Final Project I will measure whether DNA stored on paper can be recovered and remain readable after storage. In Aim 0.5, the main measured output is the presence or absence of expected PCR bands from recovered DNA fragments. Fragment A is expected to produce a ~94 bp payload PCR product, and Fragment B is expected to produce a ~166 bp product. I measured this using endpoint PCR followed by 2% E-Gel EX agarose gel electrophoresis.
A. Master Mix Component Analysis E. coli Lysate BL21 (DE3) Star Lysate: Provides the cellular machinery for transcription and translation, including ribosomes and T7 RNA polymerase. The “Star” mutation helps reduce mRNA degradation.
Salts / Buffer Potassium Glutamate: Maintains ionic strength and supports ribosome activity. HEPES-KOH pH 7.5: Keeps the reaction at a stable pH. Magnesium Glutamate: Supplies magnesium ions needed for ribosomes and enzymes. Potassium Phosphate: Acts as a buffer and supports ATP regeneration.
DNA Design Challenge 3.1 Protein Choice I chose pHluorin (superecliptic pHluorin, SEP) because it makes pH changes visible as a clear signal. pH is a broadly meaningful indicator across scales: it can reflect cellular and bodily conditions (e.g., local acidity in compartments or stress-related microenvironments) and also environmental conditions (e.g., water/soil toxicity or chemical shifts). This makes SEP useful not only as a biosensing concept, but also as an interaction metaphor. Even small changes in conditions can switch what becomes visible.
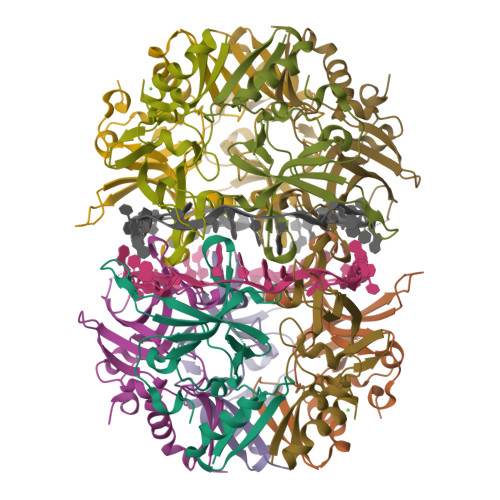
Deinococcus radiodurans. Credit USU/Michael Daly Part B: Protein Analysis and Visualization 1. Briefly describe the protein you selected and why you selected it. I selected the protein from RCSB PDB: 4NOE, which is DdrB, a single-stranded DNA-binding protein from Deinococcus radiodurans (this entry is a protein–ssDNA complex). I chose it because D. radiodurans is well known for extreme radiation tolerance, and DdrB is directly connected to DNA damage response/repair, making it a relevant example for studying UV/radiation-associated protein function with an available 3D structure.
I propose the development of Astro-Moss, a engineered strain of Physcomitrium patens designed for extreme environmental resilience. By leveraging the high genomic editability of moss, this project aims to create varieties capable of surviving extreme diurnal temperature fluctuations, high ionizing radiation, and desiccation.
Applications
Extraterrestrial Foundation: Serving as a primary “pioneer” vegetation for Martian habitats to build organic substrate for future crops.
Functional Bio-textiles: Extracting engineered cells or fibers to create “living” textiles for astronaut apparel that offer self-repairing properties and radiation shielding, contributing to long-term sustainability in space exploration.
2. Governance and Policy Goals
To ensure this application contributes to an ethical future, I have defined the following goals based on the framework of promoting constructive use while minimizing harm:
Goal 1: Ensure Biosafety and Security. Preventing the unintended release of engineered extremophiles into Earth’s ecosystems or the misuse of resilience genes.
Sub-goal 1.2: Dual-use Oversight. Screen genetic sequences related to extreme resistance to prevent their use in enhancing harmful pathogens.
Goal 2: Promote Equity and Global Autonomy. Ensuring that the foundational tools for space colonization are not monopolized by a single entity.
Sub-goal 2.1: Open Standards. Establish international protocols for safety data sharing.
Sub-goal 2.2: Technology Access. Ensure community biolabs and international researchers can contribute to and benefit from the project.
3. Governance Actions Matrix
Aspect
Action 1: Technical Biocontainment
Action 2: DNA Synthesis Screening
Action 3: Open-Source Safety Alliance
Purpose
Engineering “kill-switches” or auxotrophy, dependency on lab-only nutrients.
Mandatory screening of synthetic DNA orders.
Voluntary international consortium to share safety protocols and risk assessments.
Design
Actors: Academic researchers and firms. Must be integrated into the R&D phase.
Actors: Commercial providers and federal regulators.
Actors: International firms, NGOs, and UN.
Assumptions
Assumes mutation will not bypass the switch.
Assumes use of regulated synthesis channels.
Assumes safety over proprietary advantages.
Risks
Failure: Evolution of bypass mechanisms. Success: May limit moss adaptability in genuine emergencies.
Failure: Use of benchtop synthesizers. Success: Increases cost and time for R&D.
Failure: Soft law without enforcement may be ignored. Success: Establishes a global culture of responsibility.
4. Detailed Scoring Matrix
Score: 1 (Best/Most effective) to 3 (Least effective/Most problematic)
Does the option:
Option 1
Option 2
Option 3
Enhance Biosecurity
• By preventing incidents
2
1
2
• By helping respond
1
3
1
Foster Lab Safety
• By preventing incident
1
2
1
• By helping respond
1
N/A
1
Protect the environment
• By preventing incidents
1
2
2
• By helping respond
1
3
1
Other considerations
• Minimizing costs and burdens to stakeholders
3
2
1
• Feasibility?
2
1
3
• Not impede research
3
2
1
• Promote constructive applications
2
2
1
5. Prioritization, Recommendation, and Ethical Reflections
Target Audience: The United Nations Office for Outer Space Affairs (UNOOSA) and the International Space Exploration Coordination Group (ISECG).
Recommendation Strategy:
I prioritize a Hybrid Governance Model that mandates Technical Biocontainment (Action 1) as a prerequisite for mission approval, supported by an International Open-Source Safety Alliance (Action 3) to manage global compliance, safety standards, and equity.
Justification based on Scoring:
While DNA Screening (Action 2) provides an immediate defensive layer, our scoring matrix reveals its significant failure in Response and Equity. For a frontier technology like Astro-Moss, we cannot rely on blocking orders alone.
Action 1 is prioritized because it offers intrinsic safety; a biological kill-switch ensures that even if a containment breach occurs on Earth or a target planet, the organism’s impact is self-limiting.
Action 3 is essential to prevent the safety requirements of Action 1 from becoming a proprietary wall. By open-sourcing safety modules, we ensure that the Right to Explore remains equitable across all nations.
Ethical Trade-offs Considered:
Safety vs. Adaptability: The most robust kill-switch inherently limits the moss’s ability to evolve and survive in truly unknown extraterrestrial variables. I am consciously trading off mission success probability for guaranteed planetary protection.
Innovation Speed vs. Regulation: Mandatory international oversight through an Alliance will slow the deployment of Bio-textiles. However, the risk of a Forward Contamination event (accidentally destroying potential Martian microbial life) would set the entire field of synthetic biology back by decades due to the resulting ethical crisis and public outcry.
Assumptions and Uncertainties:
Assumption of Genetic Stability: This strategy assumes that the complex kill-switch will not be silenced by the high-radiation environments of space. If the mutation rate exceeds our containment design, the technical strategy fails.
Uncertainty of Extraterrestrial Interaction: We lack data on how engineered genes might interact with potential Martian life. This unknown unknown makes the Biological Footprint Audit (drawing from the DIYBio expert model) a critical necessity to ensure independent, peer-reviewed validation of every mission’s biocontainment integrity.
Week 2 Lecture Prep
Homework Questions from Professor Jacobson
Q1: Nature’s machinery for copying DNA is called polymerase. What is the error rate of polymerase? How does this compare to the length of the human genome. How does biology deal with that discrepancy?
Native DNA polymerase has a baseline error rate of approximately 10-7 to 10-8 per base pair. When combined with biological proofreading and mismatch repair mechanisms, the final error rate is reduced to about 10^-10. Given the human genome consists of 3.2 billion base pairs, these high fidelity repair systems ensure that fewer than one error occurs per replication cycle. Biology utilizes these multi layered verification systems to bridge the gap between high data volume and required synthesis accuracy.
Q2: How many different ways are there to code (DNA nucleotide code) for an average human protein? In practice what are some of the reasons that all of these different codes don’t work to code for the protein of interest?
For a protein of average length, the redundancy of the genetic code allows for over 10^190 different DNA sequence combinations. In practice, the vast majority of these sequences are non functional. Major constraints include codon usage bias leading to translation stalls, the formation of complex mRNA secondary structures that block ribosomal movement, and the accidental creation of regulatory motifs or splice signals that interfere with proper expression.
Homework Questions from Dr. LeProust
Q1: What’s the most commonly used method for oligo synthesis currently?
The most widely utilized method currently is phosphoramidite chemical synthesis.
Q2: Why is it difficult to make oligos longer than 200nt via direct synthesis?
The primary barrier is that the coupling efficiency of each synthesis step never reaches 100 percent. As the nucleotide chain grows, cumulative errors cause the final product yield to drop exponentially. This results in an accumulation of truncated sequences that make the target full length sequence extremely difficult to isolate.
Q3: Why can’t you make a 2000bp gene via direct oligo synthesis?
Direct chemical synthesis of a 2000bp gene results in a yield that is effectively zero. The standard industry approach involves synthesizing multiple shorter oligonucleotide fragments and then using enzymatic assembly techniques to stitch them into a complete gene.
Homework Questions from George Church
Q1: What are the 10 essential amino acids in all animals and how does this affect your view of the “Lysine Contingency”?
The ten essential amino acids for animals are phenylalanine, valine, threonine, tryptophan, isoleucine, methionine, histidine, arginine, leucine, and lysine.
Lysine contingency as a biological safety measure is fundamentally fragile. Since lysine is already a naturally essential amino acid for animals, engineering an organism to be unable to synthesize it merely replicates a natural state rather than adding a secure safeguard. In real world environments, engineered organisms can easily bypass this restraint by consuming lysine from common plants or animal tissues. Effective biocontainment should instead rely on synthetic dependencies not found in nature. An example is semantic containment where organisms require non canonical amino acids provided only in a controlled lab setting to ensure they cannot survive in the wild.
I will measure whether DNA stored on paper can be recovered and remain readable after storage. In Aim 0.5, the main measured output is the presence or absence of expected PCR bands from recovered DNA fragments. Fragment A is expected to produce a ~94 bp payload PCR product, and Fragment B is expected to produce a ~166 bp product. I measured this using endpoint PCR followed by 2% E-Gel EX agarose gel electrophoresis.
I will also compare DNA recovered from APTMS-treated paper and untreated paper. In the current pilot, this comparison is qualitative: whether each condition produces the expected band. In future work, I would measure recovery more quantitatively using qPCR Ct values, digital PCR copy number, or fluorometric DNA quantification.
For the next stage of the project, I would measure whether recovered SspCA-encoding DNA can drive functional protein production in a cell-free expression system. This would include measuring SspCA protein expression, expected protein size, and carbonic anhydrase activity. Possible technologies include SDS-PAGE or Western blot for protein size and expression, fluorescence or colorimetric assays for enzyme activity, and DNA sequencing to confirm that recovered DNA remains sequence-correct.
Technologies used or proposed:
Endpoint PCR: tests whether recovered DNA remains amplifiable.
E-Gel EX agarose gel electrophoresis: visualizes expected DNA bands and confirms approximate amplicon size.
qPCR or digital PCR: future quantitative measurement of DNA recovery.
DNA sequencing: future verification of sequence integrity and encoded data.
Cell-free protein expression: tests whether recovered DNA can produce SspCA.
SDS-PAGE / Western blot: verifies protein expression and approximate size.
Carbonic anhydrase activity assay: measures whether produced SspCA remains functional.
2. Waters Part I
Q1:The sequence provided includes the eGFP core, an LE linker and a 6x-His purification tag. Calculated Molecular Weight: 27,988.97 Da
Q2:
Peak 1 (m/z_n): 875.4421
Peak 2 (m/z_n+1): 903.7148
z for each adjacent pair of peaks: the charge state for the 903.7 peak is 31+, and the 875.4 peak is 32+.
MW of the protein: 27,981.90 Da
Accuracy of the measurement: 252.6 ppm
Q3: No.
3. Waters Part II
Q1:
Denatured state: The protein unfolds because of heat or chemicals. More amino acids become exposed, so the protein gains more charges and shows lower m/z values.
Native state: The protein stays folded in its normal shape. Fewer amino acids are exposed, so the protein gains fewer charges and shows higher m/z values.
Q2:
Yes. The charge state is 10+. The isotope peaks are spaced by 0.1 m/z, and since spacing = 1/z, 0.1 means z = 10.
Week 11 HW: BIOPRODUCTION & CLOUD LABS
A. Master Mix Component Analysis
E. coli Lysate
BL21 (DE3) Star Lysate: Provides the cellular machinery for transcription and translation, including ribosomes and T7 RNA polymerase. The “Star” mutation helps reduce mRNA degradation.
Salts / Buffer
Potassium Glutamate: Maintains ionic strength and supports ribosome activity.
HEPES-KOH pH 7.5: Keeps the reaction at a stable pH.
Magnesium Glutamate: Supplies magnesium ions needed for ribosomes and enzymes.
Potassium Phosphate: Acts as a buffer and supports ATP regeneration.
Energy / Nucleotide System
Ribose & Glucose: Provide energy for ATP and GTP regeneration.
AMP, CMP, GMP, UMP: Building blocks used to make RNA nucleotides for transcription.
Guanine: Helps regenerate GTP through the salvage pathway.
Translation Mix (Amino Acids)
17 Amino Acid Mix: Supplies amino acids for protein synthesis.
Tyrosine: Added separately because it dissolves poorly.
Cysteine: Added separately because it is reactive and easily oxidized.
Additives & Backfill
Nicotinamide: Supports metabolic reactions by helping maintain NAD+/NADH cofactors.
Nuclease Free Water: Brings the reaction to the correct volume without introducing nucleases.
–
B. Master Mix Component Analysis
The 1-hour PEP-NTP mix is designed for fast protein production using high-energy PEP and ready-made NTPs. The 20-hour NMP-Ribose-Glucose mix is designed for long-term production, using cheaper precursors and the lysate’s metabolism to slowly regenerate energy and nucleotides over time.
–
C. Planning the Global Experiment | Cell-Free Master Mix Design
Fluorescent Protein Properties
sfGFP: Folds quickly and reliably, so it gives strong fluorescence in many cell-free conditions.
mRFP1: Matures slowly, so fluorescence appears later than protein production.
mKO2: Very bright orange protein, but repeated reads may reduce signal through photobleaching.
mTurquoise2: Has high brightness, so it can be detected even at low protein yield.
mScarlet_I: Very bright red protein, but needs oxygen for chromophore maturation.
Electra2: Designed for fast maturation and stability, so it can track protein production quickly.
Hypothesis: Increasing magnesium glutamate will improve ribosome activity and translation efficiency, producing more mRFP1 over the 36-hour incubation. Because mRFP1 matures slowly, higher total protein production should lead to stronger final fluorescence after enough maturation time.
Week 2: DNA Read, Write, & Edit
DNA Design Challenge
3.1 Protein Choice
I chose pHluorin (superecliptic pHluorin, SEP) because it makes pH changes visible as a clear signal. pH is a broadly meaningful indicator across scales: it can reflect cellular and bodily conditions (e.g., local acidity in compartments or stress-related microenvironments) and also environmental conditions (e.g., water/soil toxicity or chemical shifts). This makes SEP useful not only as a biosensing concept, but also as an interaction metaphor. Even small changes in conditions can switch what becomes visible.
3.2 Reverse Translate: Protein (amino acid) sequence to DNA (nucleotide) sequence.
To obtain the precise nucleotide sequence for superecliptic pHluorin (SEP), I performed a targeted search and retrieval process using the NCBI:
Protein Identification: I accessed the official NCBI Protein record for superecliptic pHluorin (SEP) under accession number AAS66682.1.
