Week 4 HW: ProteinDesign
Part A. Conceptual Questions
Question: How many molecules of amino acids do you take with a piece of 500 grams of meat?
(On average an amino acid is ~100 Daltons.)
Answer:
5 mol × 6.02 × 10²³ ≈ 3 × 10²⁴ amino acid molecules.
So eating 500 g of meat gives you on the order of 10²⁴ (about three septillion) amino acid molecules.
Question: Why do humans eat beef but do not become a cow, eat fish but do not become fish?
Answer: These free amino acids are absorbed and reused as generic building blocks to make human proteins, under the instructions of human DNA and our own gene expression programs.
Part B. Protein Analysis and Visualization
In this part, pick any protein (with a known 3D structure) and answer the following.
B1. Protein choice
Briefly describe the protein you selected and why you selected it.
Answer: I chose the spider dragline silk protein, specifically a major ampullate spidroin (MaSp1). This is one of the main structural proteins that spiders use to spin their dragline silk, the “lifeline” they hang from and use as a safety rope. Dragline silk is famous for combining very high tensile strength (comparable to steel) with remarkable toughness and elasticity, and these mechanical properties come directly from its unusual amino acid sequence: long, repetitive blocks that form a mix of crystalline β-sheet nanodomains and more disordered, elastic regions.
I selected MaSp1 because it sits at the intersection of protein sequence, hierarchical structure, and macroscopic material behavior. It’s a natural example of how repeating sequence motifs can encode a programmable mechanical material, which fits nicely with the theme of protein design and with thinking about proteins as engineerable structural materials, not just enzymes or receptors.
B2. Amino acid sequence
Identify the amino acid sequence of your protein.
https://www.uniprot.org/uniprotkb/P19837/entry
B3. Length and amino acid frequencies
How long is it? What is the most frequent amino acid?
Answer:
Sequence Length: 748 Amino Acid Frequencies: G: 317 A: 193 Q: 75 L: 40 S: 38 Y: 22 R: 20 V: 16 N: 8 I: 8 E: 3 P: 3 T: 3 C: 1 D: 1
Observations about composition:
This sequence is heavily enriched in glycine (G) and alanine (A), which together make up the majority of residues. Glycine is extremely small and flexible, while alanine is small and slightly hydrophobic. In spider silk, repeated (Gly–Ala)n motifs are known to stack into tightly packed β-sheet nanocrystals that provide the fiber’s high stiffness and tensile strength.
B4. Sequence homologs
How many protein sequence homologs are there for your protein?
Answer: UniProt BLAST returns hundreds of homologous sequences, mostly other spider silk proteins from related species, showing that MaSp1 belongs to a large and diversified silk protein family.
B5. Protein family
Does your protein belong to any protein family?
Answer:
Belongs to the silk fibroin family
B6. Structure identification (RCSB PDB)
Identify the structure page of your protein in RCSB.
Answer:
- PDB ID: 5IZ2
- RCSB URL: https://www.rcsb.org/structure/5IZ2
B7. Structure quality
When was the structure solved? Is it a good quality structure?
Answer:
- Year solved: 2016
- Resolution: 2.02A
- Comment on quality: Good
B8. Other molecules in the structure
Are there any other molecules in the solved structure apart from the protein?
Answer:
Ligands:
None – there are no bound small-molecule ligands or cofactors reported.Ions:
None – no metal ions are annotated in this structure.Water / other components:
Only crystallographic water molecules (HOH) are present as part of the solvent.Comments:
The 5IZ2 structure is essentially just the N-terminal domain of MaSp1A plus bulk water, with no extra cofactors or metal ions. This makes it a relatively “clean” system for analyzing the intrinsic protein–protein interface and packing.
B9. Structural classification
Does your protein belong to any structure classification family?
(e.g., SCOP, CATH)
Answer:
- 5IZ2 A:6–128 (SCOP ID: 8046115),
labeled “Major ampullate spidroin 1A (fragment)” from Trichonephila clavipes.
This shows that the N-terminal domain of MaSp1 belongs to a specific spidroin N-terminal structural family in SCOP.
