form three hydrogen bonds, a higher GC content increases Tm
2. Presence of mismatches: Tm decrease, if the primer is not perfectly complementary
3. Primer length: Longer primers tend to have higher Tm, but the optimal range is usually 18-22 bp.
4. Salt concentration: Especially Mg 2+ and monovalent cations (Na+, K+) in the buffer, higher concentrations stabilize binding and raise Tm.
3. There are two methods from this class that create linear fragments of DNA: PCR, and restriction enzyme digests. Compare and contrast these two methods, both in terms of protocol as well as when one may be preferable to use over the other.
Aspect
PCR
Restriction enzyme digestion
Protocol
Requires specific primers, master mix and thermocycler
Uses restrcition enzymes that cut DNA at specific sites
Flexibility
Generate fragments of virtually any sequence with appropriate primers.
Less flexible for generating custom fragments without suitable sites.
4. How can you ensure that the DNA sequences that you have digested and PCR-ed will be appropriate for Gibson cloning?
The several conditions must be met:
Purity: After PCR or digestion, DNA is purified to remove salts, enzymes, and residual primers.
Proper concentration: DNA concentration must be quantified to calculate the correct molar ratio.
Integrity: Verify by gel electrophoresis that fragments are not degraded.
5. How does the plasmid DNA enter the E. coli cells during transformation?
Plasmid DNA enters E. coli cells during transformation with artificially induced process:
Preparation of competent cells: bacteria are treated with calcium chloride
Heat shock : Exposing to 42 °C for a short time.
Passive diffusion: Plasmid DNA enters by diffusion through the pores.
6. Describe another assembly method in detail (such as Golden Gate Assembly)
1. Explain the other method in 5 - 7 sentences plus diagrams (either handmade or online).
Golden Gate Assembly is a molecular cloning method that uses type IIS restriction enzymes, such as BsaI, which recognize non-palindromic DNA sequences but cut outside of their recognition sites. This unique property allows researchers to design custom overhangs that determine the precise order in which DNA fragments are assembled. By placing these recognition sites at the ends of multiple DNA fragments, several pieces can be joined together in a single, one-pot restriction-ligation reaction. During the process, the restriction enzyme cuts the DNA while a ligase simultaneously joins the fragments, enabling seamless and scarless assembly. This method is highly efficient and reduces the need for multiple cloning steps compared to traditional techniques. Golden Gate Assembly is widely used for constructing gene libraries, assembling synthetic biological circuits, and combining multiple genetic elements into a destination vector. However, one limitation is that internal restriction sites matching the enzyme used must be removed or avoided to prevent unwanted cutting
2. Model this assembly method with Benchling or Asimov Kernel!
Assignment: Asimov Kernel
Create a Repository for your work
Create a blank Notebook entry to document the homework and save it to that Repository
Explore the devices in the Bacterial Demos Repo to understand how the parts work together by running the Simulator on various examples, following the instructions for the simulator found in the “Info” panel (click the “i” icon on the right to open the Info panel)
Create a blank Construct and save it to your Repository
Recreate the Repressilator in that empty Construct by using parts from the
Characterized Bacterial Parts repository
Search the parts using the Search function in the right menu
Drag and drop the parts into the Construct
Confirm it works as expected by running the Simulator (“play” button) and compare your results with the Repressilator Construct found in the Bacterial Demos repository
Document all of this work in your Notebook entry - you can copy the glyph image and the simulator graphs, and paste them into your Notebook
Build three of your own Constructs using the parts in the Characterized Bacterials Parts Repo
Explain in the Notebook Entry how you think each of the Constructs should function
Run the simulator and share your results in the Notebook Entry
If the results don’t match your expectations, speculate on why and see if you can adjust the simulator settings to get the expected outcome
REFERENCES:
PromptS ChatGPT
Explain Golden Gate Assembly in detail, including how type IIS restriction enzymes function, why they cut outside their recognition sites, and how this enables seamless DNA assembly. Provide a step-by-step explanation and include advantages over traditional cloning methods.
Describe how Golden Gate Assembly can be used to construct synthetic genetic circuits. Include an example with multiple DNA fragments (e.g., promoter, coding sequence, terminator) and explain how their order is controlled during assembly.
week-07-hw-genetic-circuits-part-ii
Homework — DUE BY Mar 31 2PM ET
Assignment Part 1: Intracellular Artificial Neural Networks (IANNs)
1. What advantages do IANNs have over traditional genetic circuits, whose input/output behaviors are Boolean functions?
Tradicional genetic rely on Boolean logic (AND, OR, NOT), producing discrete outputs. In contrast, IAANS are inspired by the perceptron by the perceptron model, where inputs correspond to : Activation is the gene expression threshold, Inputs are molecular concentrations,Weights is regulatory strength. Some advanges are: the continuous signal precessing, wich allows graded rather than binary responses, scalability through multilayer architectures, improved classification of complex cellular states, as multiple biological signals can be integrated and weighted simultaneously.