Nucleotide Record Location: On the protein record page, I utilized the “Nucleotide sequence from coding region” link found under the Related information sidebar. This link redirected me to the NCBI Nucleotide (nuccore) entry AY533296.1, titled “Synthetic construct superecliptic pHluorin mRNA, complete cds”.
CDS Feature Selection: Within the nuccore page, I specifically identified the CDS (coding region) segment to isolate the exact sequence responsible for protein translation.
FASTA Retrieval: I displayed the sequence in FASTA format (ensuring the header line starts with >) to maintain compatibility with downstream design tools.
Sequence Capture: I copied the nucleotide CDS sequence (bases 1..717) as the foundational DNA template for the SEP protein.
3.2 & 3.3 Reverse Translation & Optimization: The sequence was reverse-translated and codon-optimized for E. coli expression to ensure high yield while avoiding Type IIs restriction sites (BsaI, BsmBI, BbsI).
DNA Sequence:
Prepare a Twist DNA Synthesis Order
I designed an expression cassette to be synthesized as a clonal gene.
Vector Selection: pTwist Amp High Copy. This circular backbone allows for direct transformation into E. coli, speeding up the experimental cycle by 1-2 weeks.
Figure 1: Annotated sfGFP expression cassette designed in Benchling.
Part 5: DNA Read/Write/Edit
Inspired by Marina Otero Verzier’s theory, this documentation explores a move away from the Cartesian enclosure of traditional data centers. I propose using Engineered Moss as a living fabric and information archive for extraterrestrial exploration.
5.1 DNA Read: Material Entanglement
Technology: Nanopore Sequencing.
Vision: Reading the Moss. This transcends binary retrieval; it is an act of “touching a leaf” to listen to the archive. Nanopore allows us to decode how cosmic radiation and human interaction have “co-written” the DNA, creating a material entanglement where the building, the document, and the environment are one.
5.2 DNA Write: The Breathing Library
Technology: Enzymatic DNA Synthesis.
Vision: Encoding human collective memory into moss spores.
Theory: This breaks the “myth of endless growth.” Moss archives are non-hierarchical; as they flourish on alien regolith, they capture CO2 and produce oxygen. It is an architecture that “makes breath possible”—an archive that is both alive and gives life.
5.3 DNA Edit: Ontological Freedom
Technology: CRISPR/Cas9.
Vision: Editing moss for extreme desiccation and radiation tolerance.
Ethics: This edit is not an extractive tool for profit. We grant the moss ontological freedom to express its agency in a new world. It is an act of solidarity and cohabitation between species, allowing the “Data Forest” to germinate in the inconceivable futures of space.
Week 4 HW: PROTEIN DESIGN PART I
Deinococcus radiodurans. Credit USU/Michael Daly
Part B: Protein Analysis and Visualization
1. Briefly describe the protein you selected and why you selected it.
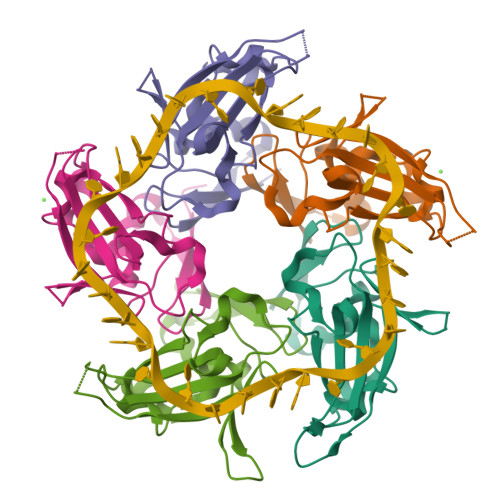
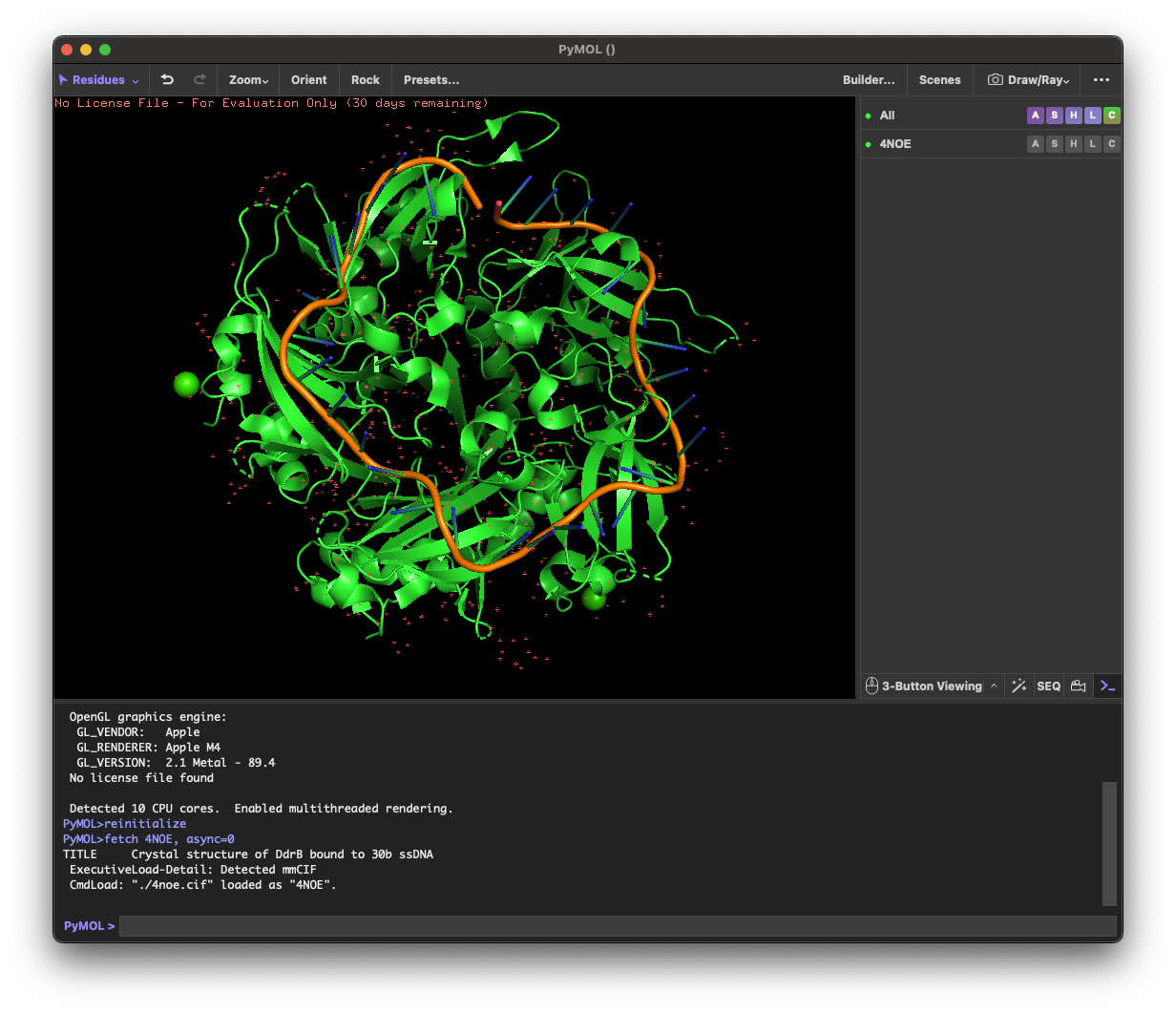
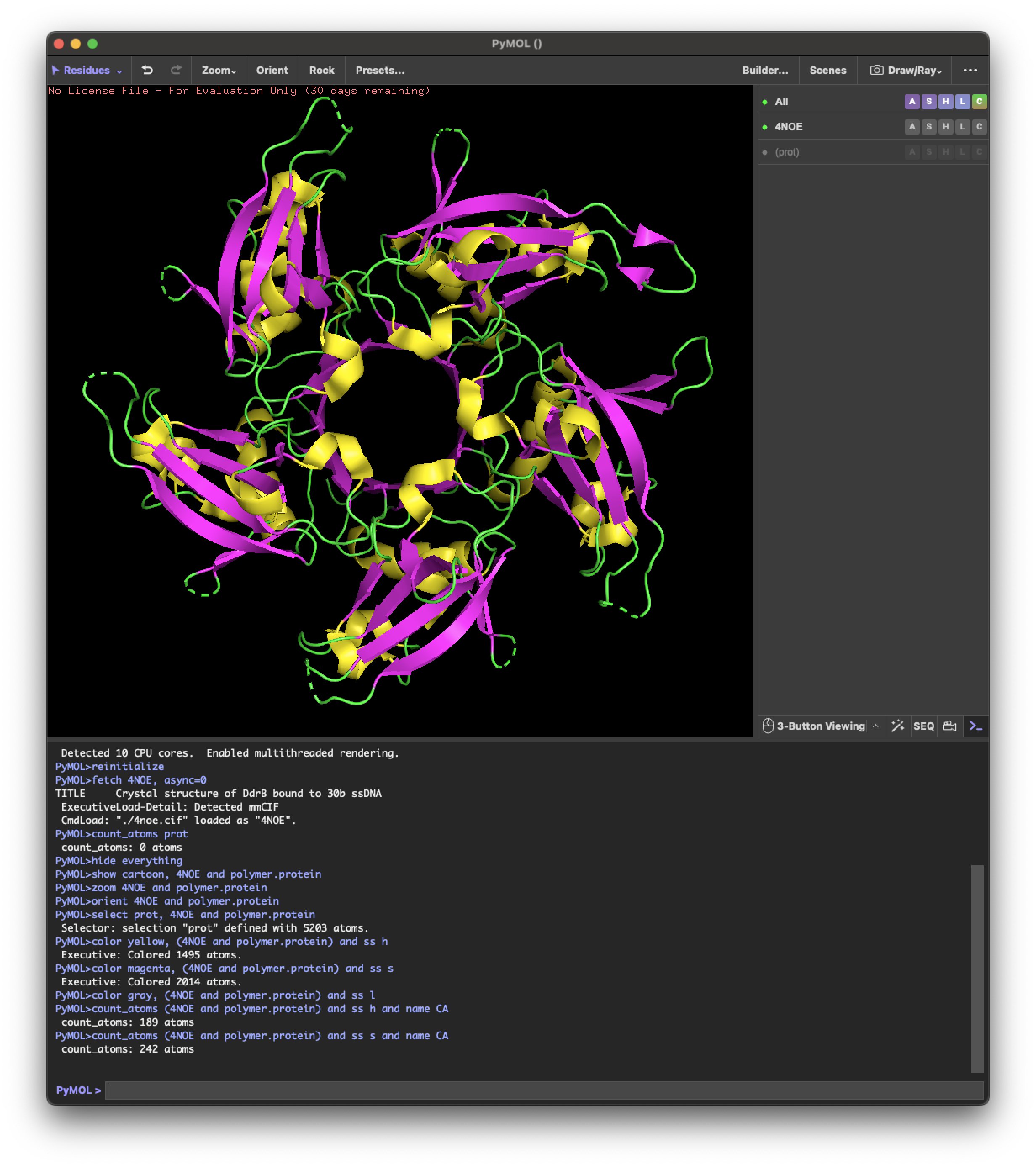
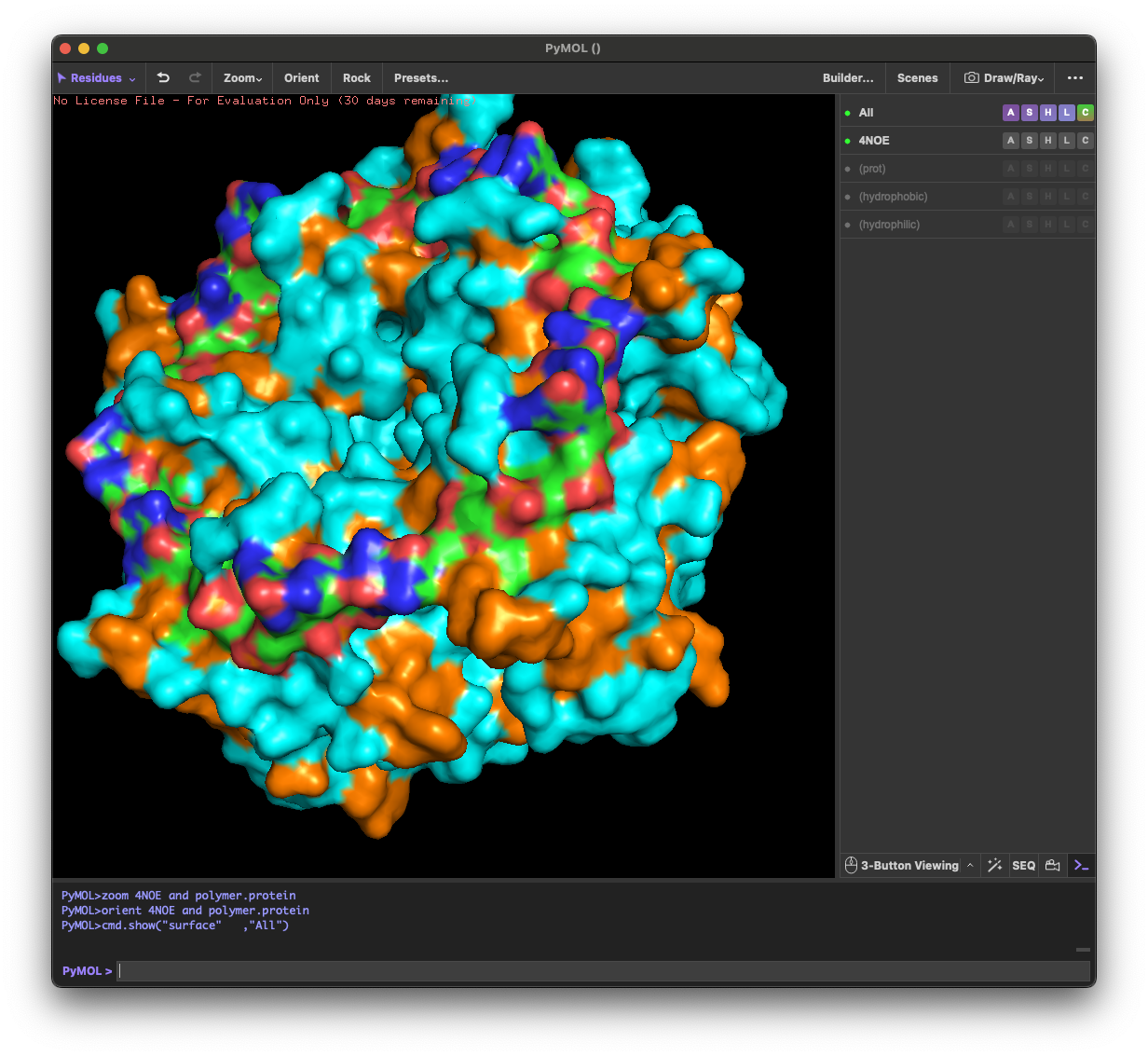
I selected the protein from RCSB PDB: 4NOE, which is DdrB, a single-stranded DNA-binding protein from Deinococcus radiodurans (this entry is a protein–ssDNA complex). I chose it because D. radiodurans is well known for extreme radiation tolerance, and DdrB is directly connected to DNA damage response/repair, making it a relevant example for studying UV/radiation-associated protein function with an available 3D structure.