B10. 3D visualization
B10.1 Cartoon / ribbon / ball-and-stick views
In PyMOL, I loaded the N-terminal domain of MaSp1 (PDB 5IZ2) and visualized it with different representations:
Cartoon view (secondary structure):
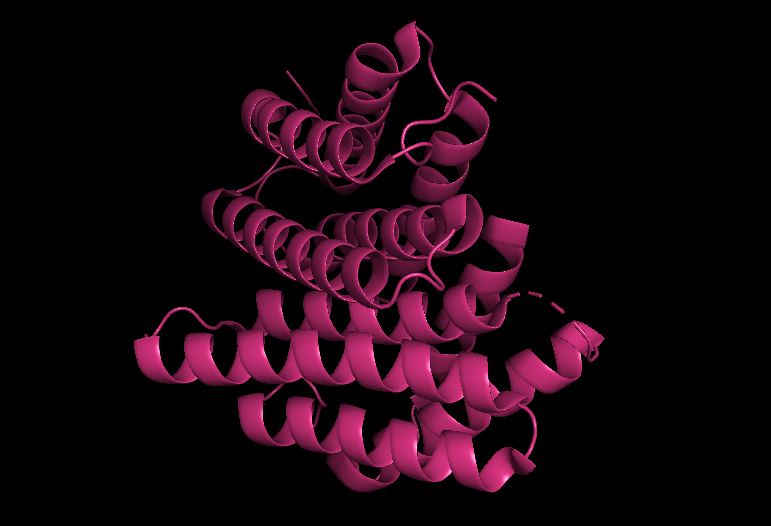
Ribbon view:

Ball-and-stick view (side chains):
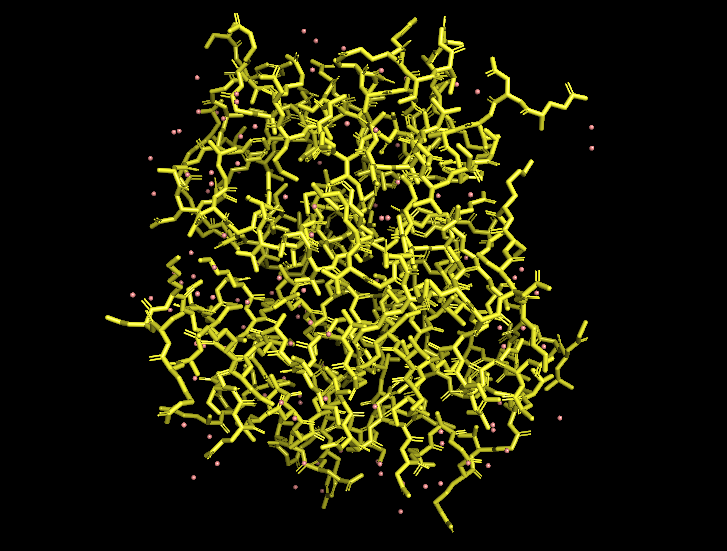
B10.2 Hydrophobic vs hydrophilic residue distribution
When I color the protein by residue type / hydrophobicity, I observe a typical hydrophobic-core, hydrophilic-surface pattern:
- Many hydrophobic residues (e.g., Leu, Val, Ile, Phe) are buried in the interior of each helical bundle and at the dimer interface.
- Polar and charged residues (e.g., Gln, Glu, Lys, Arg, Ser) are mostly exposed on the outside, where they can interact with solvent and potentially mediate pH-dependent behavior.
This distribution is consistent with the idea that the MaSp1 N-terminal domain is a soluble, dimerizing helical bundle, with its hydrophobic core shielded from water and its polar residues decorating the surface.
B10.3 Surface and potential “holes” (pockets)
Finally, I visualized the molecular surface of the dimer:
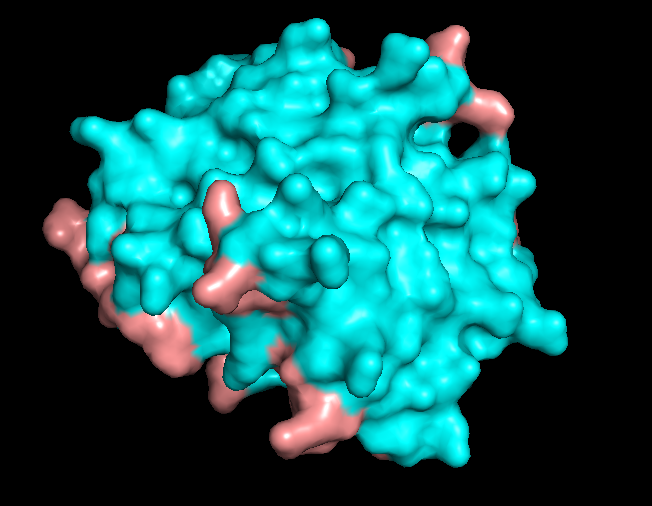
The overall shape is compact and smooth, but there are some shallow grooves and one noticeable depression on the surface. This depression looks like a small, shallow pocket rather than a deep enzyme-like active site or tunnel. It is more likely to be a protein–protein interaction groove (or simply a consequence of how the helices pack) than a dedicated small-molecule binding pocket.