2. Describe a useful application for an IANN; include a detailed description of input/output behavior, as well as any limitations an IANN might face to achieve your goal.
An IANN can be effectively applied to model complex biological systems such as the development of engineerred cells capable of detecting and selectively eliminating cancer cells based on multiple biological inputs.
In this system, the inputs consist of abnormal levels of messeger RNA, cancer-specific proteins, and metabolic changes, which are sensed by genetic circuits as the first layer of the network.These signals are then integrated through intracellular processing layers that mimic the weighted summation and decision-making behavior of artificial neural networks, allowing the cell to classify whether a target cell is cancerous or not. The output is a targered response, such as inducing apoptosis or releasing therapeutic molecules, while on action is taken if the cell is healthy.
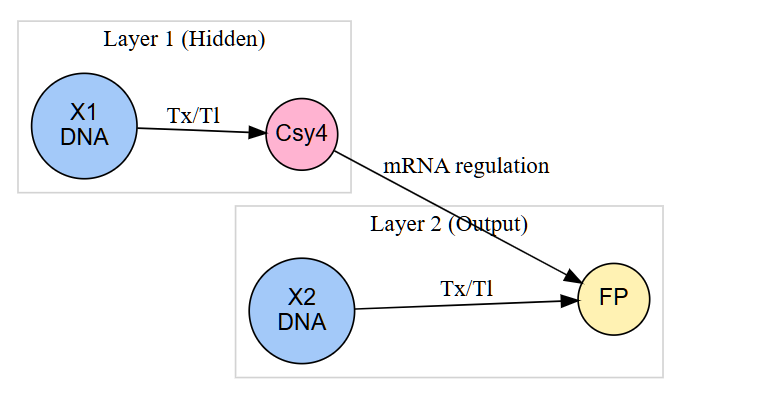
3. Below is a diagram depicting an intracellular single-layer perceptron where the X1 input is DNA encoding for the Csy4 endoribonuclease and the X2 input is DNA encoding for a fluorescent protein output whose mRNA is regulated by Csy4. Tx: transcription; Tl: translation.
Draw a diagram for an intracellular multilayer perceptron where layer 1 outputs an endoribonuclease that regulates a fluorescent protein output in layer 2.
Assignment Part 2: Fungal Materials
1. What are some examples of existing fungal materials and what are they used for? What are their advantages and disadvantages over traditional counterparts?
Mycelium composites are primarily used for sustainable packaging and thermal insulation, serving as alternatives to petroleum-based plastics like polystyrene. Mycelium leather is applied in the textile industry as eco-friendly subsite for animal leather. Also bio-bricks are using agricultural waste enhancing their sustainability.
Some advantages include biodegradability, wich reduces environmental impact and low energy requirements during production compared to tradicional materials. The disadvantages are low mechanical strength and environmental sensitivity to environmental conditions like moisture and longer production times.
2. What might you want to genetically engineer fungi to do and why? What are the advantages of doing synthetic biology in fungi as opposed to bacteria?
I want to explore more about the development of living biosensors and bioremediation sytems capable of detecting and degrading environmental pollutants or pathogens. For instance, fungi can be engineered to express florescent reporter proteins in response to toxins, heavy metlas or infection biomarkers in livestock. ALso fungi can be modified to produce degradative enzymes such as ligninases ans cellulases, enebling the breakdown of persistent organix pollutants.Compared to bacteria, fungi offer several key advantages in synthetic biology: The eukaryotic organization is enable complex post-traslational modifications required for functional proteins. 3D mycelial growth distributed sensing ans biofabrication. High protein secretion capacity for industrial enzyme production.
REFERENCES:
Pandi, A., Koch, M., Voyvodic, P.L. et al. Metabolic perceptrons for neural computing in biological systems. Nat Commun 10, 3880 (2019). https://doi.org/10.1038/s41467-019-11889-0
Nilsson, A., Peters, J.M., Meimetis, N. et al. Artificial neural networks enable genome-scale simulations of intracellular signaling. Nat Commun 13, 3069 (2022). https://doi.org/10.1038/s41467-022-30684-y
IA PROMBTS: I use deepseek and ChatGPT for generate the image 1
Analyze the current state of fungal engineering in synthetic biology. What are the main applications, limitations, and future directions?