2. Identify the amino acid sequence of your protein.
FASTA Sequence
>4NOE_1|Chains A, B, C, D, E|Single-stranded DNA-binding protein DdrB|Deinococcus radiodurans (1299)
DPFTMLQIEFITDLGARVTVNVEHESRLLDVQRHYGRLGWTSGEIPSGGYQFPIENEADFDWSLIGARKWKSPEGEELVIHRGHAYRRRELEAVDSRKLKLPAAIKYSRGAKVSDPQHVREKADGDIEYVSLAIFRGGKRQERYAVPG
>4NOE_2|Chain F|5'-D(*TP*TP*GP*CP*GP*CP*TP*TP*GP*CP*GP*CP*TP*TP*GP*CP*GP*CP*TP*TP*GP*CP*GP*CP*TP*TP*GP*CP*GP*CP)-3'|
TTGCGCTTGCGCTTGCGCTTGCGCTTGCGC
2.1. How long is it? What is the most frequent amino acid?
Sequence Length(DdrB): 148 aa
2.2. How many protein sequence homologs are there for your protein?
BLAST Input:
>4NOE_1|Chains A, B, C, D, E|Single-stranded DNA-binding protein DdrB|Deinococcus radiodurans (1299)
DPFTMLQIEFITDLGARVTVNVEHESRLLDVQRHYGRLGWTSGEIPSGGYQFPIENEADFDWSLIGARKWKSPEGEELVIHRGHAYRRRELEAVDSRKLKLPAAIKYSRGAKVSDPQHVREKADGDIEYVSLAIFRGGKRQERYAVPG
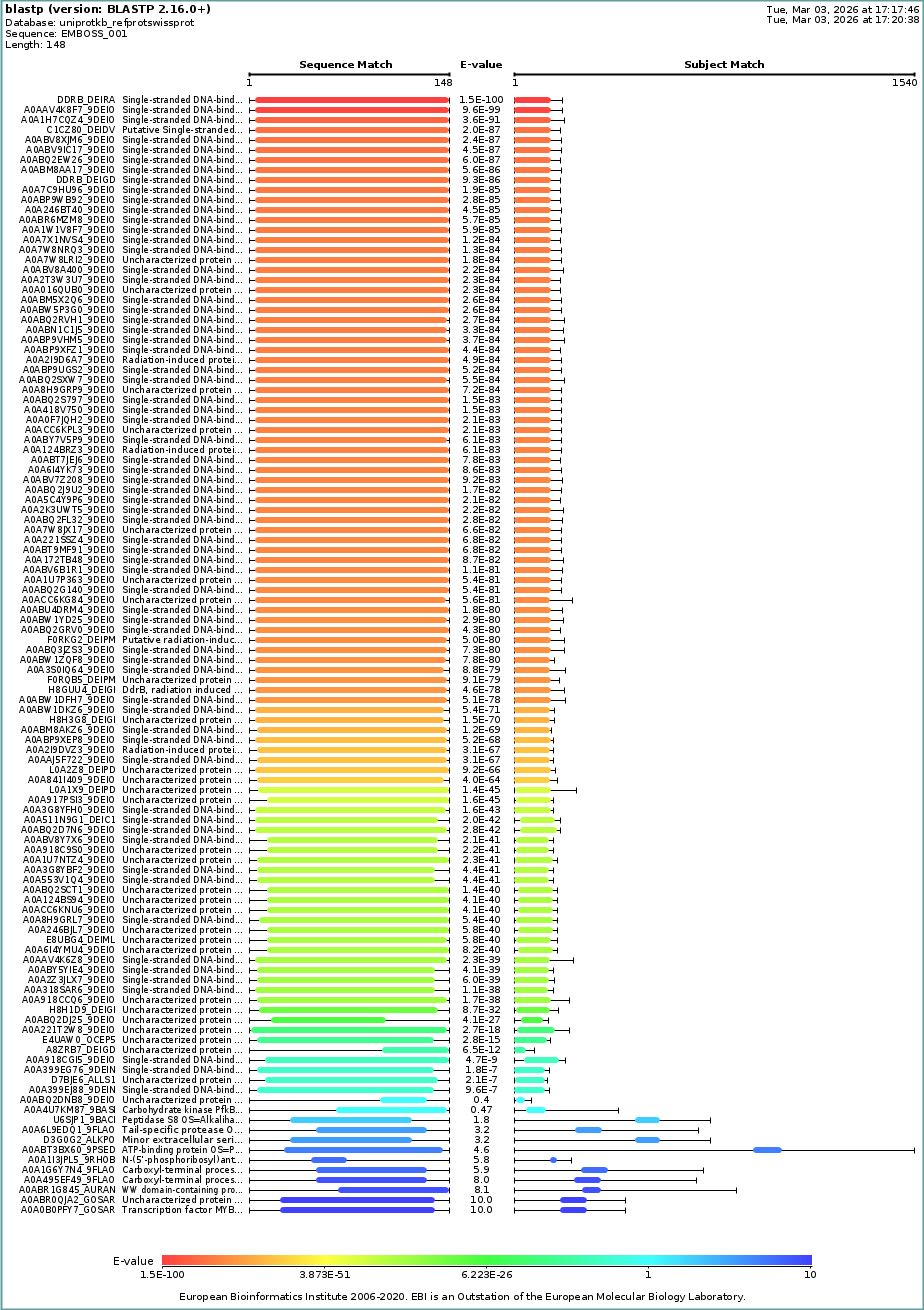
I used UniProt BLAST with default settings and reported the number of hits as an estimate of sequence homologs (based on BLAST similarity/E-value criteria).
112 hits
2.3. Does your protein belong to any protein family?
PF12747 — DdrB-like protein (DdrB)
3. Identify the structure page of your protein in RCSB
3.1. When was the structure solved? Is it a good quality structure?
Deposited: 2013-11-19
Released: 2015-05-20
Resolution: 2.20 Å (2.20 Å < 2.70 Å, good quality)
3.2. Are there any other molecules in the solved structure apart from protein?
Yes.
Nucleic acid: 30-nt ssDNA (Crystal structure of DdrB bound to 30b ssDNA)
CALCIUM ION (CA)
