Week 4 HW: PROTEIN DESIGN PART I

Deinococcus radiodurans. Credit USU/Michael Daly
Part B: Protein Analysis and Visualization
1. Briefly describe the protein you selected and why you selected it.
I selected the protein from RCSB PDB: 4NOE, which is DdrB, a single-stranded DNA-binding protein from Deinococcus radiodurans (this entry is a protein–ssDNA complex). I chose it because D. radiodurans is well known for extreme radiation tolerance, and DdrB is directly connected to DNA damage response/repair, making it a relevant example for studying UV/radiation-associated protein function with an available 3D structure.
2. Identify the amino acid sequence of your protein.
FASTA Sequence
2.1. How long is it? What is the most frequent amino acid?
Sequence Length(DdrB): 148 aa
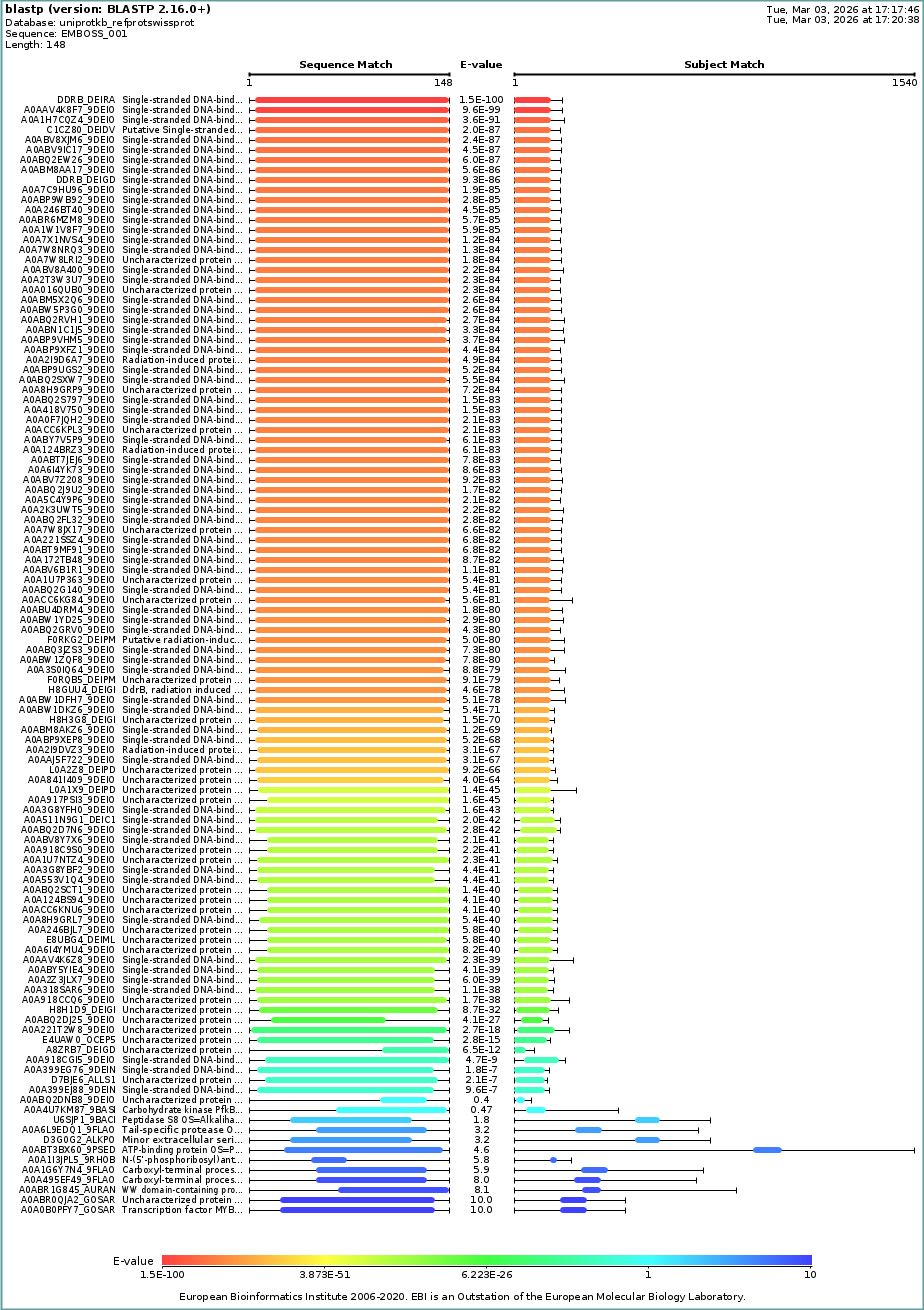
2.2. How many protein sequence homologs are there for your protein?
BLAST Input:
I used UniProt BLAST with default settings and reported the number of hits as an estimate of sequence homologs (based on BLAST similarity/E-value criteria).
112 hits
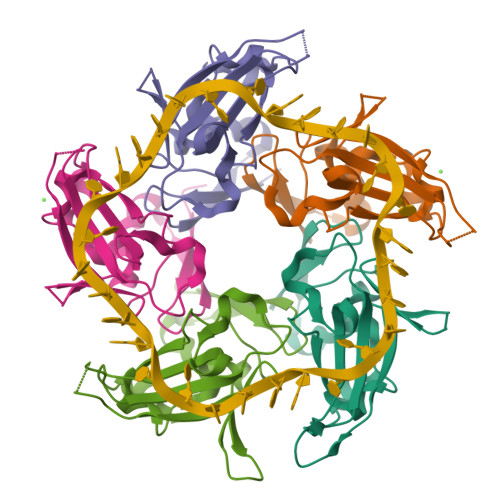
2.3. Does your protein belong to any protein family?
PF12747 — DdrB-like protein (DdrB)
3. Identify the structure page of your protein in RCSB
3.1. When was the structure solved? Is it a good quality structure?
Deposited: 2013-11-19
Released: 2015-05-20
Resolution: 2.20 Å (2.20 Å < 2.70 Å, good quality)
3.2. Are there any other molecules in the solved structure apart from protein?
Yes.