Compare genetically engineered fungi and bacteria in terms of metabolic capacity, protein production, and environmental robustness. What are the key advantages of fungi?
Critically analyze the scalability of fungal-based materials. What are the technical and economic barriers to industrial production?
Explain how intracellular artificial neural networks (IANNs) are implemented in biological systems. How do they differ from traditional genetic circuits?
Describe how molecular components (e.g., promoters, ribozymes, endoribonucleases) can be used to implement weights and activation functions in IANNs.
Assignment Part 3: First DNA Twist Order
Review the Individual Final Project documentation guidelines.
Submit this Google Form with your draft Aim 1, final project summary, HTGAA industry council selections, and shared folder for DNA designs. DUE MARCH 20 FOR MIT/HARVARD/WELLESLEY STUDENTS
Review Part 3: DNA Design Challenge of the week 2 homework. Design at least 1 insert sequence and place it into the Benchling/Kernel/Other folder you shared in the Google Form above. Document the backbone vector it will be synthesized in on your website.
Week 9 HW: Cell free systems
General homework questions
1. Explain the main advantages of cell-free protein synthesis over traditional in vivo methods, specifically in terms of flexibility and control over experimental variables. Name at least two cases where cell-free expression is more beneficial than cell production.
2. Describe the main components of a cell-free expression system and explain the role of each component.
3. Why is energy provision regeneration critical in cell-free systems? Describe a method you could use to ensure continuous ATP supply in your cell-free experiment.
4. Compare prokaryotic versus eukaryotic cell-free expression systems. Choose a protein to produce in each system and explain why.
5. How would you design a cell-free experiment to optimize the expression of a membrane protein? Discuss the challenges and how you would address them in your setup.
6. Imagine you observe a low yield of your target protein in a cell-free system. Describe three possible reasons for this and suggest a troubleshooting strategy for each.
Homework question from Kate Adamala
Design an example of a useful synthetic minimal cell as follows:
1. Pick a function and describe it.
a. What would your synthetic cell do? What is the input and what is the output?
b. Could this function be realized by cell-free Tx/Tl alone, without encapsulation?
c. Could this function be realized by genetically modified natural cell?
d. Describe the desired outcome of your synthetic cell operation.
2. Design all components that would need to be part of your synthetic cell.
What would be the membrane made of?
What would you encapsulate inside? Enzymes, small molecules.
Which organism your Tx/Tl system will come from? Is bacterial OK, or do you need a mammalian system for some reason? (hint: for example, if you want to use small molecule modulated promotors, like Tet-ON, you need mammalian)
How will your synthetic cell communicate with the environment? (hint: are substrates permeable? or do you need to express the membrane channel?)
3. Experimental details
List all lipids and genes. (bonus: find the specific genes; for example, instead of just saying “small molecule membrane channel” pick the actual gene.)
How will you measure the function of your system?
Homework question from Peter Nguyen
Freeze-dried cell-free systems can be incorporated into all kinds of materials as biological sensors or as inducible enzymes to modify the material itself or the surrounding environment. Choose one application field — Architecture, Textiles/Fashion, or Robotics — and propose an application using cell-free systems that are functionally integrated into the material. Answer each of these key questions for your proposal pitch:
- Write a one-sentence summary pitch sentence describing your concept.
- How will the idea work, in more detail? Write 3-4 sentences or more.
- What societal challenge or market need will this address?
- How do you envision addressing the limitation of cell-free reactions (e.g., activation with water, stability, one-time use)?
Homework question from Ally Huang
Freeze-dried cell-free reactions have great potential in space, where resources are constrained. As described in my talk, the Genes in Space competition challenges students to consider how biotechnology, including cell-free reactions, can be used to solve biological problems encountered in space. While the competition is limited to only high school students, your assignment will be to develop your own mock Genes in Space proposal to practice thinking about biotech applications in space!
For this particular assignment, your proposal is required to incorporate the BioBits® cell-free protein expression system, but you may also use the other tools in the Genes in Space toolkit (the miniPCR® thermal cycler and the P51 Molecular Fluorescence Viewer). For more inspiration, check out https://www.genesinspace.org/ .