3.3 Does your protein belong to any structure classification family?
RCSB Classification: DNA BINDING PROTEIN/DNA
ECOD family: DdrB
ECOD architecture: beta barrels
4. 3D molecule visualization
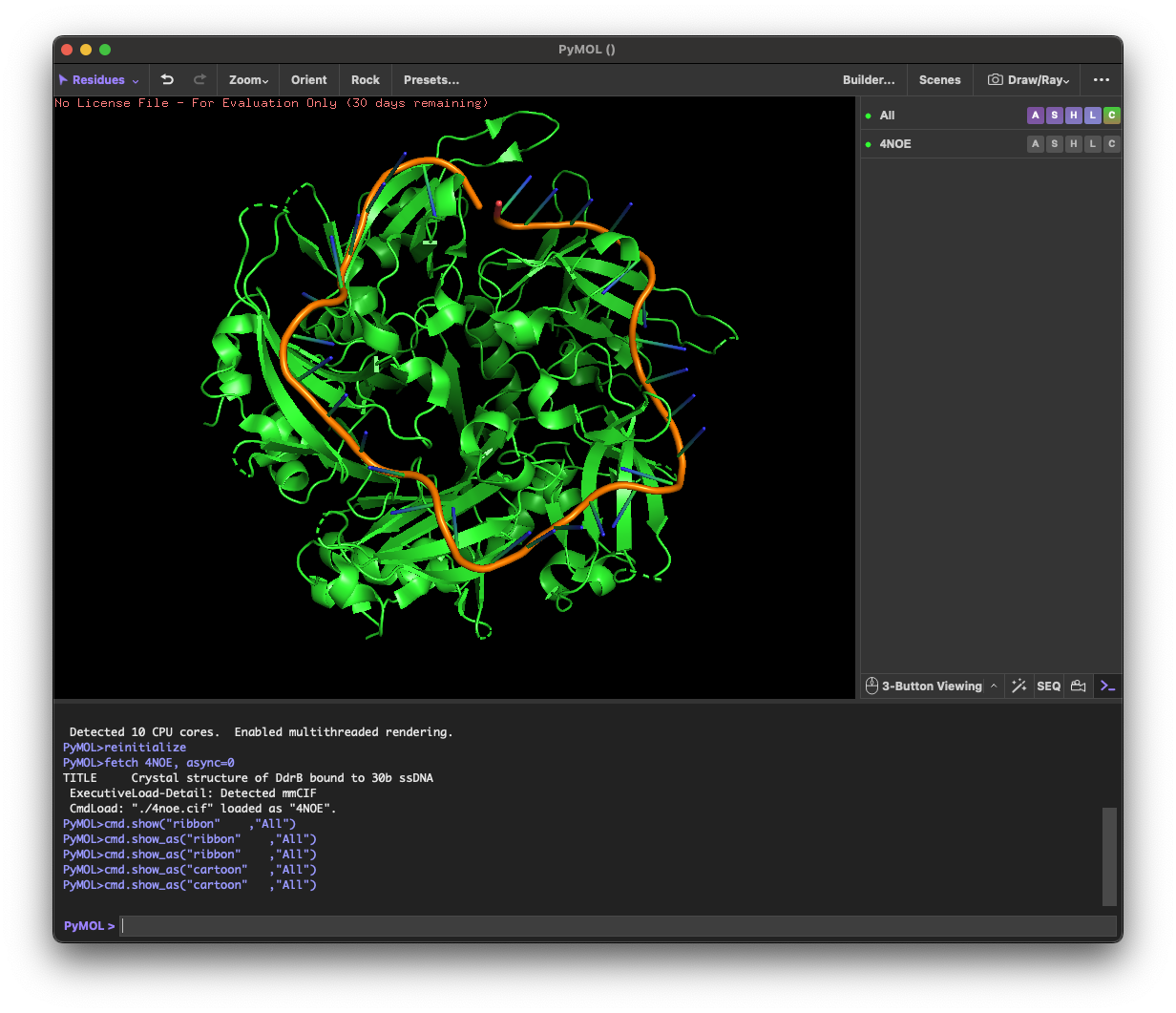
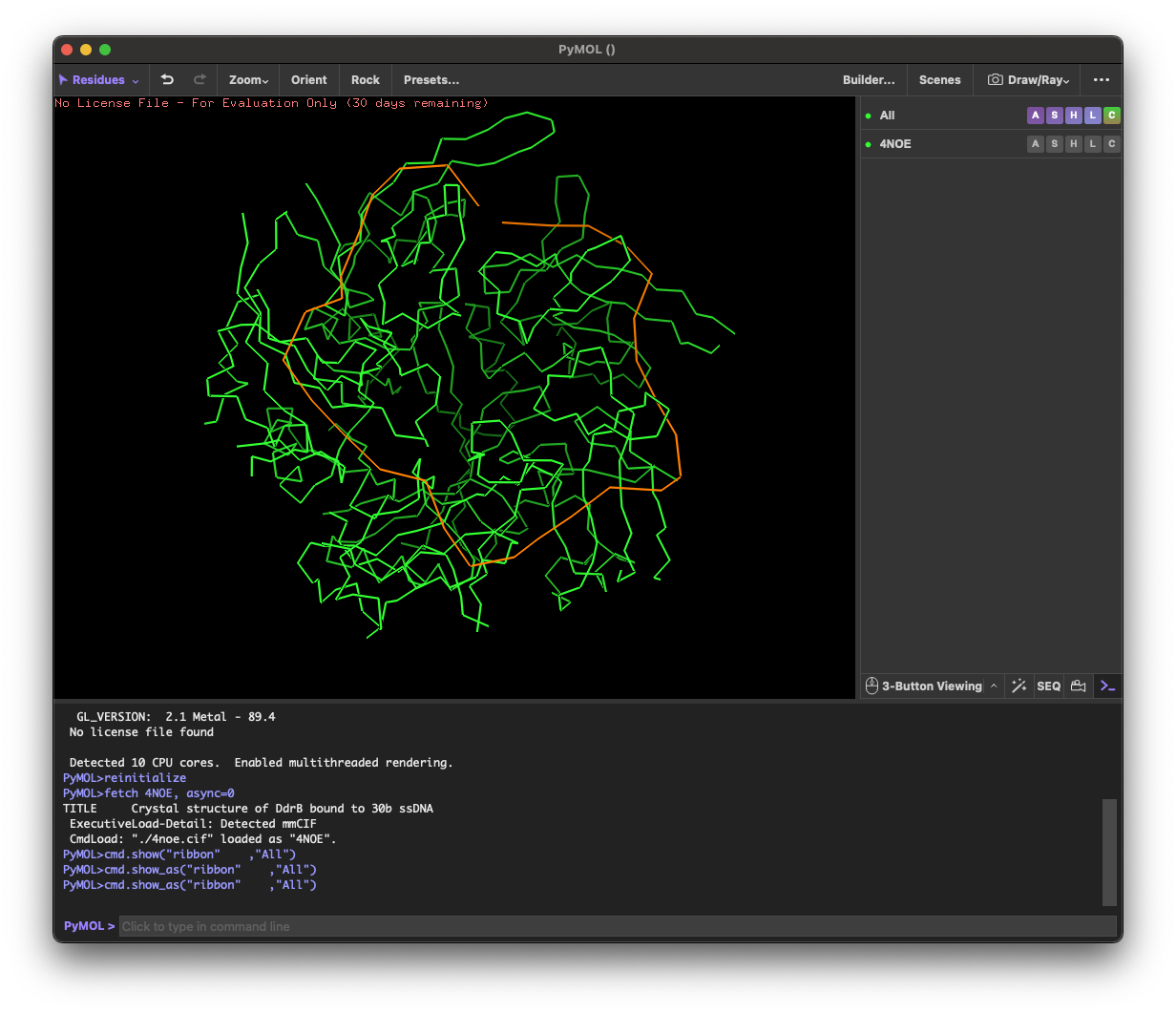
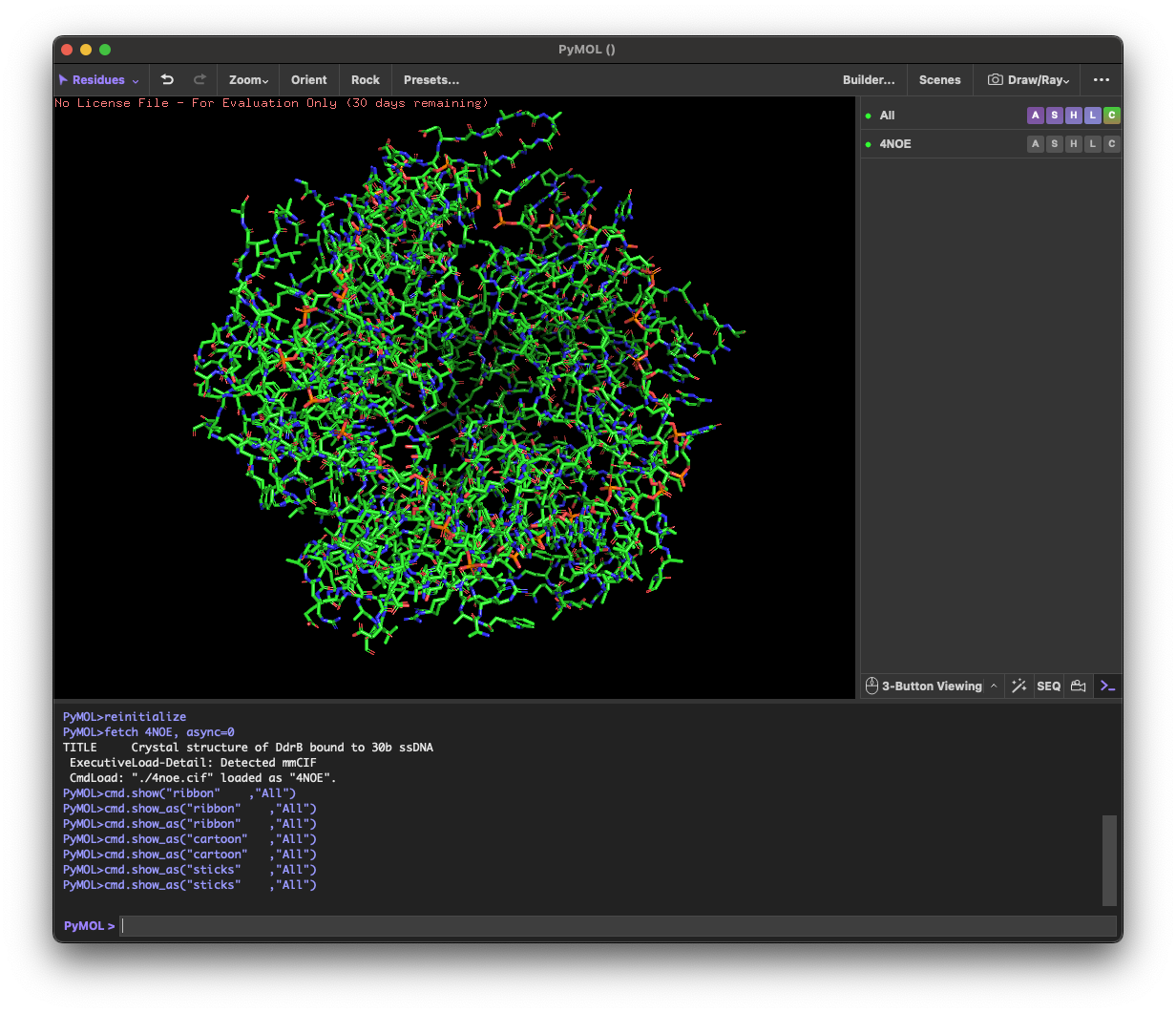
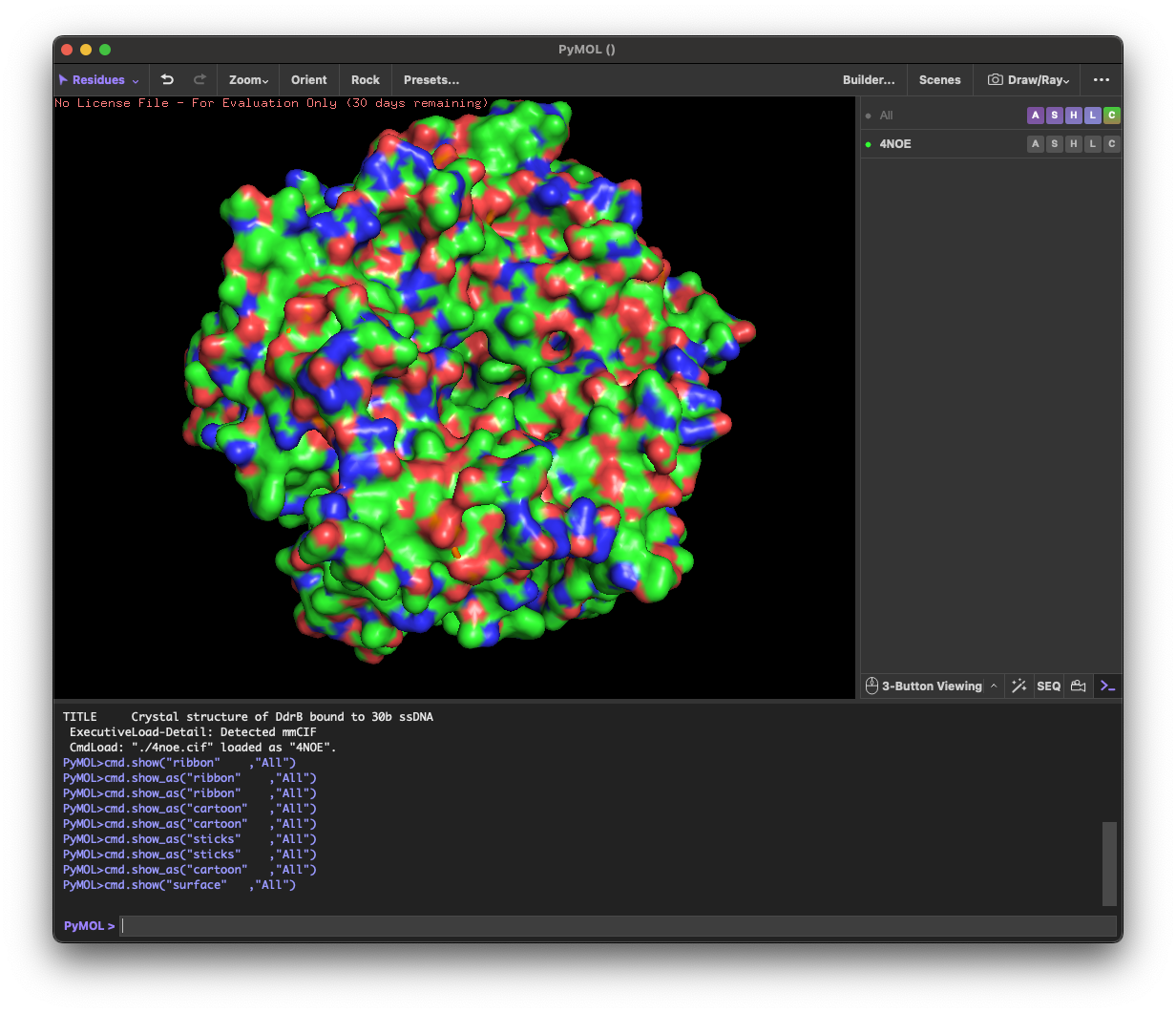
4.1. Visualize the protein as “cartoon”, “ribbon” and “ball and stick”.
4.2.Color the protein by secondary structure. Does it have more helices or sheets?
helix: 189
sheet: 242
It has more β-sheets than α-helices.
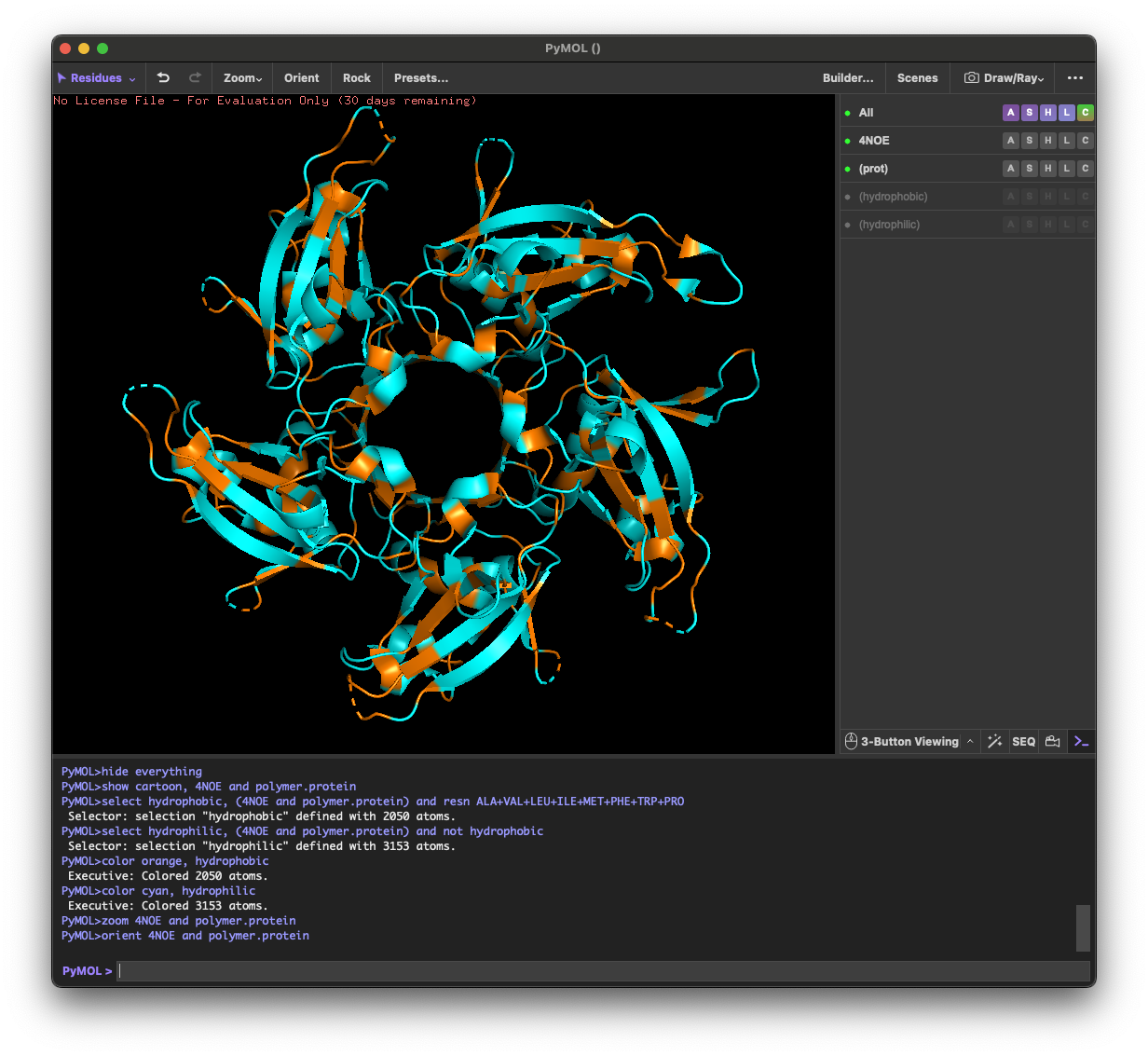
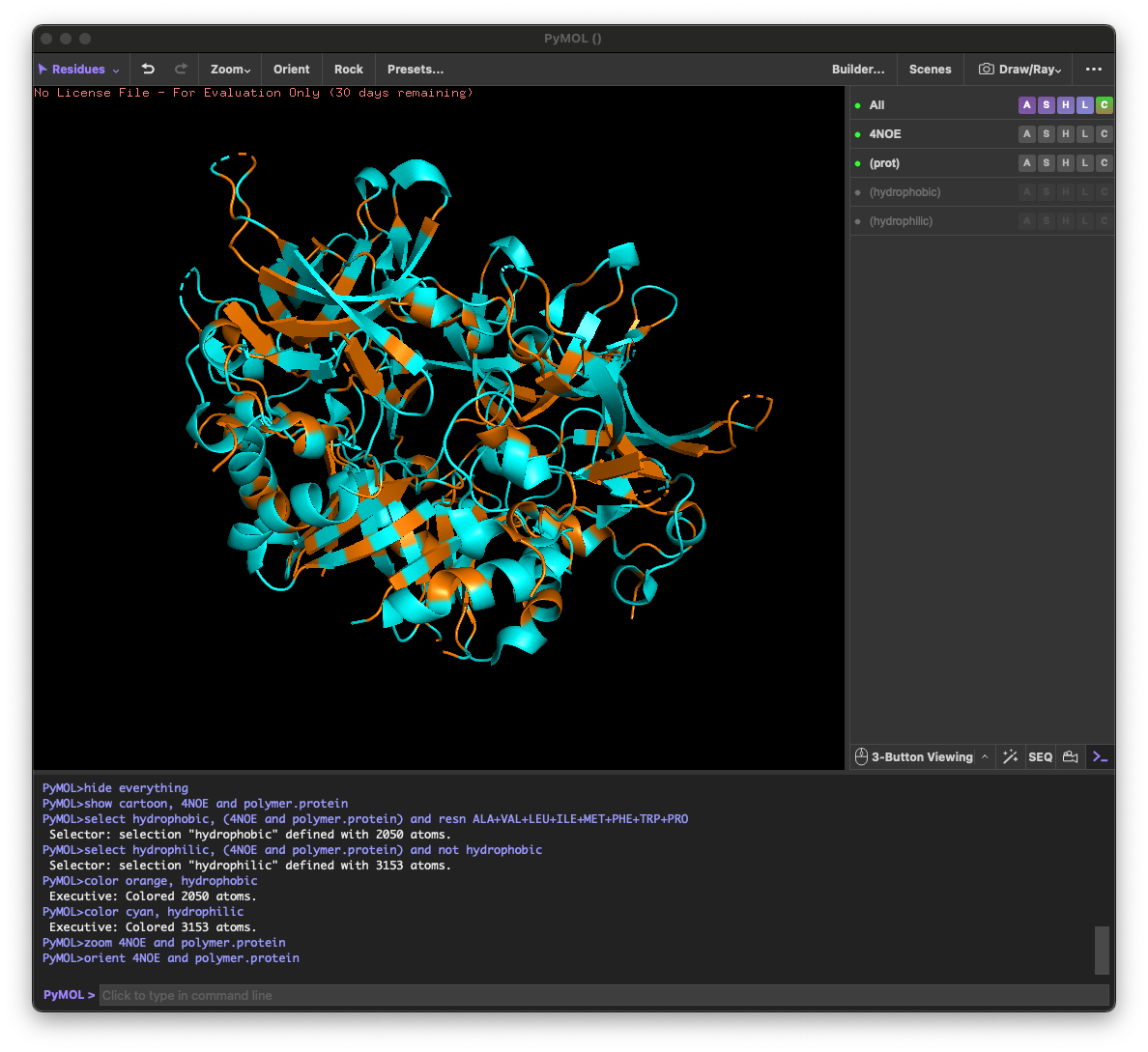
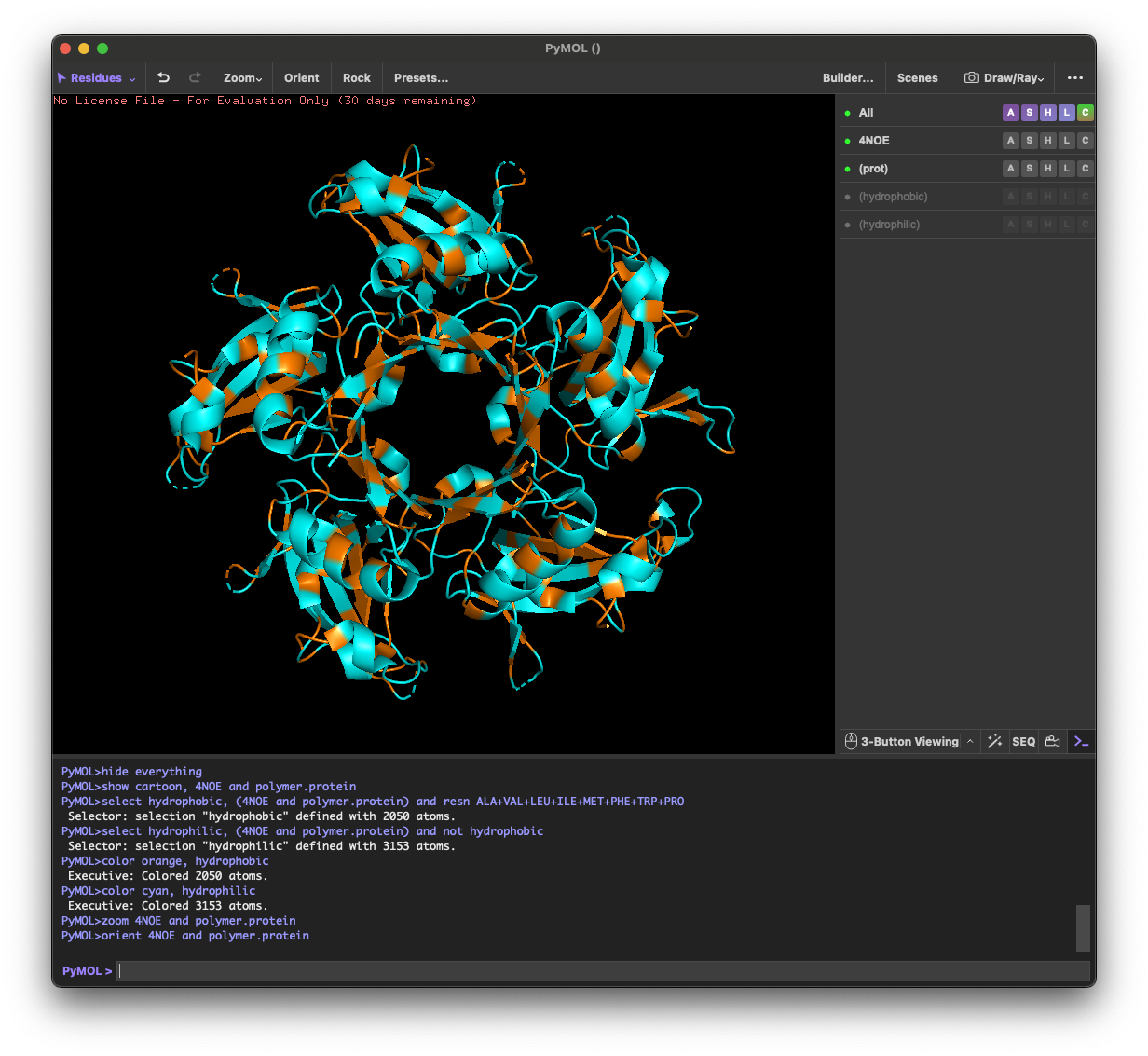
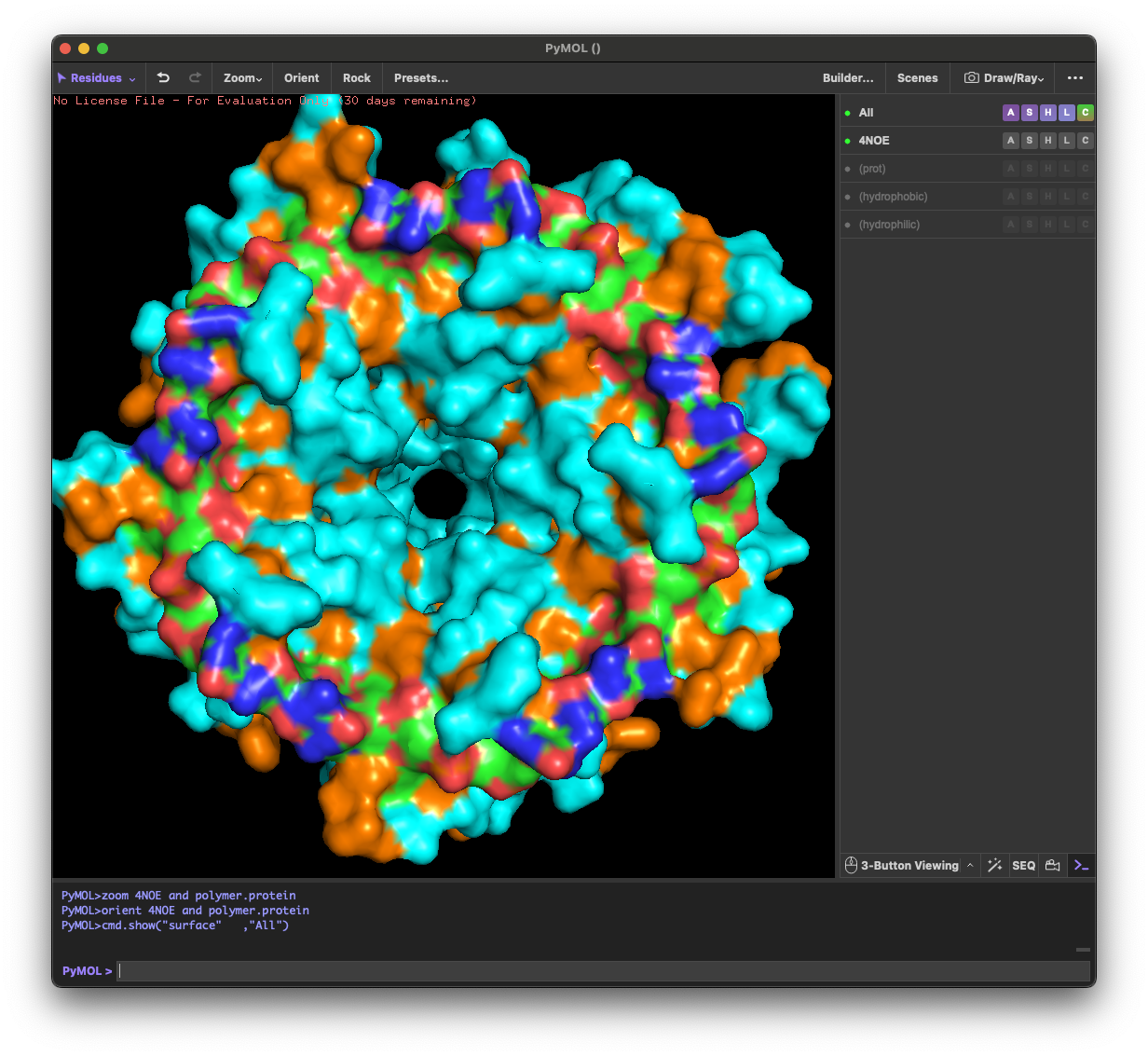
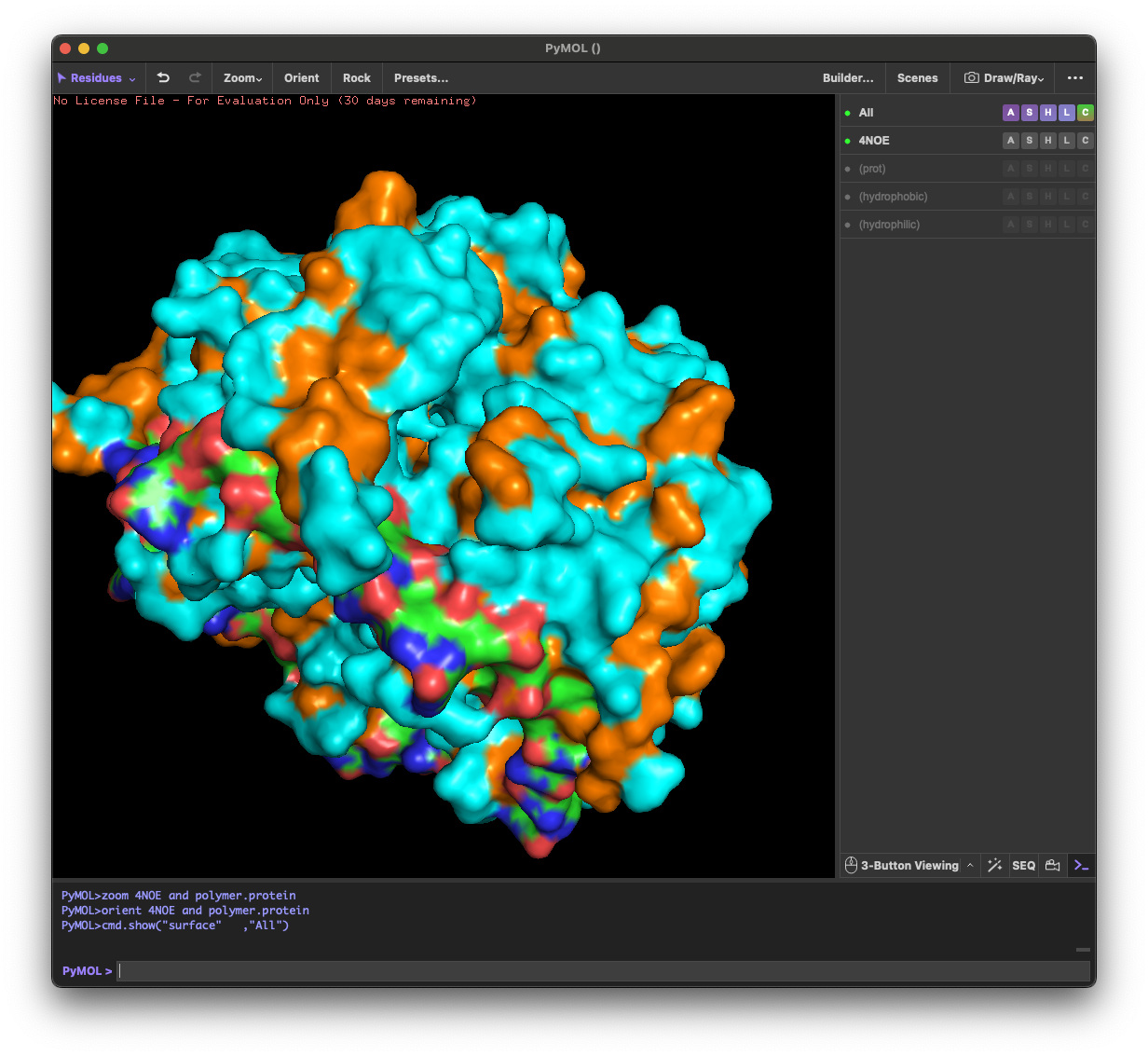
4.3. Color the protein by residue type. What can you tell about the distribution of hydrophobic vs hydrophilic residues?