Nucleic acid: 30-nt ssDNA (Crystal structure of DdrB bound to 30b ssDNA)
CALCIUM ION (CA)
3.3 Does your protein belong to any structure classification family?
RCSB Classification: DNA BINDING PROTEIN/DNA
ECOD family: DdrB
ECOD architecture: beta barrels
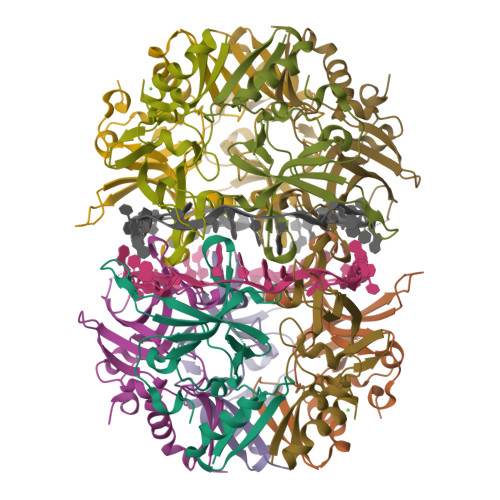
4. 3D molecule visualization
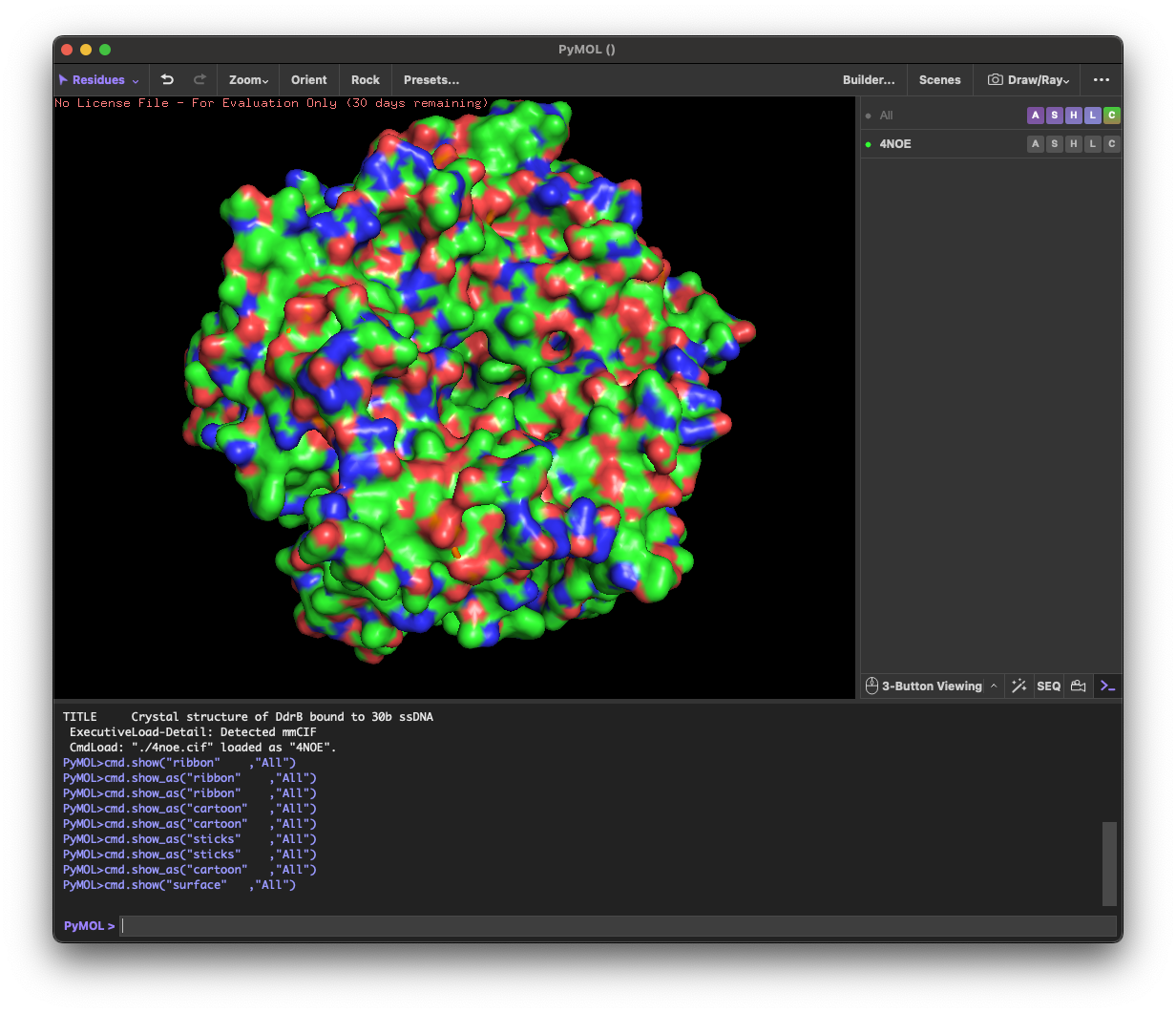
4.1. Visualize the protein as “cartoon”, “ribbon” and “ball and stick”.
4.2.Color the protein by secondary structure. Does it have more helices or sheets?
helix: 189
sheet: 242
It has more β-sheets than α-helices.
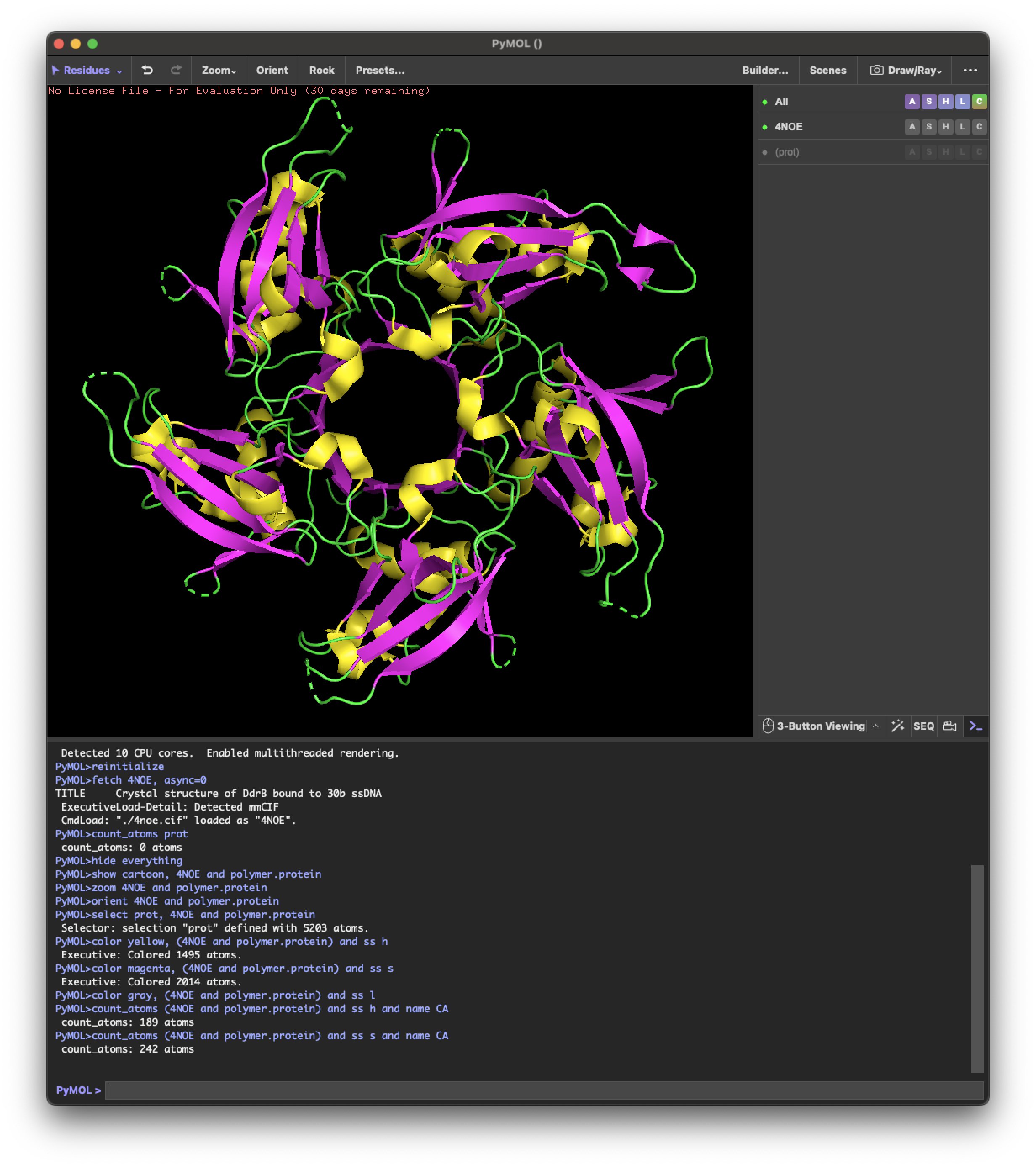