Provide background information that describes the space biology question or challenge you propose to address. Explain why this topic is significant for humanity, relevant for space exploration, and scientifically interesting. (Maximum 100 words)
Name the molecular or genetic target that you propose to study. Examples of molecular targets include individual genes and proteins, DNA and RNA sequences, or broader -omics approaches. (Maximum 30 words)
Describe how your molecular or genetic target relates to the space biology question or challenge your proposal addresses. (Maximum 100 words)
Clearly state your hypothesis or research goal and explain the reasoning behind it. (Maximum 150 words)
Outline your experimental plan - identify the sample(s) you will test in your experiment, including any necessary controls, the type of data or measurements that will be collected, etc. (Maximum 100 words)
Week 1 HW: imaging and measurement
Homework — DUE BY START OF Apr 14 LECTURE
Homework is partly based on data that will be generated in the Waters Immerse Lab in Cambridge, MA. Students will characterize green fluorescent protein (eGFP, a recombinant protein standard) structure (primary, secondary/tertiary) in the lab using liquid chromatography and mass spectrometry, as well as Keyhole Limpet Hemocyanin (KLH) oligomeric states using charge detection mass spectrometry (CDMS). Data generated in the lab needed to do the homework is included both within this document and in the Appendix of the laboratory protocol.
Homework: Final Project
For your final project:
Please identify at least one (ideally many) aspect(s) of your project that you will measure. It could be the mass or sequence of a protein, the presence, absence, or quantity of a biomarker, etc.
Please describe all of the elements you would like to measure, and furthermore describe how you will perform these measurements.
What are the technologies you will use (e.g., gel electrophoresis, DNA sequencing, mass spectrometry, etc.)? Describe in detail.
Homework: Waters Part I — Molecular Weight
We will analyze an eGFP standard on a Waters Xevo G3 QTof MS system to determine the molecular weight of intact eGFP and observe its charge state distribution in the native and denatured (unfolded) states. The conditions for LC-MS analysis of intact protein cause it to unfold and be detected in its denatured form (due to the solvents and pH used for analysis).
Based on the predicted amino acid sequence of eGFP (see below) and any known modifications, what is the calculated molecular weight? You can use an online calculator like the one at https://web.expasy.org/compute_pi/
Calculate the molecular weight of the eGFP using the adjacent charge state approach described in the recitation. Select two charge states from the intact LC-MS data (Figure 1) and:
Determine
for each adjacent pair of peaks
using
Determine the MW of the protein using the relationship between
,
, and
Calculate the accuracy of the measurement using the deconvoluted MW from 2.2 and the predicted weight of the protein from 2.1 using:
Can you observe the charge state for the zoomed-in peak in the mass spectrum for the intact eGFP? If yes, what is it? If no, why not?
Homework: Waters Part II — Secondary/Tertiary structure
Homework: Waters Part III — Peptide Mapping - primary structure
We will digest the eGFP protein standard into peptides using trypsin (an enzyme that selectively cleaves the peptide bond after Lysine (K) and Arginine (R) residues. The resulting peptides will be analyzed on the Waters BioAccord LC-MS to measure their molecular weights and fragmented to confirm the amino acid sequence within each peptide – generating a “peptide map”. This process is used to confirm the primary structure of the protein.
There are a variety of tools available online to calculate protein molecular weight and predict a list of peptides generated from a tryptic digest. We will be using tools within the online resource Expasy (the bioinformatics resource portal of the Swiss Institute of Bioinformatics (SIB)) to predict a list of tryptic peptides from eGFP.
How many Lysines (K) and Arginines (R) are in eGFP? Please circle or highlight them in the eGFP sequence given in Waters Part I question 1 above. (Note: adding the sequence to Benchling as an amino acid file and clicking biochemical properties tab will show you a count for each amino acid).
How many peptides will be generated from tryptic digestion of eGFP?
Navigate to https://web.expasy.org/peptide_mass/
Copy/paste the sequence above into the input box in the PeptideMass tool to generate expected list of peptides.
Use Figure 4 below as a guide for the relevant parameters to predict peptides from eGFP.
Click “Perform the Cleavage” button in the PeptideMass tool and report the number of peptides generated when using trypsin to perform the digest.
Based on the LC-MS data for the Peptide Map data generated in lab (please use Figure 5a as a reference) how many chromatographic peaks do you see in the eGFP peptide map between 0.5 and 6 minutes? You may count all peaks that are >10% relative abundance.
Assuming all the peaks are peptides, does the number of peaks match the number of peptides predicted from question 2 above? Are there more peaks in the chromatogram or fewer?
Identify the mass-to-charge (
) of the peptide shown in Figure 5b. What is the charge (
) of the most abundant charge state of the peptide (use the separation of the isotopes to determine the charge state). Calculate the mass of the singly charged form of the peptide (
) based on its
and
.