Across the different views, hydrophilic residues (cyan) dominate and appear broadly distributed over the outer surface, especially along loops and exposed regions. Hydrophobic residues (orange) are more patchy and discontinuous, showing up as scattered stripes/spots across β-strands and some helical segments rather than forming one continuous hydrophobic face. Overall, the protein looks like a typical soluble complex: a largely hydrophilic exterior with localized hydrophobic patches that likely contribute to core stabilization and/or subunit–subunit interfaces.
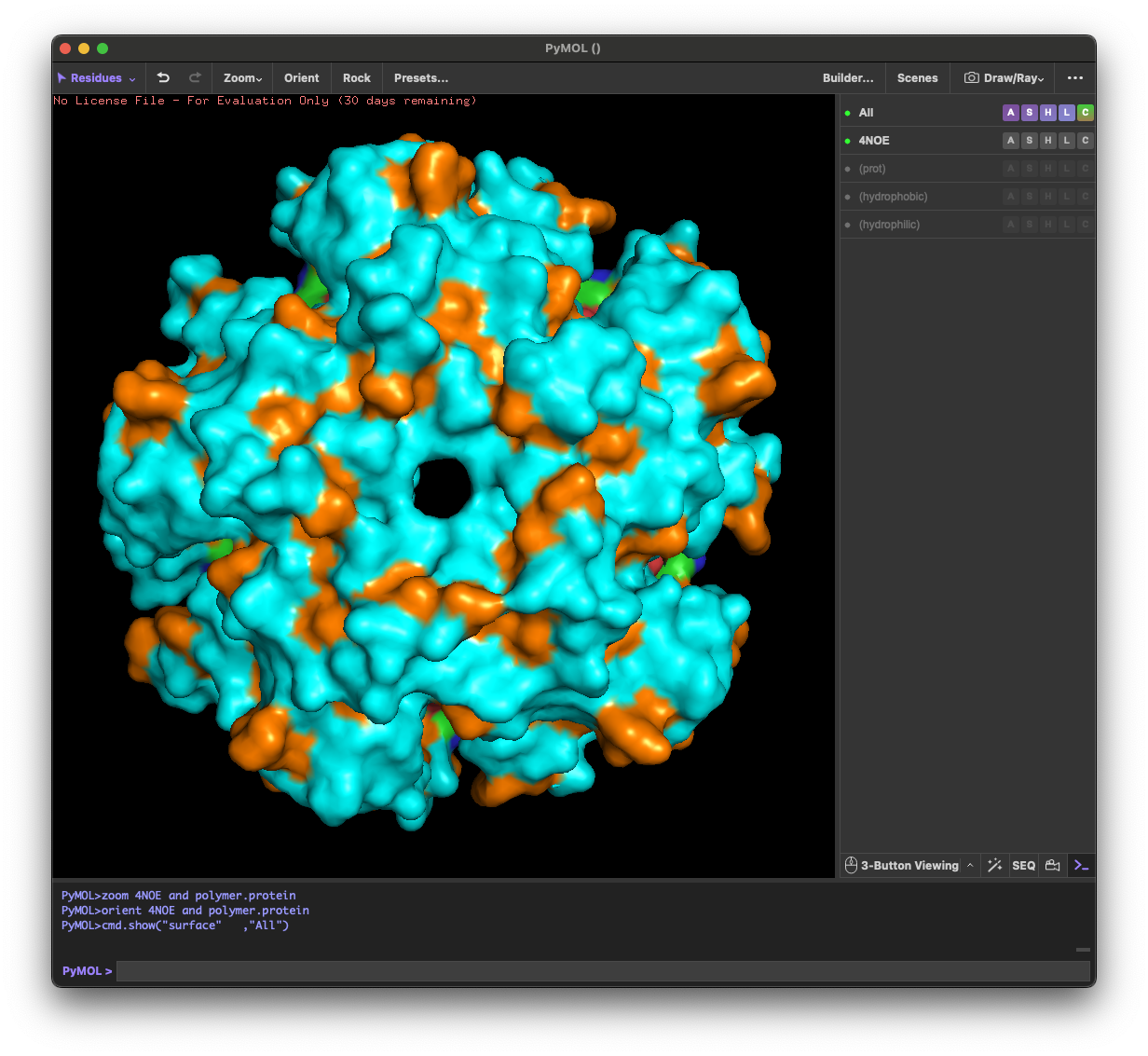
4.4. Visualize the surface of the protein. Does it have any “holes” (aka binding pockets)?
Yes. The surface rendering shows a clear pore-like hole near the center that remains visible across angles. There are also multiple smaller grooves and depressions on the surface, which could be potential binding pockets.
Part C: Using ML-Based Protein Design Tools
C1. Protein Language Modeling
1. Deep Mutational Scans
a. Use ESM2 to generate an unsupervised deep mutational scan of your protein based on language model likelihoods.