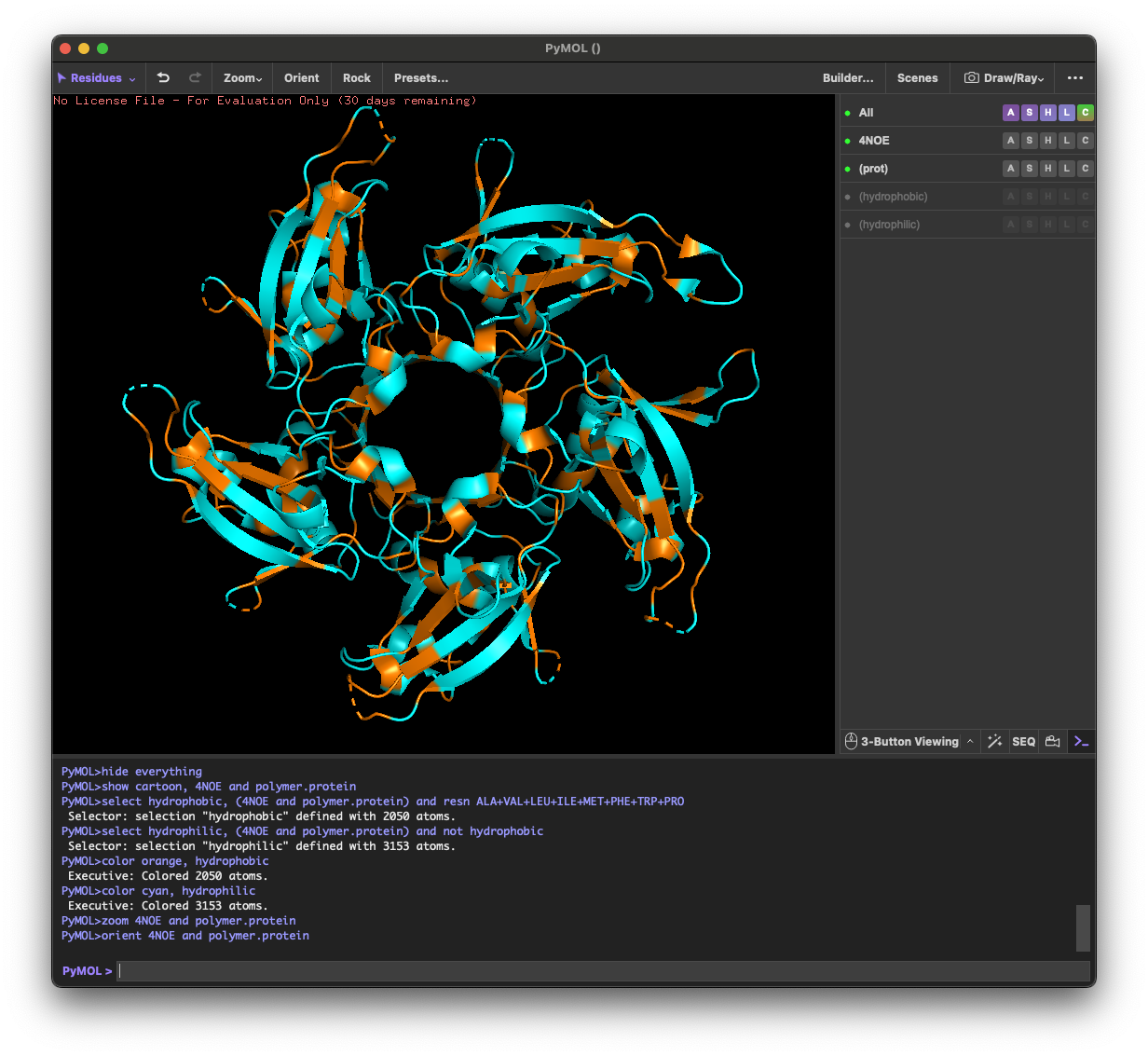
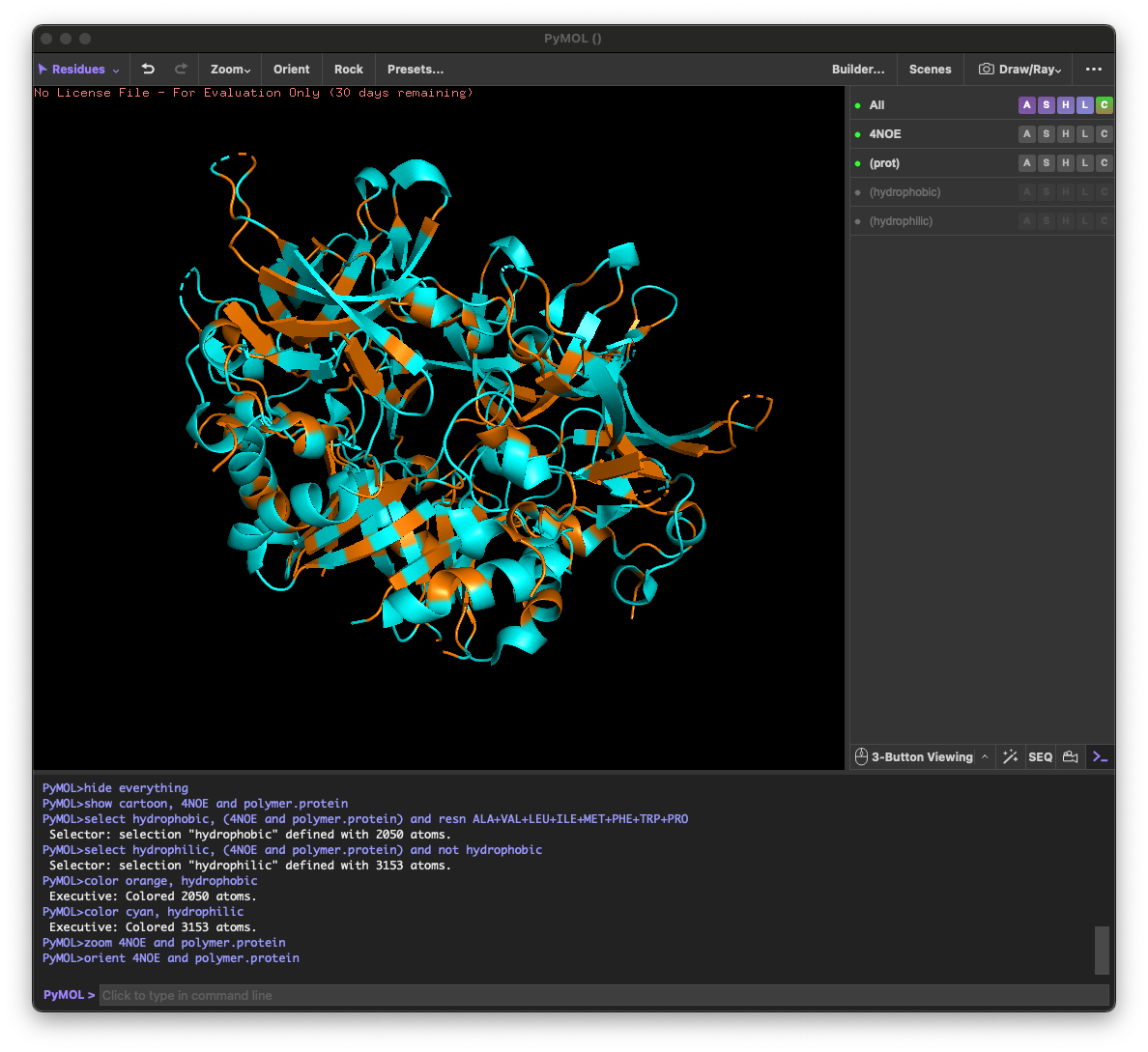
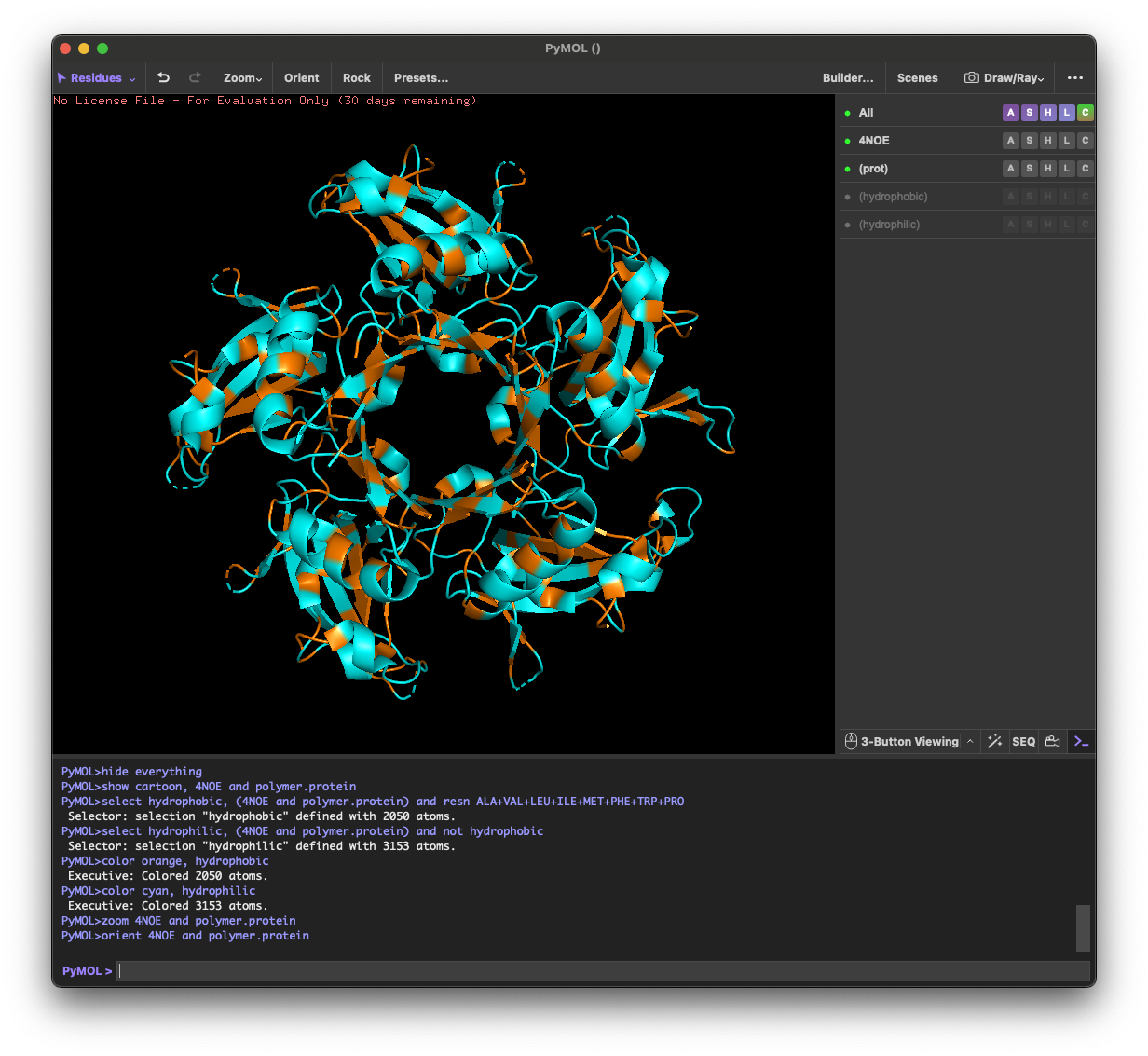
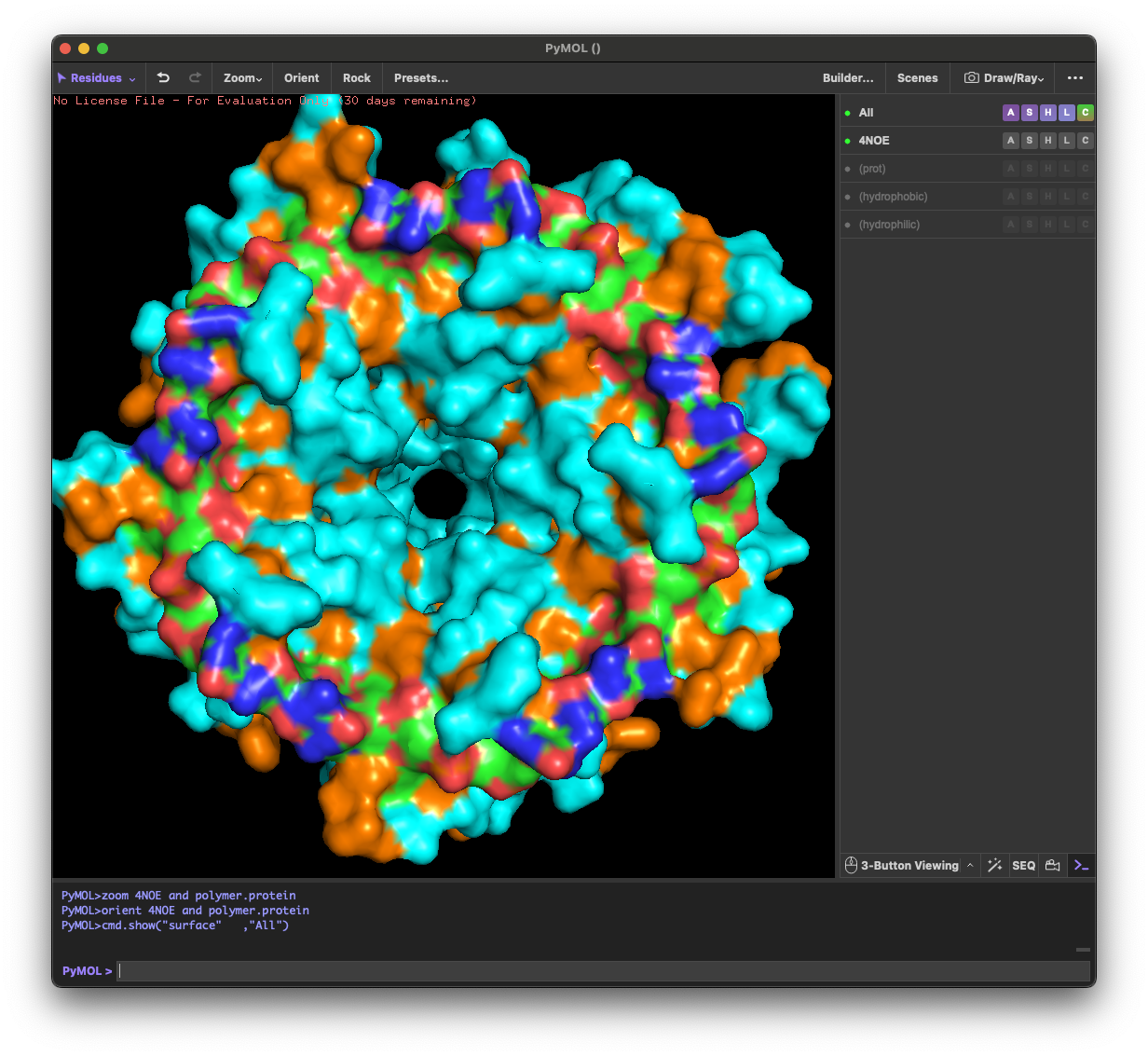
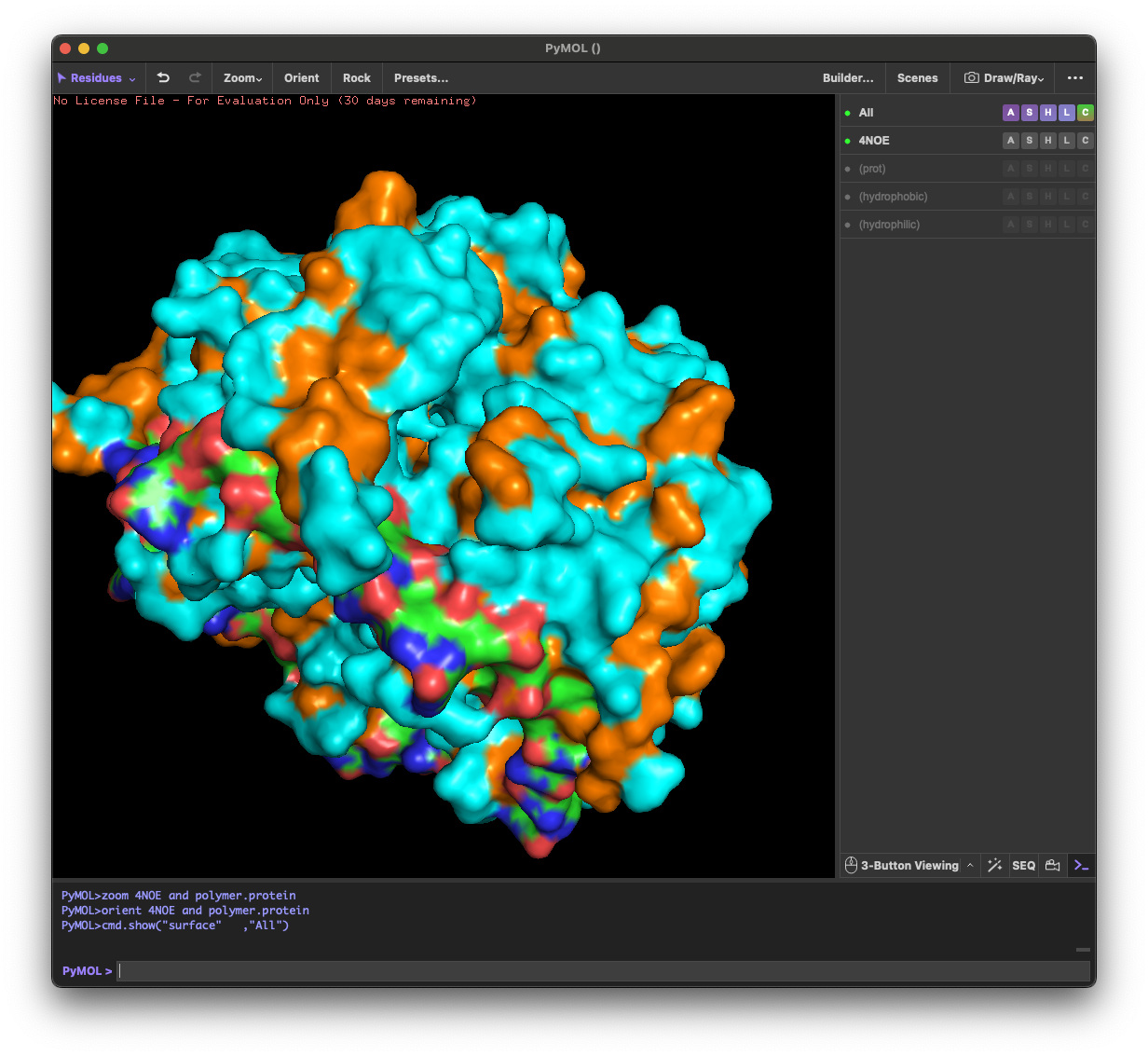
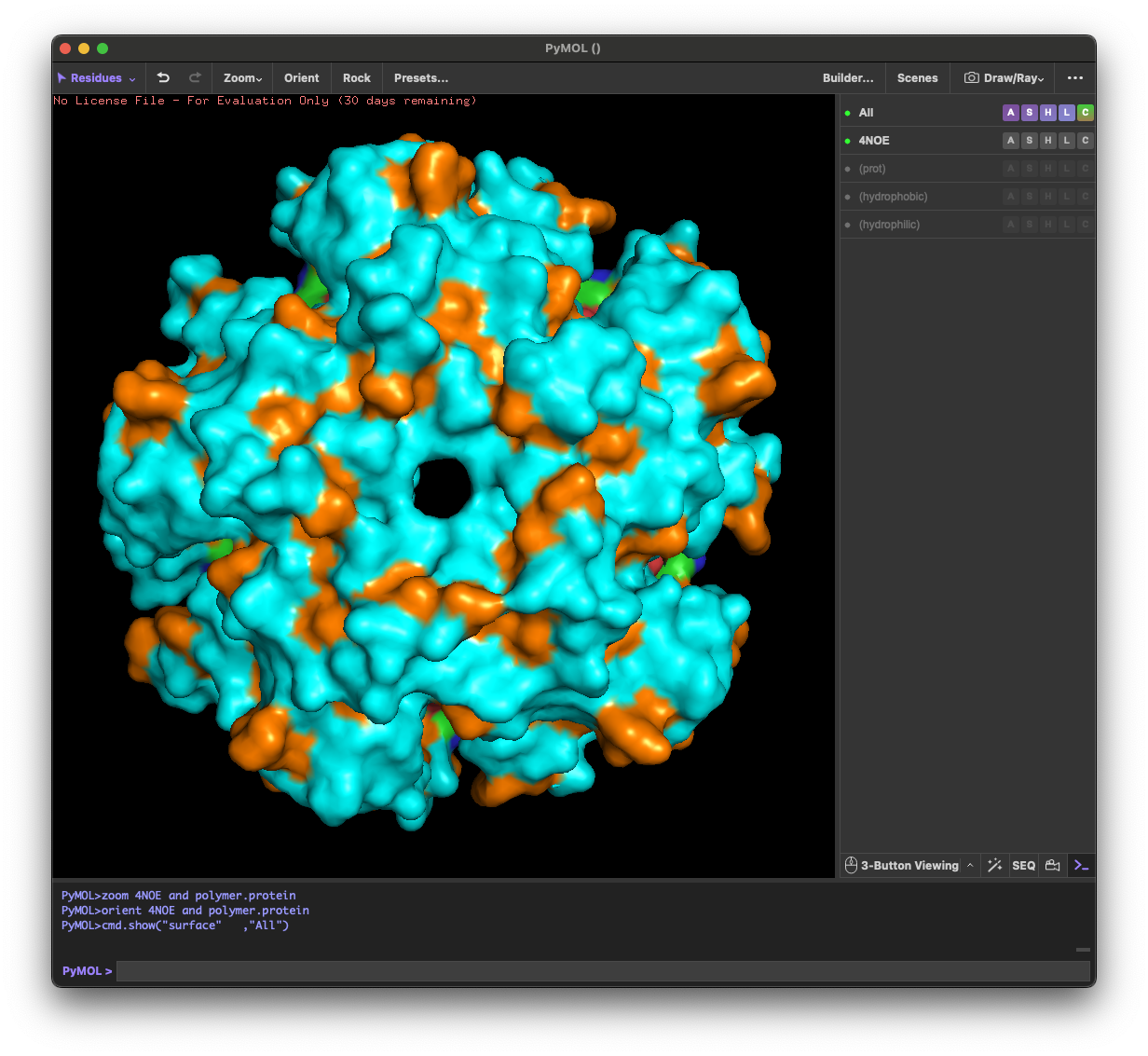
4.3. Color the protein by residue type. What can you tell about the distribution of hydrophobic vs hydrophilic residues?
Across the different views, hydrophilic residues (cyan) dominate and appear broadly distributed over the outer surface, especially along loops and exposed regions. Hydrophobic residues (orange) are more patchy and discontinuous, showing up as scattered stripes/spots across β-strands and some helical segments rather than forming one continuous hydrophobic face. Overall, the protein looks like a typical soluble complex: a largely hydrophilic exterior with localized hydrophobic patches that likely contribute to core stabilization and/or subunit–subunit interfaces.