Identify the peptide based on comparison to expected masses in the PeptideMass tool. What is mass accuracy of measurement? Please calculate the error in ppm. (Recall that
)
What is the percentage of the sequence that is confirmed by peptide mapping? (see Figure 6
Bonus Peptide Map Questions
Can you determine the peptide sequence for the peptide fragmentation spectrum shown in Figure 5c? (HINT: Use your results from Question 2 above to match the peptide molecular weight that is closest to that shown in Figure 5b. Copy and paste its sequence into this tool online to predict the fragmentation pattern based on its amino acid sequence: http://db.systemsbiology.net/proteomicsToolkit/FragIonServlet.html. What is the sequence of the eGFP peptide that best matches the fragmentation spectrum in Figure 5c?
Does the peptide map data make sense, i.e. do the results indicate the protein is the eGFP standard? Why or why not? Consult with Figure 6, which depicts the % amino acid coverage of peptides positively identified using their calculated mass and fragmentation pattern.
Homework: Waters Part IV — Oligomers
We will determine Keyhole Limpet Hemocyanin (KLH)’s oligomeric states using charge detection mass spectrometry (CDMS). CDMS single-particle measurements of KLH allow us to make direct mass measurements to determine what oligomeric states (that is, how many protein subunits combine) are present in solution. Using the known masses of the polypeptide subunits (Table 1) for KLH, identify where the following oligomeric species are on the spectrum shown below from the CDMS (Figure 7):
Homework: Waters Part V — Did I make GFP?
Please fill out this table with the data you acquired from the lab work done at the Waters Immerse Lab in Cambridge, or else the data screenshots in this document if you were unable to have lab work done at Waters.
Week 11 HW: Building-genomes
Part A: The 1,536 Pixel Artwork Canvas | Collective Artwork
Contribute at least one pixel to this global artwork experiment before the editing ends on Sunday 4/19 at 11:59 PM EST.
A personalized URL was sent to the email address associated with your Discourse account, and you can discuss the artwork on the Discourse.
If you did not have a chance to contribute, it’s okay, just make sure you become a TA this fall! 😉
Make a note on your HTGAA webpages including:
what you contributed to the community bioart project (e.g., “I made part of the DNA on the bottom right plate”)
what you liked about the project, and
what about this collaborative art experiment could be made better for next year.
Part B: Cell-Free Protein Synthesis | Cell-Free Reagents
Referencing the cell-free protein synthesis reaction composition (the middle box outlined in yellow on the image above, also listed below), provide a 1-2 sentence description of what each component’s role is in the cell-free reaction.
E. coli Lysate
BL21 (DE3) Star Lysate (includes T7 RNA Polymerase)
Salts/Buffer
Potassium Glutamate
HEPES-KOH pH 7.5
Magnesium Glutamate
Potassium phosphate monobasic
Potassium phosphate dibasic
Energy / Nucleotide System
Nuclease Free Water
Describe the main differences between the 1-hour optimized PEP-NTP master mix and the 20-hour NMP-Ribose-Glucose master mix shown in the Google Slide above. (2-3 sentences)
Bonus question: How can transcription occur if GMP is not included but Guanine is?
Part C: Planning the Global Experiment | Cell-Free Master Mix Design
Given the 6 fluorescent proteins we used for our collaborative painting, identify and explain at least one biophysical or functional property of each protein that affects expression or readout in cell-free systems. (Hint: options include maturation time, acid sensitivity, folding, oxygen dependence, etc) (1-2 sentences each)
sfGFP
mRFP1
mKO2
mTurquoise2
mScarlet_I
Electra2
The amino acid sequences are shown in the HTGAA Cell-Free Benchling folder.
Create a hypothesis for how adjusting one or more reagents in the cell-free mastermix could improve a specific biophysical or functional property you identified above, in order to maximize fluorescence over a 36-hour incubation. Clearly state the protein, the reagent(s), and the expected effect.
The second phase of this lab will be to define the precise reagent concentrations for your cell-free experiment. You will be assigned artwork wells with specific fluorescent proteins and receive an email with instructions this week (by April 24). You can begin composing master mix compositions here.
The final phase of this lab will be analyzing the fluorescence data we collect to determine whether we can draw any conclusions about favorable reagent compositions for our fluorescent proteins. This will be due a week after the data is returned (date TBD!). The reaction composition for each well will be as follows:
6 μL of Lysate
10 μL of 2X Optimized Master Mix from above
2 μL of assigned fluorescent protein DNA template
2 μL of your custom reagent supplements
Total: 20 μL reaction