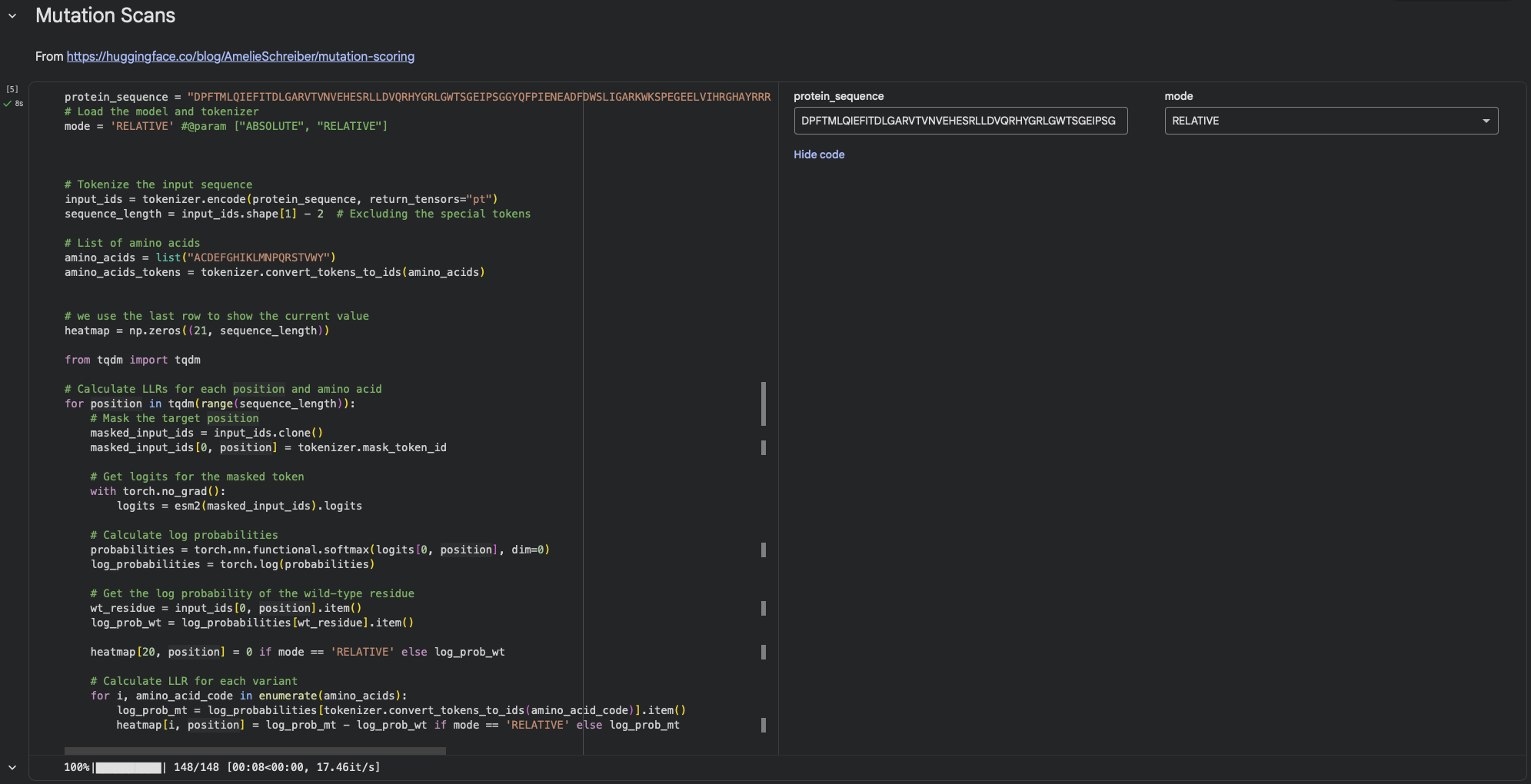
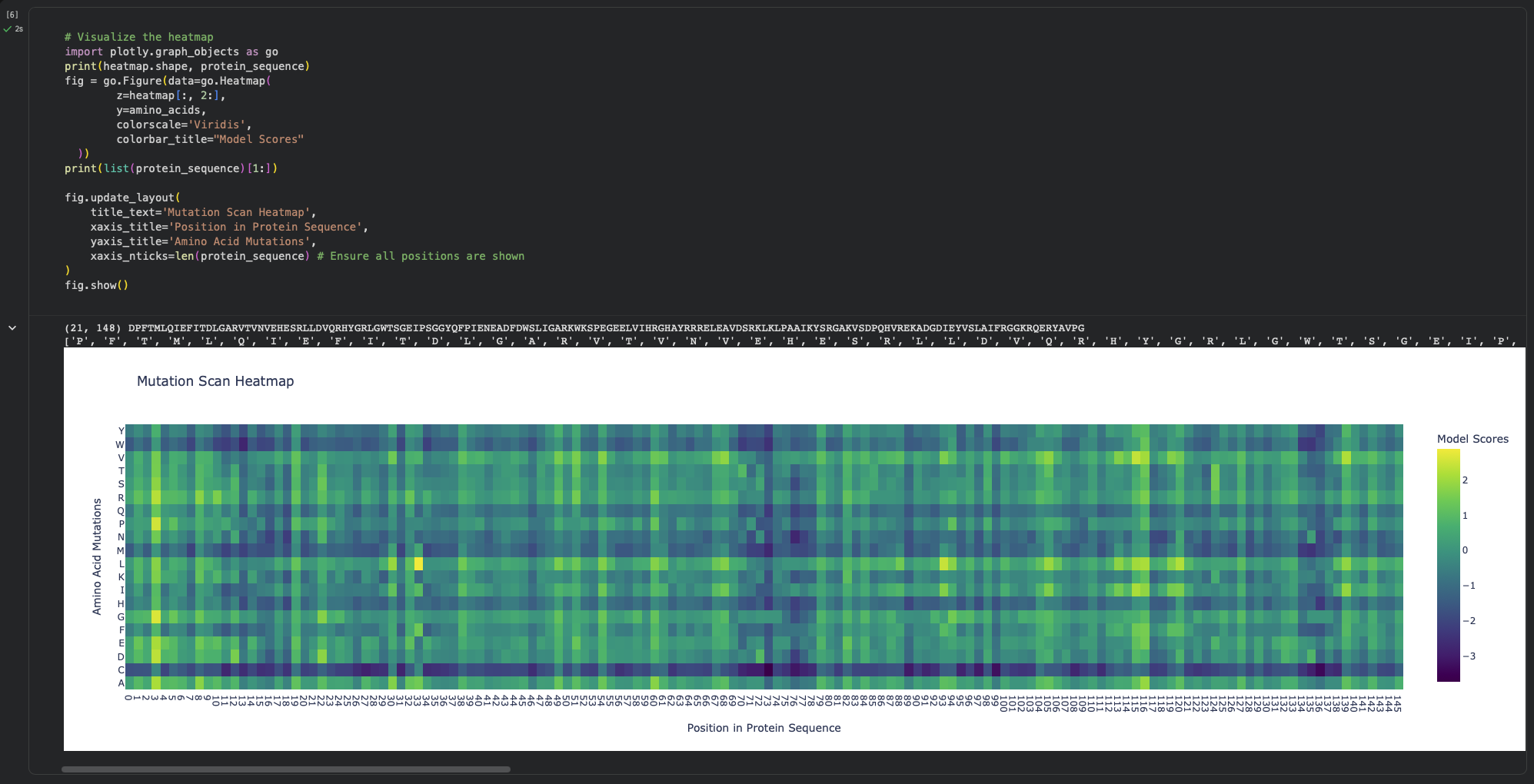
Using ESM2’s unsupervised likelihood-based scan on DdrB, most single–amino-acid substitutions have relatively modest effects, but a few positions show strong outliers, suggesting that some residues are much more constrained than others.
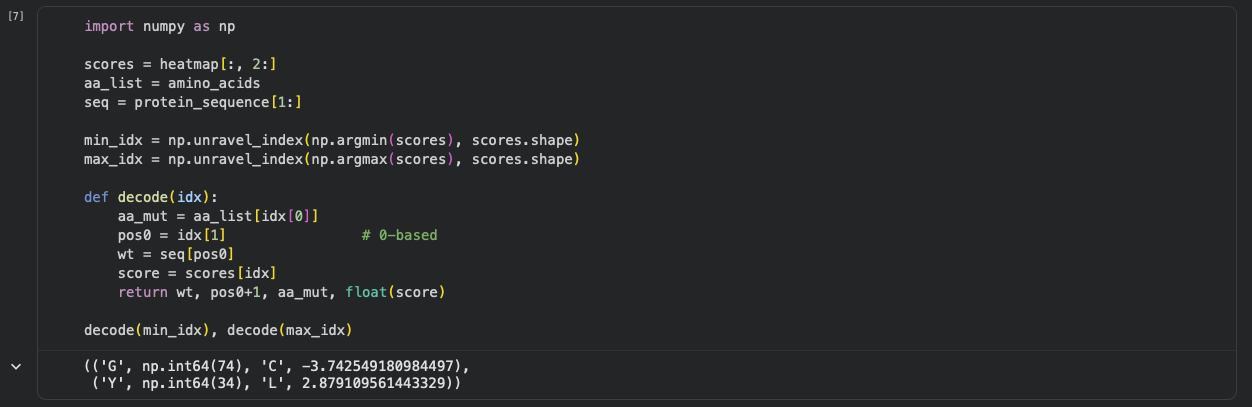
b. Can you explain any particular pattern? (choose a residue and a mutation that stands out)
One mutation that stands out is G74→C, which has the most unfavorable score (-3.74), indicating that position 74 is highly constrained and likely important for maintaining the protein’s stability/assembly. A strongly favorable outlier is Y34→L (+2.88).
2. Latent Space Analysis
In the latent space analysis section, the run failed with the error “ValueError: perplexity must be less than n_samples.”At that point the embedding array had shape (2, 320) (only two sequences were embedded), while the t-SNE settings used perplexity = 30, which requires a larger number of samples. As a result, the projection step did not complete and neighborhood interpretation was not possible in this run.
2. Protein Folding Folding a protein
1. Fold your protein with ESMFold. Do the predicted coordinates match your original structure?
I folded the DdrB sequence (length 148 aa) with ESMFold. The run completed with pTM = 0.204 and mean pLDDT = 34.332. These confidence metrics are low, so the predicted coordinates are not expected to reliably match the experimental PDB structure (4NOE), and any apparent similarity should be treated as uncertain.
Q1. What are some components in the Phusion High-Fidelity PCR Master Mix and what is their purpose?
Components in Phusion High-Fidelity PCR Master Mix:
DNA polymerase: a high-fidelity enzyme that synthesizes new DNA strands based on the template sequence.
dNTPs: the molecular building blocks used to make the new DNA strands.
Buffer: provides the proper chemical environment, such as pH and ionic strength, to maintain polymerase activity and stability.
MgCl₂ (Mg²⁺): an essential cofactor for DNA polymerase and is required for its catalytic activity.
Q2. What are some factors that determine primer annealing temperature during PCR?
In PCR, primer annealing temperature (Ta) is primarily determined by the primer melting temperature (Tm), usually 2-5°C below the lowest Tm of the pair.
Key factors influencing this include:
primer length
guanine-cytosine (GC) content
base sequence (nearest-neighbor interactions)
salt concentration (e.g., Na⁺)
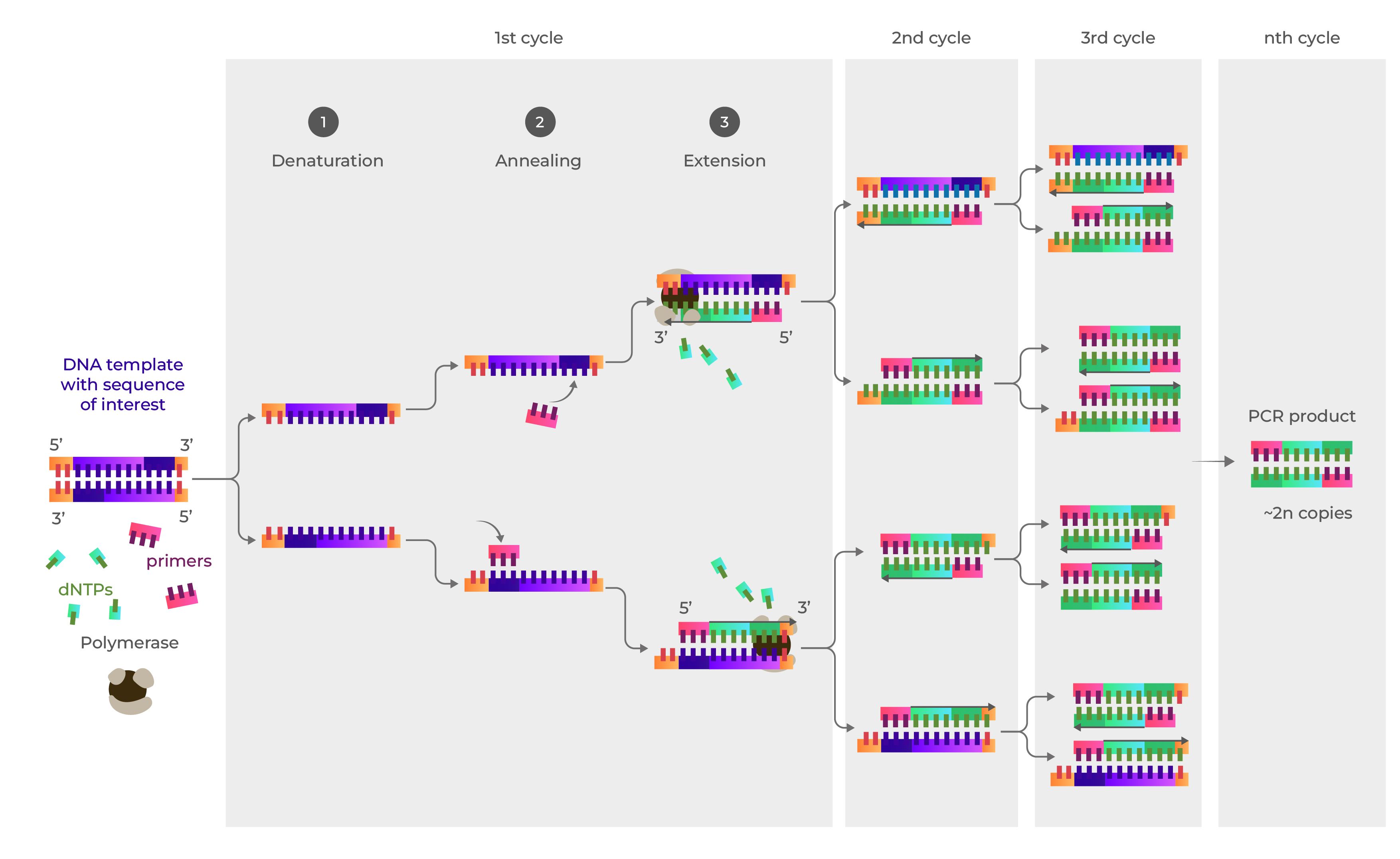
Q3. There are two methods from this class that create linear fragments of DNA: PCR, and restriction enzyme digests. Compare and contrast these two methods, both in terms of protocol as well as when one may be preferable to use over the other.
PCR amplifies a specific DNA sequence from a template using heat-stable polymerase and primers, while restriction enzymes (endonucleases) cut double-stranded DNA at specific recognition sites. PCR is ideal for generating large quantities of DNA from small amounts of starting material, while restriction digests are often used for cutting out known fragments from existing plasmids or genomic DNA.
Q4. How can you ensure that the DNA sequences that you have digested and PCR-ed will be appropriate for Gibson cloning?
The vector is properly linearized.
Each digested or PCR-amplified fragment has the correct 20–40 bp overlapping homology with the adjacent fragment.
All DNA fragments are clean and of the expected size.
After assembly, confirm the final construct by sequencing.
Q5. How does the plasmid DNA enter the E. coli cells during transformation?
Plasmid DNA enters E. coli when the cells are shocked by heat shock or electrical shock, which temporarily opens the cell membrane and allows the plasmid to enter by diffusion.
Q6. Describe another assembly method in detail (such as Golden Gate Assembly)
Explain the other method in 5 - 7 sentences plus diagrams (either handmade or online).