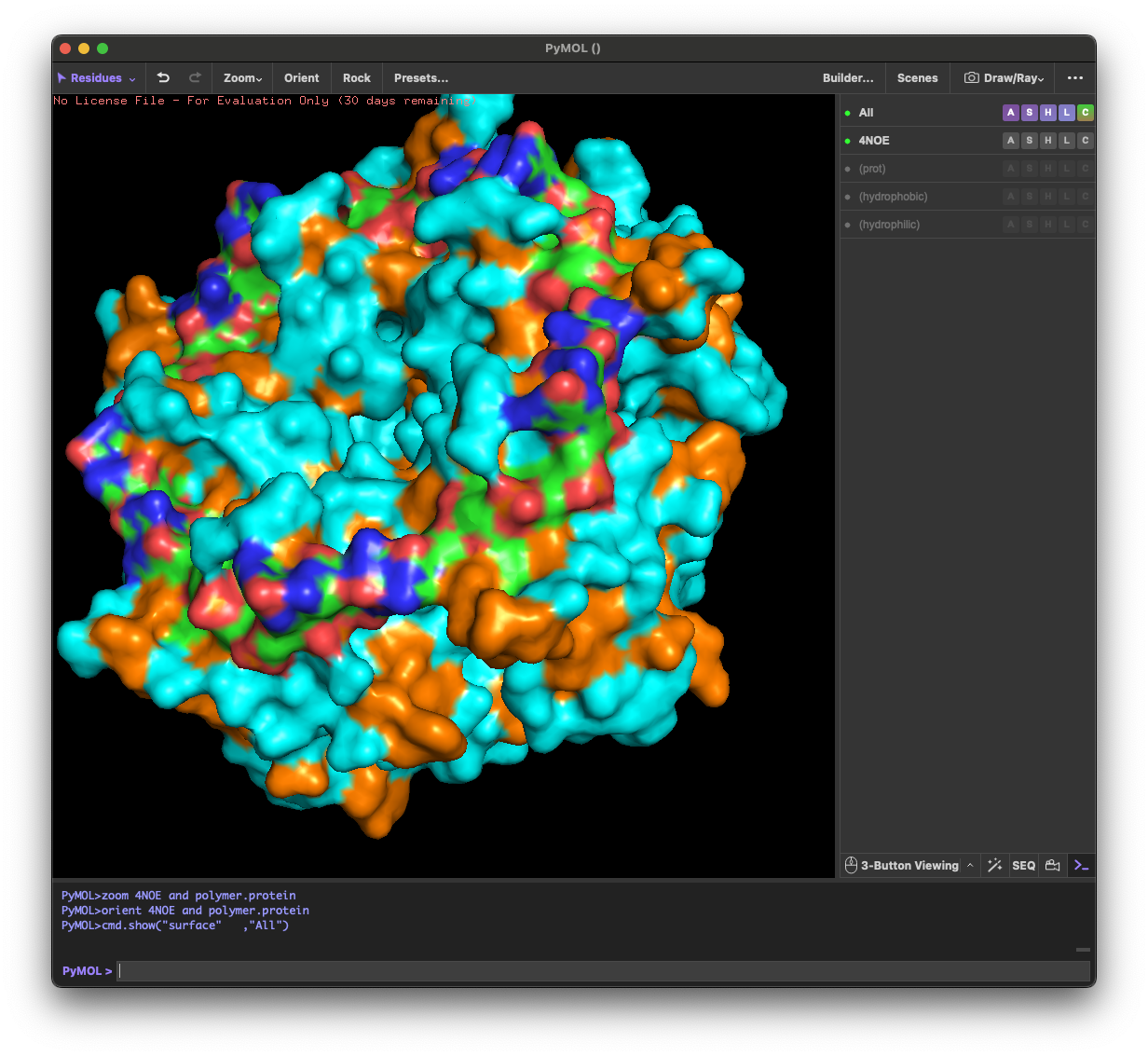
4.4. Visualize the surface of the protein. Does it have any “holes” (aka binding pockets)?
Yes. The surface rendering shows a clear pore-like hole near the center that remains visible across angles. There are also multiple smaller grooves and depressions on the surface, which could be potential binding pockets.
Part C: Using ML-Based Protein Design Tools
C1. Protein Language Modeling
1. Deep Mutational Scans
a. Use ESM2 to generate an unsupervised deep mutational scan of your protein based on language model likelihoods.
Using ESM2’s unsupervised likelihood-based scan on DdrB, most single–amino-acid substitutions have relatively modest effects, but a few positions show strong outliers, suggesting that some residues are much more constrained than others.
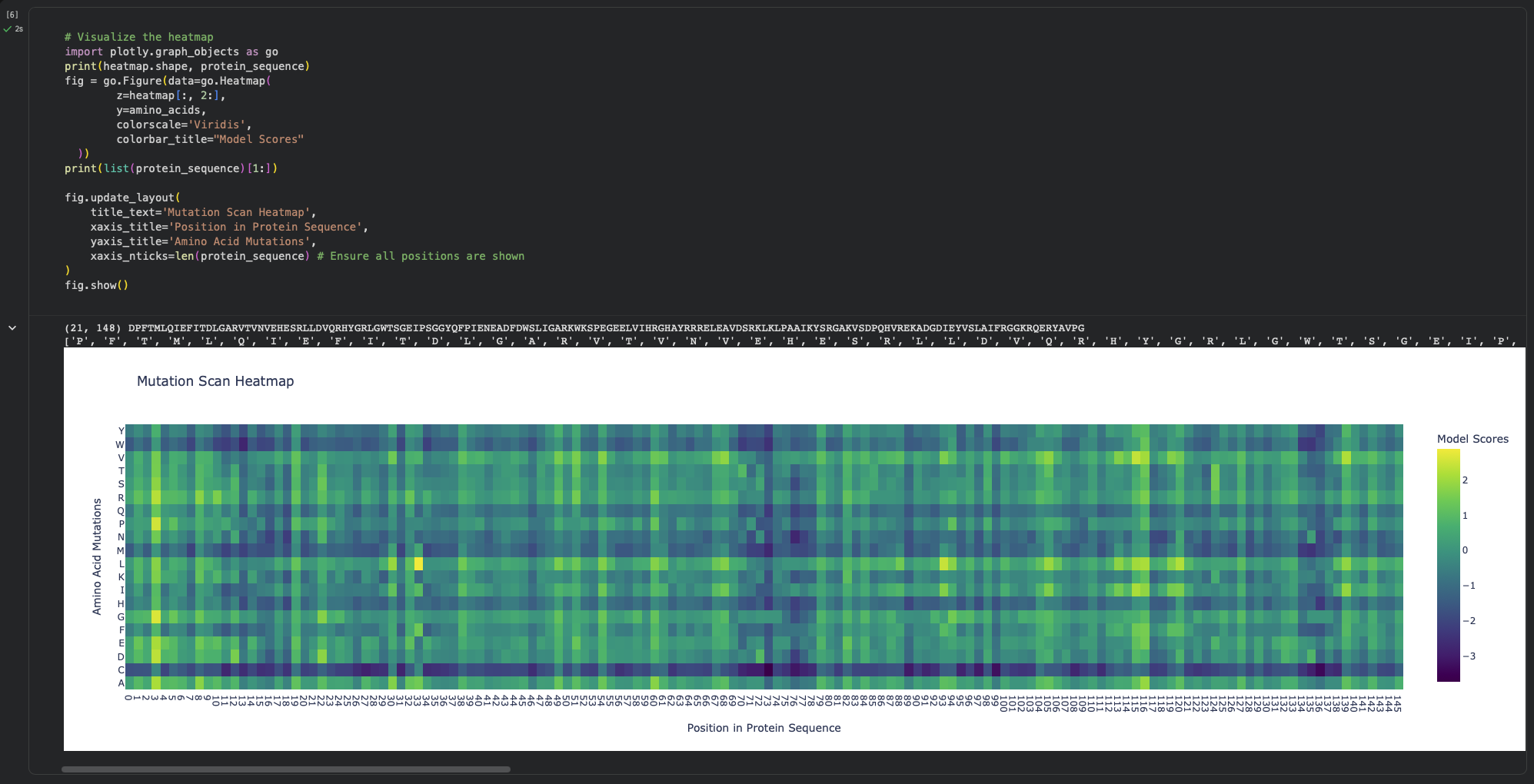
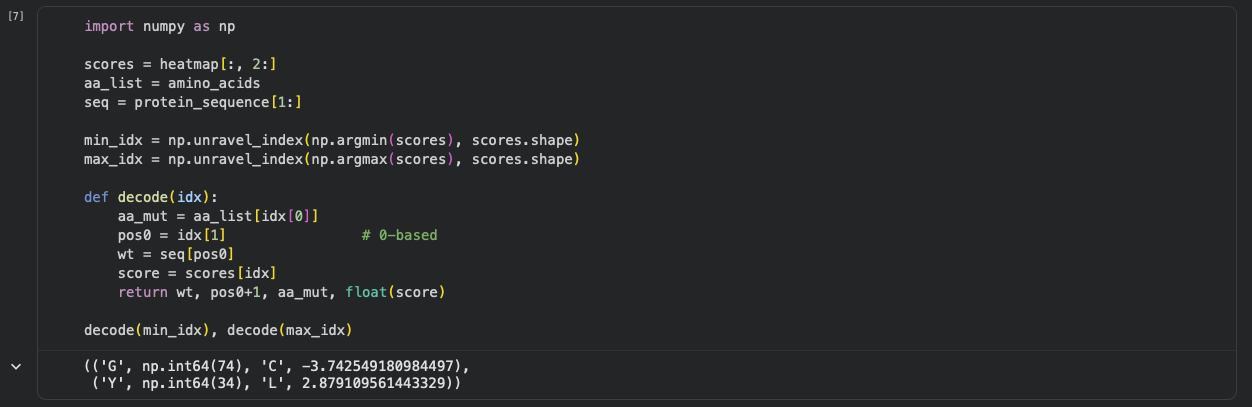
b. Can you explain any particular pattern? (choose a residue and a mutation that stands out)