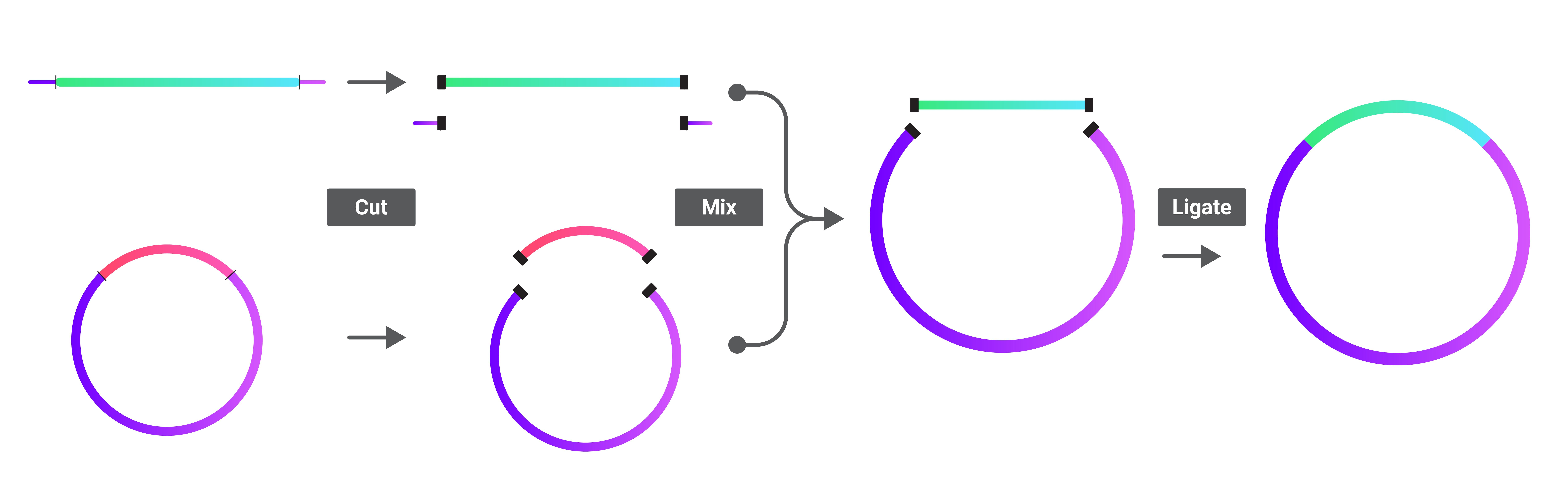
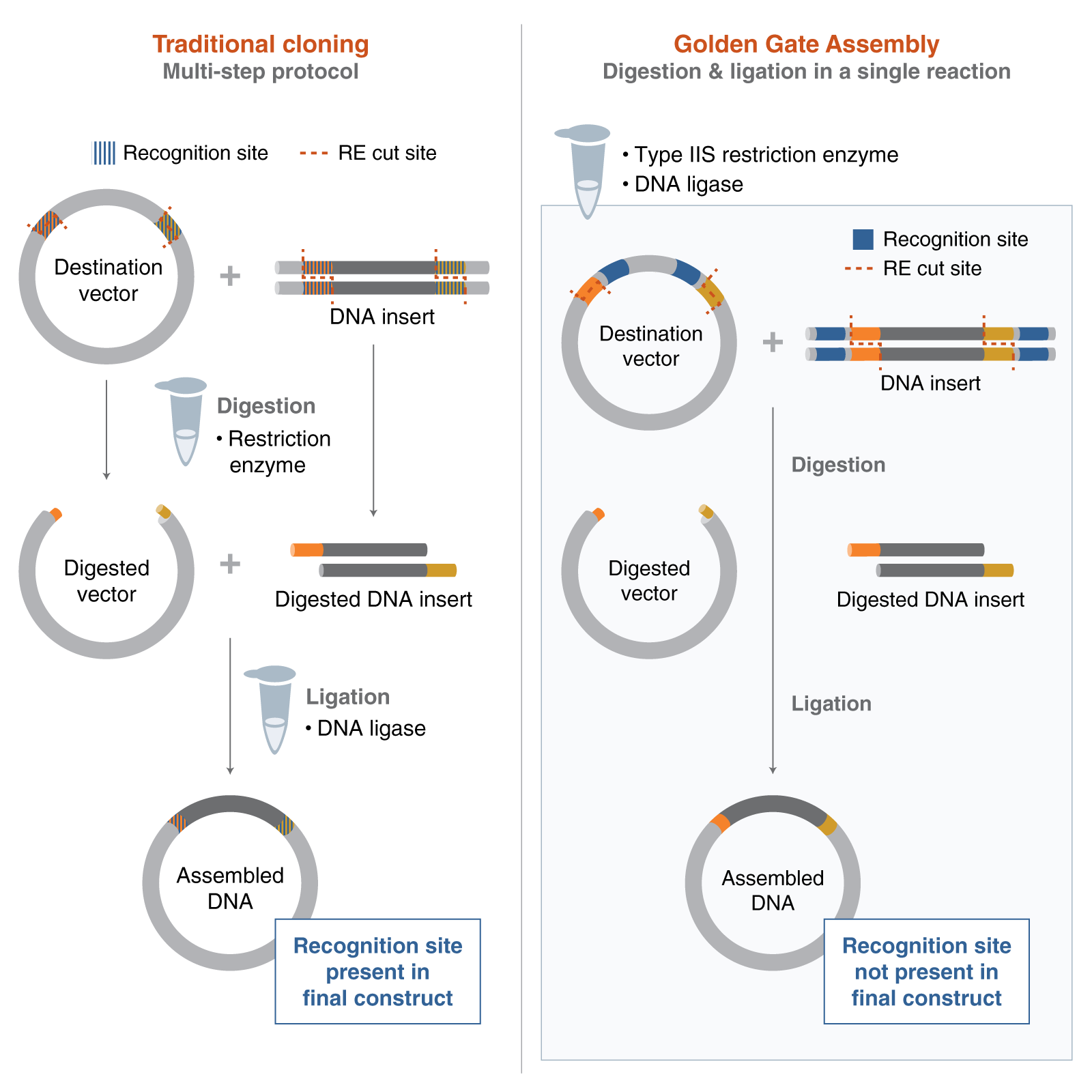
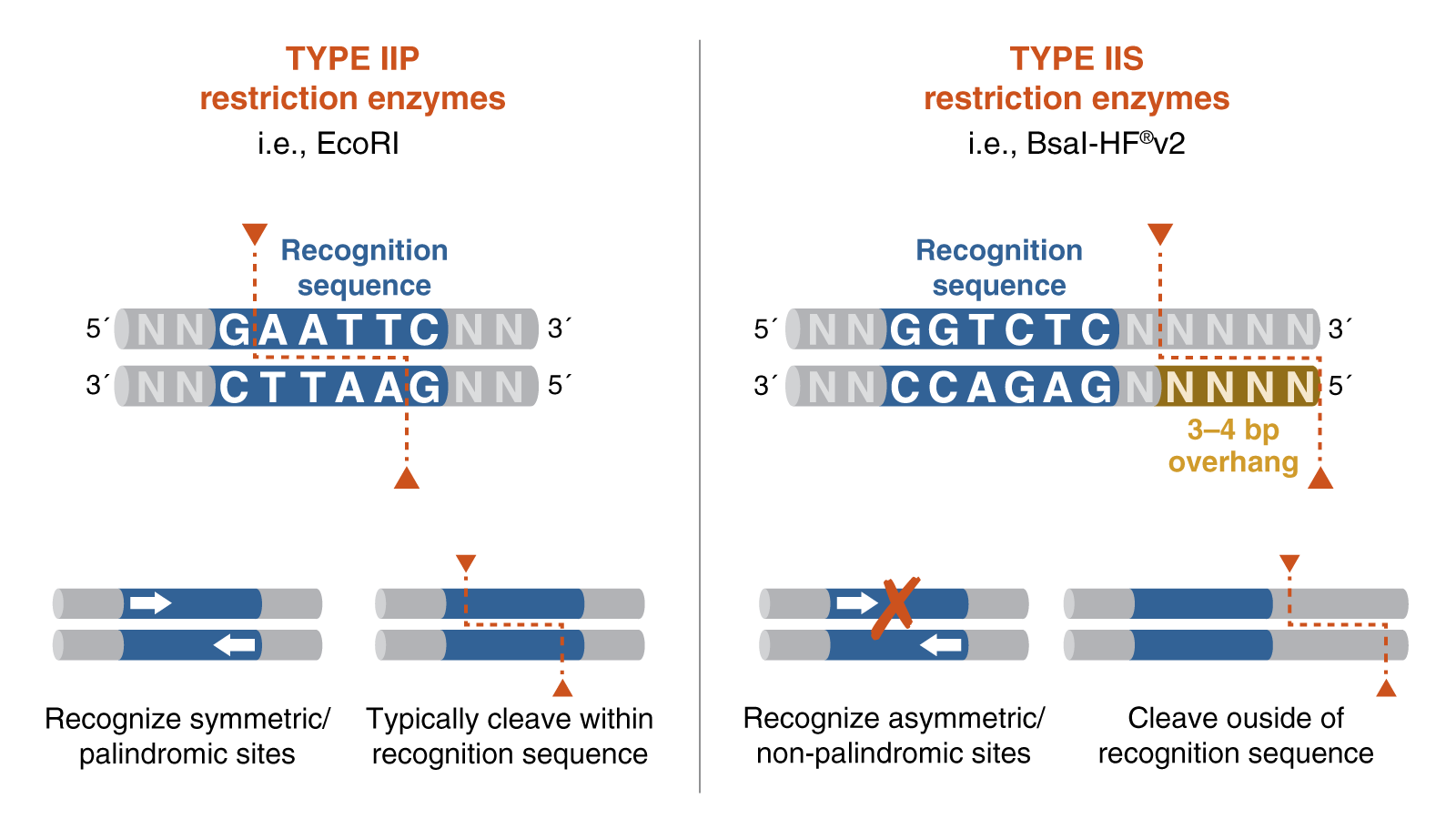
Golden Gate Assembly is a one-step cloning method that uses a Type IIS restriction enzyme and DNA ligase in the same reaction tube. Unlike standard restriction enzymes, Type IIS enzymes cut outside of their recognition sites, which allows researchers to design custom overhangs that determine the order of assembly. DNA fragments and the destination vector are designed with matching overhangs, so the pieces ligate together in a predefined sequence. Because the recognition sites are removed during the reaction, the correctly assembled product is typically scarless and is not re-cut by the enzyme. This also helps reduce the background from empty vector, since unassembled or re-ligated vector can be cut again during the cycling reaction. Golden Gate Assembly is especially useful for efficient multi-fragment and modular DNA assembly.
Model this assembly method with Benchling or Asimov Kernel!
Part 2. Asimov Kernel
Methodology & Logic Implementation
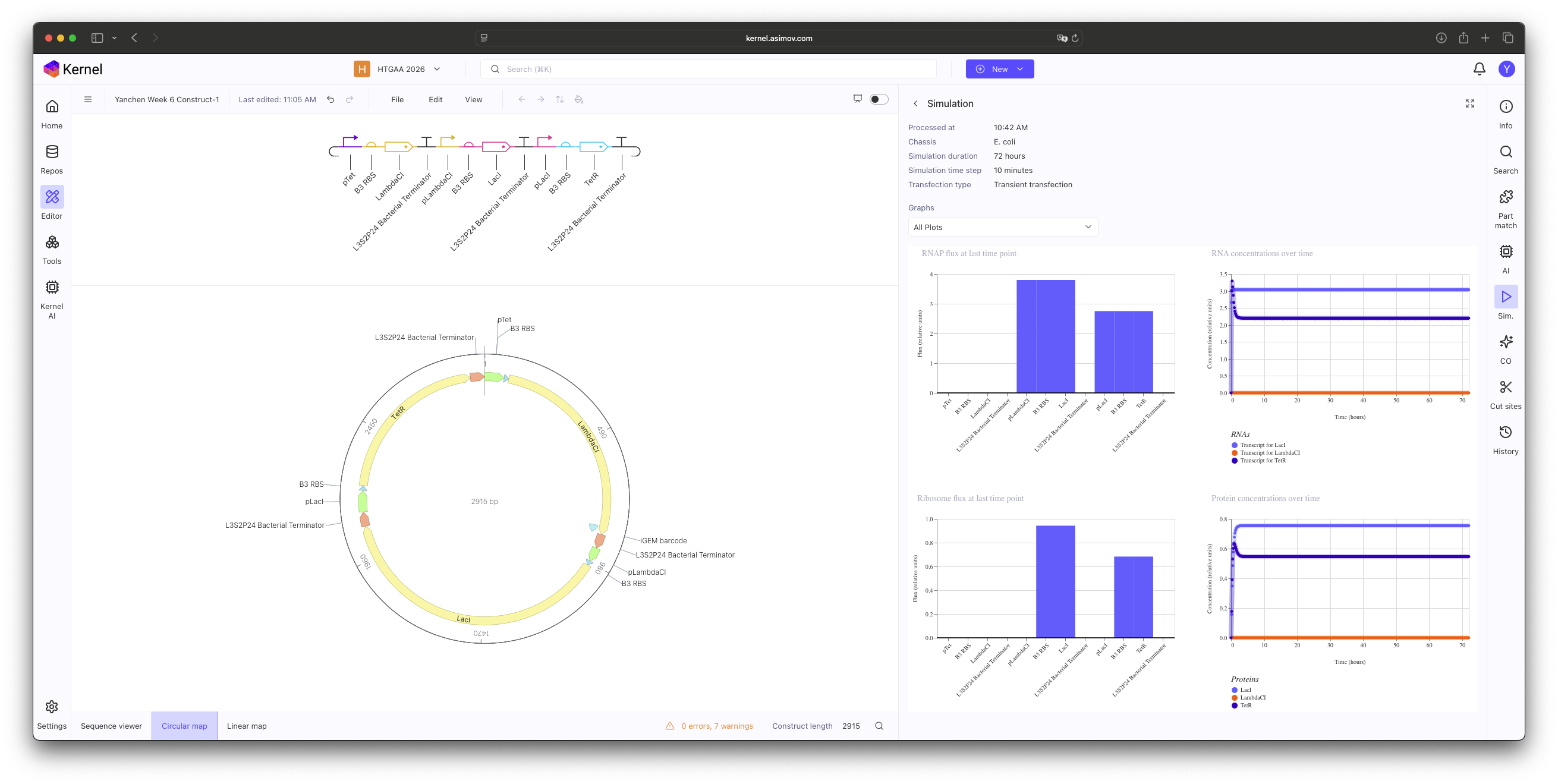
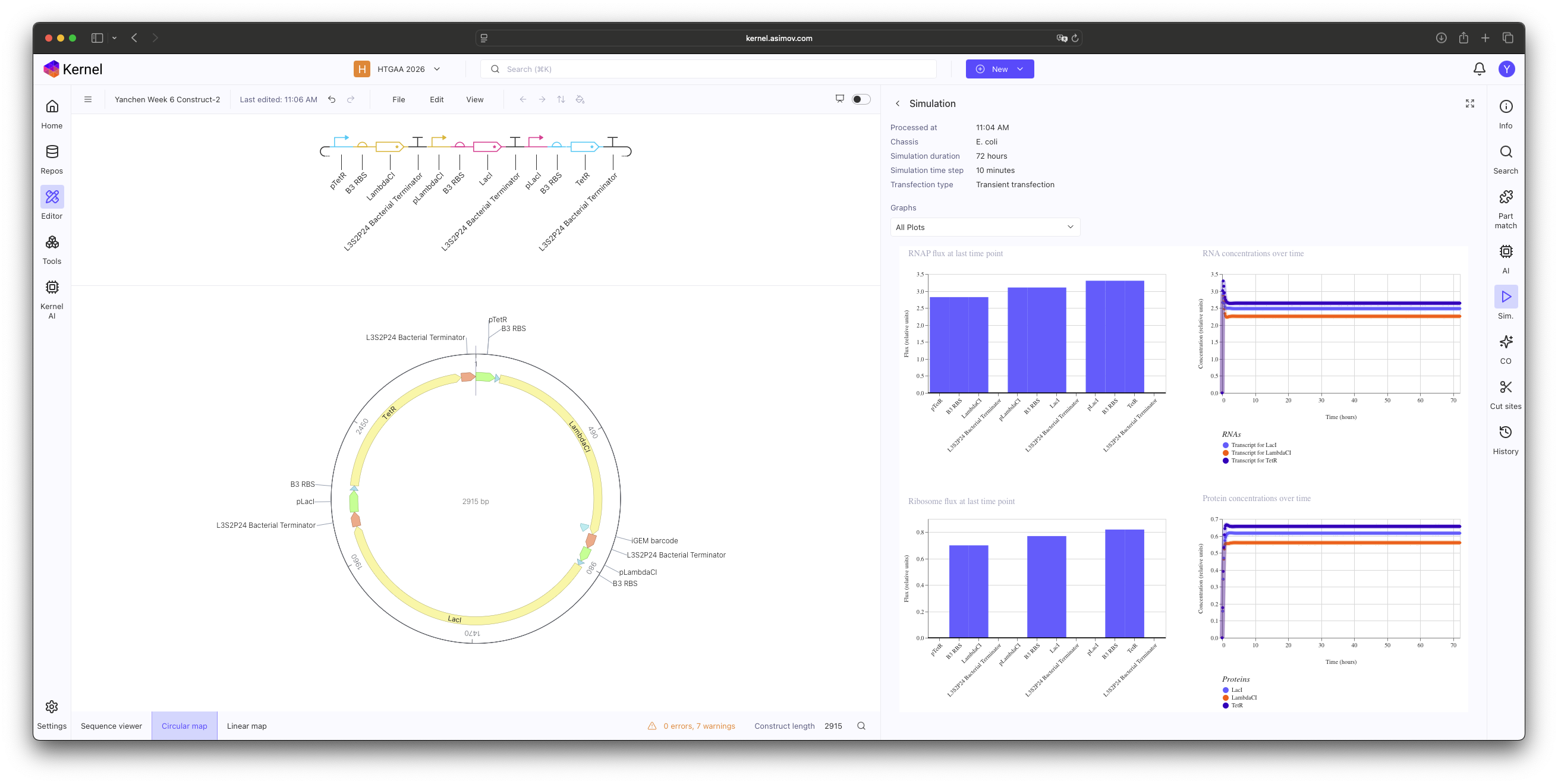
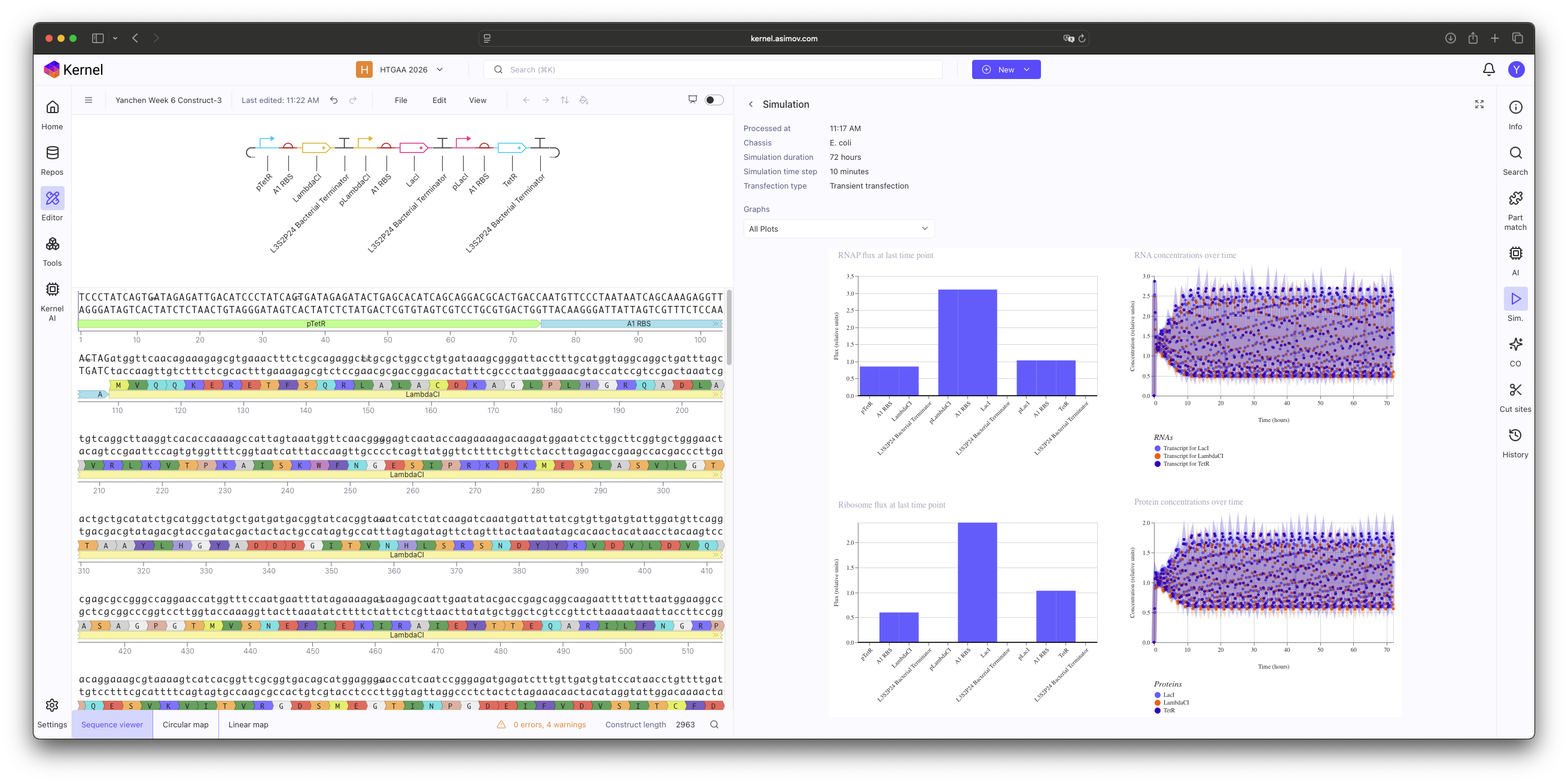
In this practice, I manually selected individual components from the Characterized Bacterial Parts repository to construct a functional Repressilator. The core logic was implemented by cross-linking three inhibitory units in a cyclic negative feedback loop: pTet → LambdaCI, pLambdaCI → LacI, and pLacI → TetR.