One mutation that stands out is G74→C, which has the most unfavorable score (-3.74), indicating that position 74 is highly constrained and likely important for maintaining the protein’s stability/assembly. A strongly favorable outlier is Y34→L (+2.88).
2. Latent Space Analysis
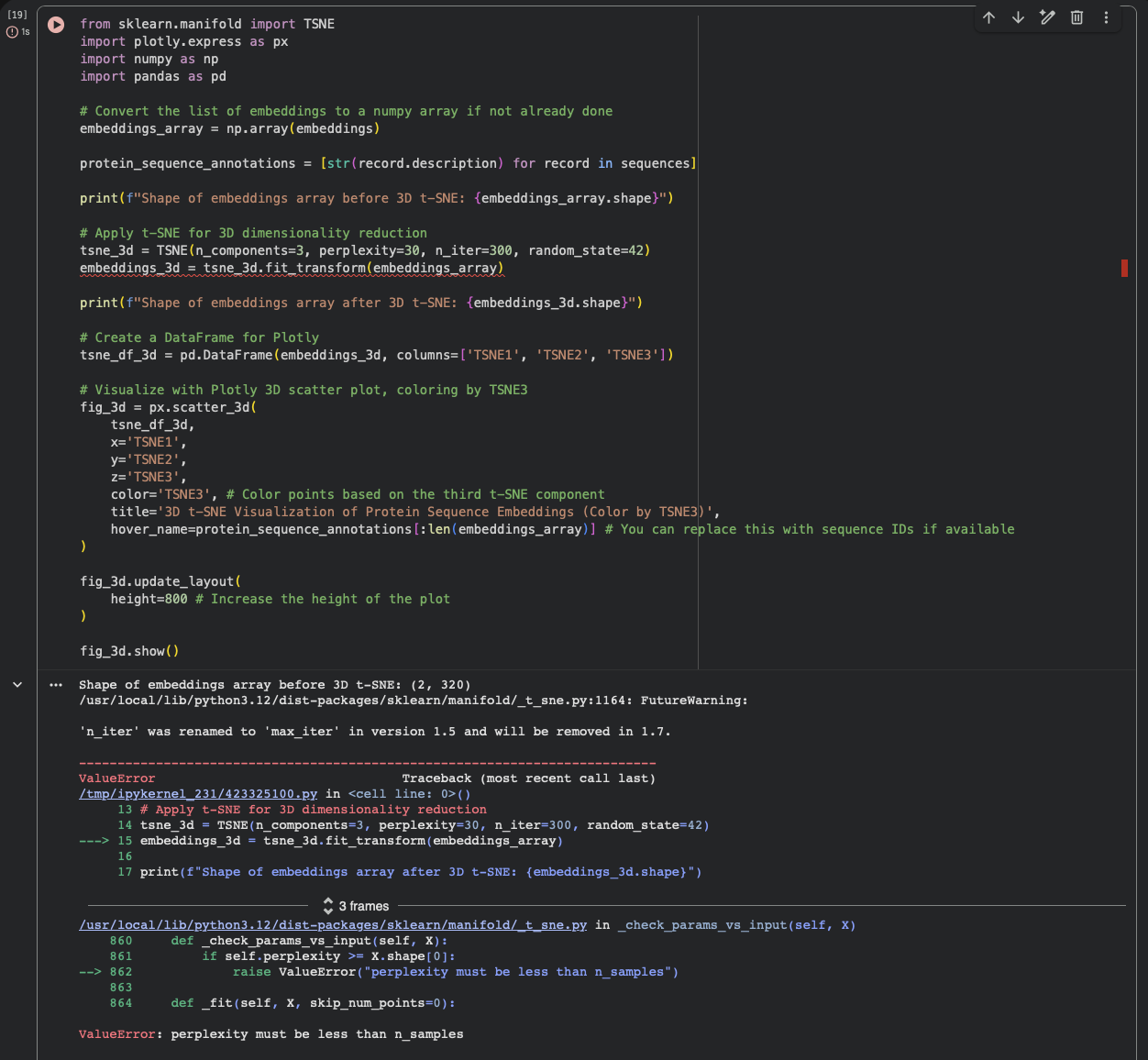
In the latent space analysis section, the run failed with the error “ValueError: perplexity must be less than n_samples.”At that point the embedding array had shape (2, 320) (only two sequences were embedded), while the t-SNE settings used perplexity = 30, which requires a larger number of samples. As a result, the projection step did not complete and neighborhood interpretation was not possible in this run.
2. Protein Folding Folding a protein
1. Fold your protein with ESMFold. Do the predicted coordinates match your original structure?
I folded the DdrB sequence (length 148 aa) with ESMFold. The run completed with pTM = 0.204 and mean pLDDT = 34.332. These confidence metrics are low, so the predicted coordinates are not expected to reliably match the experimental PDB structure (4NOE), and any apparent similarity should be treated as uncertain.