My primary focus was to explore how specific biological factors impact the simulation outcome, including:"
RNAP & Ribosome Flux: Assessing the transcriptional and translational efficiency at each junction.
RNA & Protein Concentrations: Monitoring the temporal dynamics of gene expression.
Component Strength: Evaluating if the specific Characterized Parts selected (such as the B3 RBS) are kinetically compatible with the desired oscillatory behavior.
Construct 1: Promoter mismatch.
Construct 2: Ribosome binding site (B3 RBS). Initial simulations showed no oscillations; this is likely due to the B3 RBS being too potent, which leads to an excessive accumulation of repressor proteins that the system cannot degrade quickly enough to maintain a periodic rhythm.
Construct 3: Change Ribosome binding site (B3 RBS) to A1 RBS.
Results & Analysis
The simulation initially resulted in stable, non-oscillatory steady states. Through this exercise, I identified that using standard Characterized Parts requires more than just correct logic; it requires kinetic tuning. Specifically, the B3 RBS proved to be too potent, leading to a protein synthesis rate that overwhelmed the system’s degradation capacity. This ’locked’ the Protein and RNA concentrations at a high-level equilibrium, preventing the periodic ‘dip’ necessary for oscillation.
Week 7 HW: Genetic Circuits Part II
Part 1. Intracellular Artificial Neural Networks (IANNs)
Q1. What advantages do IANNs have over traditional genetic circuits, whose input/output behaviors are Boolean functions?
IANNs can process inputs in a more continuous and analog way. This allows them to respond to gradual changes in input levels and produce more complex behaviors, such as weighted integration, thresholding, and pattern classification. Because of this, IANNs are better for tasks where biological signals are noisy and not strictly binary. They can also represent more complicated decision boundaries than traditional logic-gate-based circuits.
Q2. Describe a useful application for an IANN; include a detailed description of input/output behavior, as well as any limitations an IANN might face to achieve your goal.
An IANN can be used for disease sensing in mammalian cells. It could integrate multiple signals, such as inflammation, hypoxia, and cancer-related RNAs, and produce an output only when the overall pattern matches a disease state. The output could be a fluorescent reporter or a therapeutic protein. A key limitation is that IANNs can be difficult to design and tune because of noise, cell-to-cell variability, and unpredictable interactions between components.
Q3. Draw a diagram for an intracellular multilayer perceptron where layer 1 outputs an endoribonuclease that regulates a fluorescent protein output in layer 2.
Part 2. Fungal Materials
1. What are some examples of existing fungal materials and what are they used for? What are their advantages and disadvantages over traditional counterparts?
Existing fungal materials include mycelium packaging, insulation, leather-like materials, and mycelium composites for products or building panels. They are used as alternatives to plastic foam, synthetic leather, and some lightweight construction materials. Their advantages are that they are renewable, biodegradable, lightweight, and can be grown from waste. Their disadvantages are that they may have lower durability, lower water resistance, and less consistent mechanical properties than traditional materials.
2. What might you want to genetically engineer fungi to do and why? What are the advantages of doing synthetic biology in fungi as opposed to bacteria?
I would want to genetically engineer fungi to create a higher-performance biofilament for 3D printing. For example, fungi could be engineered to improve the filament’s mechanical strength, flexibility, printability, and stability. This would be useful because it could make fungal materials more practical for fabrication while still being sustainable. Fungi are advantageous over bacteria because they naturally grow as filamentous networks, which makes them more suitable for forming continuous material structures. They are therefore a promising platform for developing stronger and more functional bio-based materials for 3D printing.
Week 9 HW: Cell Free Systems
Part A: General and Lecturer-Specific Questions
General Homework Questions
Q1. Explain the main advantages of cell-free protein synthesis over traditional in vivo methods, specifically in terms of flexibility and control over experimental variables. Name at least two cases where cell-free expression is more beneficial than cell production.
Cell-free protein synthesis (CFPS) is more flexible than in vivo expression because it removes cell growth and viability constraints and allows the reaction environment to be adjusted directly. In a cell-free system, researchers can independently tune DNA template concentration, salts, cofactors, energy substrates, pH, redox conditions, crowding agents, and added folding or membrane-assisting components. This makes it easier to study protein production as a controllable biochemical process.
CFPS is especially beneficial when:
(1) Producing toxic proteins or peptides, because the product does not need to accumulate inside a living host.
(2) When producing complex proteins such as membrane proteins or post-translationally modified proteins, because the reaction can be supplemented with vesicles, microsomes, chaperones or glycosylation machinery.
Q2. Describe the main components of a cell-free expression system and explain the role of each component.
A cell-free expression system usually contains:
(1) Cell extract or purified translation/transcription machinery: this provides ribosomes, translation factors, tRNAs, enzymes, and sometimes endogenous metabolic activities needed for protein synthesis.
(2) DNA or RNA template: this encodes the target protein and includes the regulatory elements required for transcription and/or translation.
(3) RNA polymerase and nucleotides: RNA polymerase transcribes DNA into mRNA, and nucleotides supply the building blocks for RNA synthesis.
(4) Amino acids, tRNAs, and translation factors: these are required to decode mRNA and assemble the protein chain.
(5) Energy system: substrates such as PEP, creatine phosphate, pyruvate, glucose, or maltodextrin support ATP regeneration for sustained transcription and translation.
(6) Salts, buffer, and cofactors: magnesium, potassium, ammonium, NAD, CoA, and buffering agents maintain ionic balance, enzyme activity, and pH stability.
(7) Optional additives such as chaperones, redox agents, detergents, liposomes, nanodiscs, or microsomes can be added for difficult proteins.
Q3. Why is energy provision regeneration critical in cell-free systems? Describe a method you could use to ensure continuous ATP supply in your cell-free experiment.
Energy regeneration is critical because transcription and translation consume large amounts of ATP and GTP, and a batch cell-free reaction quickly loses productivity if energy is depleted or inhibitory byproducts accumulate. Without regeneration, protein yield drops because the system can no longer support RNA synthesis, amino acid activation, and ribosome function. One practical method is to use a continuous-exchange or fed-batch setup together with an ATP-regenerating substrate such as PEP or maltodextrin. In this design, the reaction chamber is supplied with fresh small molecules from a feeding solution while waste products diffuse away, which prolongs reaction lifetime and maintains ATP production.
Q4. Compare prokaryotic versus eukaryotic cell-free expression systems. Choose a protein to produce in each system and explain why.
A prokaryotic cell-free system, especially E. coli-based CFPS, is usually cheaper, faster, and higher-yielding, but it has limited native capacity for complex eukaryotic post-translational modifications. I would choose sfGFP or a simple bacterial enzyme for an E. coli system because these proteins do not require glycosylation and are ideal for rapid, low-cost, high-yield expression.
A eukaryotic cell-free system, such as CHO, wheat germ, or tobacco BY-2 extract, is more suitable for proteins that need more complex folding or post-translational processing. I would choose a full-length IgG antibody for a CHO-based system because CHO extracts can support production of functional antibodies and are better suited for proteins that benefit from eukaryotic folding and glycosylation-related machinery.
Q5. How would you design a cell-free experiment to optimize the expression of a membrane protein? Discuss the challenges and how you would address them in your setup.
To optimize a membrane protein, I would set up a screening experiment in parallel conditions using the same DNA template but different membrane-mimicking environments. For example, I would compare nanodiscs, liposomes, mild detergents, and, if available, microsome-containing extracts. I would also vary temperature, magnesium concentration, and lipid composition, then measure both total yield and functional activity.
The main challenges are aggregation of hydrophobic transmembrane domains, incorrect insertion or orientation, and loss of activity due to the absence of a natural membrane environment. I would address these by adding membrane mimics during expression, choosing lipid compositions that better match the protein’s hydrophobic thickness, and using chaperones or lower-temperature conditions to improve folding. For activity testing, I would use ligand binding or transport assays rather than relying only on total protein yield, because a membrane protein can be expressed but still be nonfunctional.
Q6. Imagine you observe a low yield of your target protein in a cell-free system. Describe three possible reasons for this and suggest a troubleshooting strategy for each.
Reason 1: Poor template quality or template degradation.
Linear DNA can be degraded by nucleases, and low-quality DNA can reduce transcription efficiency. A good troubleshooting strategy is to switch to a plasmid template, improve template purification, or protect linear DNA with strategies such as GamS-like inhibition or terminal protection.
Reason 2: Energy limitation or inhibitory byproduct accumulation.
If ATP regeneration is insufficient, or if phosphate and acidic byproducts accumulate, transcription and translation will slow down. I would troubleshoot this by optimizing the energy substrate, adjusting magnesium and buffer conditions, or moving from a simple batch reaction to fed-batch or continuous exchange.
Reason 3: Protein-specific folding or solubility problems.
Some proteins misfold, aggregate, or require special environments such as oxidizing conditions, chaperones, or membranes. I would troubleshoot this by lowering the reaction temperature, adding folding chaperones, adjusting redox conditions, or supplementing membrane mimics if the target is membrane-associated. If the protein is highly complex, I would also consider switching from a prokaryotic to a eukaryotic cell-free system.
Homework question from Peter Nguyen
Freeze-dried cell-free systems can be incorporated into all kinds of materials as biological sensors or as inducible enzymes to modify the material itself or the surrounding environment. Choose one application field — Architecture, Textiles/Fashion, or Robotics — and propose an application using cell-free systems that are functionally integrated into the material. Answer each of these key questions for your proposal pitch:
Write a one-sentence summary pitch sentence describing your concept.
A topology-optimized architectural substrate is coated with a biopolymer-based surface layer made from freeze-dried cell-free manufactured ingredients to improve durability while adding a lower-carbon functional finish.
How will the idea work, in more detail? Write 3-4 sentences or more.
The base material would first be designed to use less material while increasing surface area for coating adhesion. A freeze-dried cell-free system would be used off-site to produce a dry biopolymer ingredient, such as a polysaccharide- or protein-based additive. This ingredient would then be blended into a coating and applied to the surface by spraying, dipping, or brushing. After curing, the coating would help improve adhesion, bridge microcracks, and create a tougher protective layer on top of the structural substrate.
What societal challenge or market need will this address?
This idea addresses the need for more sustainable building materials and longer-lasting surface finishes. Many architectural coatings and repair layers rely on carbon-intensive ingredients and still fail through cracking, abrasion, or moisture damage. A bio-derived coating could reduce maintenance frequency, extend service life, and support lower-carbon construction without requiring a complete change in structural material systems.
How do you envision addressing the limitation of cell-free reactions (e.g., activation with water, stability, one-time use)?
The cell-free system would be used only during the manufacturing stage to produce the biopolymer ingredient for the coating. Water activation would take place in a controlled setting off-site, where the freeze-dried reaction can be rehydrated, run once, and converted into a stable dry product. That finished biopolymer would then be blended into the coating formula, so the final architectural coating would function as a passive material layer and would not depend on ongoing biological activity after application. This approach improves stability, avoids maintenance challenges linked to repeated activation, and makes one-time use acceptable because the reaction is completed before the material is installed on the building.
Homework question from Ally Huang
How does deep-space radiation affect the integrity of stored DNA templates, and can that damage reduce the reliability of freeze-dried cell-free protein production systems such as BioBits® in future space missions?
Background information
Deep-space radiation damages DNA and is a major risk for astronauts beyond Earth’s protective atmosphere. Future missions may also depend on stored DNA templates and freeze-dried cell-free systems to manufacture sensors, medicines, or enzymes on demand with minimal equipment. My proposal asks whether radiation exposure degrades those DNA instructions enough to reduce later protein production. This matters for human exploration because damaged DNA stockpiles could weaken in-flight diagnostics and biomanufacturing, and it is scientifically interesting because it tests how the space environment affects the information layer of synthetic biology, not only living cells.
Molecular or genetic target
A GFP reporter DNA cassette used as a model stored genetic template for later cell-free protein production in space.
How the target relates to the challenge
The reporter DNA is a stand-in for any DNA blueprint that astronauts might store and later load into BioBits. If radiation introduces strand breaks or base damage, PCR amplification should become less efficient and the cell-free system should produce less fluorescent protein. Measuring expression from the same cassette after different exposure conditions turns DNA integrity into a simple functional readout. This connects a molecular target to a practical space question: can freeze-dried biomanufacturing remain reliable after DNA templates spend time in the space environment?
Hypothesis or research goal
My hypothesis is that DNA templates exposed to higher space-radiation dose or lower shielding will show lower PCR recoverability and lower GFP output in BioBits than protected templates. The reasoning is simple: ionizing radiation can damage DNA, and BioBits needs DNA instructions plus water to synthesize protein. If this is true, future missions should store critical DNA libraries behind better shielding or refresh them periodically. If expression remains stable despite exposure, that would support the idea that freeze-dried DNA-plus-cell-free kits are robust enough for long missions. Either result would be useful because it would test a key bottleneck between DNA storage and on-demand biomanufacturing in space.
Experimental plan
I would test identical dried GFP DNA templates stored in two conditions: shielded and minimally shielded. Matching ground controls would stay on Earth. After exposure, each sample would be amplified with miniPCR and then added to BioBits; the P51 viewer would measure endpoint GFP fluorescence. Controls would include a fresh positive-control template, a no-DNA negative control, and non-flown matched DNA. The main data would be fluorescence intensity, PCR success or yield, and expression level normalized across conditions.