Overall, the surface of 5IZ2 matches its role as a structural N-terminal dimerization domain, not a classical ligand-binding enzyme.
Part C. Using ML-Based Protein Design Tools
C1. Protein Language Modeling
C1.1 Deep mutational scan with ESM2
Use ESM2 to generate an unsupervised deep mutational scan of your protein.
Can you explain any particular pattern?
(Choose a residue and a mutation that stands out.)
Answer:
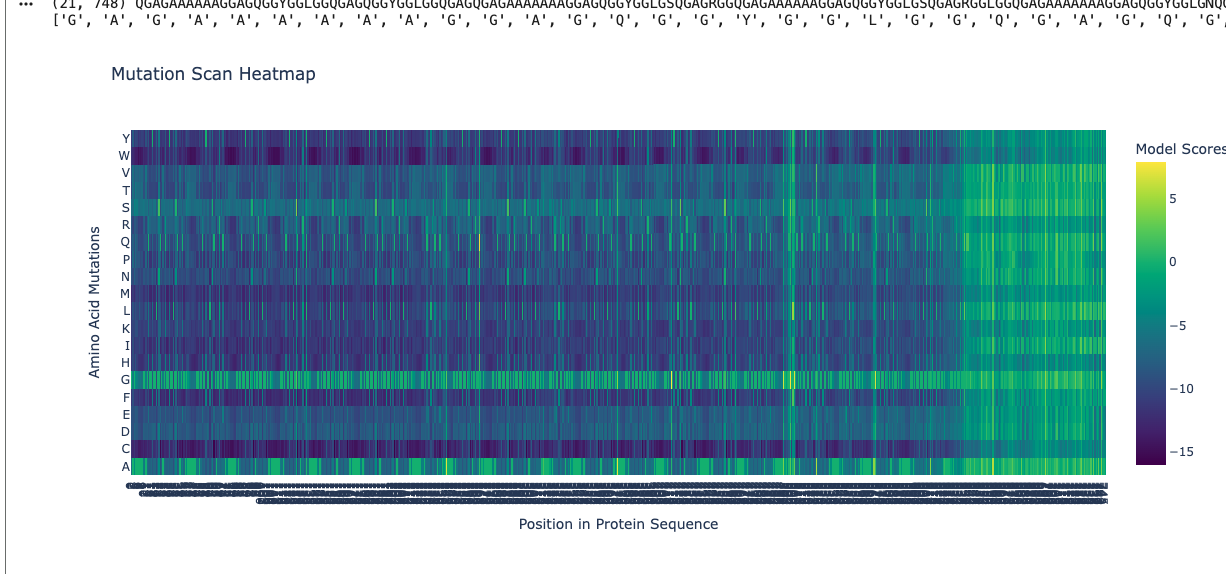
Using ESM2, I generated a deep mutational scan for the full MaSp1 sequence. A clear pattern is that the model strongly prefers small residues like glycine (G) and alanine (A) in the long repetitive core of the silk protein, and strongly dislikes large or strongly charged residues at those positions.
For example, I looked at one position in the middle of the repetitive region where the wild-type residue is glycine. In the heatmap, mutating this site to alanine (G → A) has a relatively neutral or mildly favorable score (greenish color), but mutating it to tryptophan (G → W) gives a very unfavorable score (dark purple). This makes sense: natural spider dragline silk is built from G/A-rich motifs such as (Gly–Ala)n that pack into tight β-sheet nanocrystals. Replacing a small glycine with a bulky aromatic residue like tryptophan would likely disrupt this packing, so the language model correctly treats this mutation as strongly deleterious.
C1.2 Latent Space Analysis
Using the notebook’s SCOPe ASTRAL dataset, I first embedded all protein domains with ESM2 and then reduced the embedding dimensionality to 3D using t-SNE (n_components = 3, perplexity = 30). Each point in the plot below corresponds to one protein domain in the SCOPe dataset; nearby points represent proteins that the language model considers similar in terms of sequence statistics and learned evolutionary patterns.
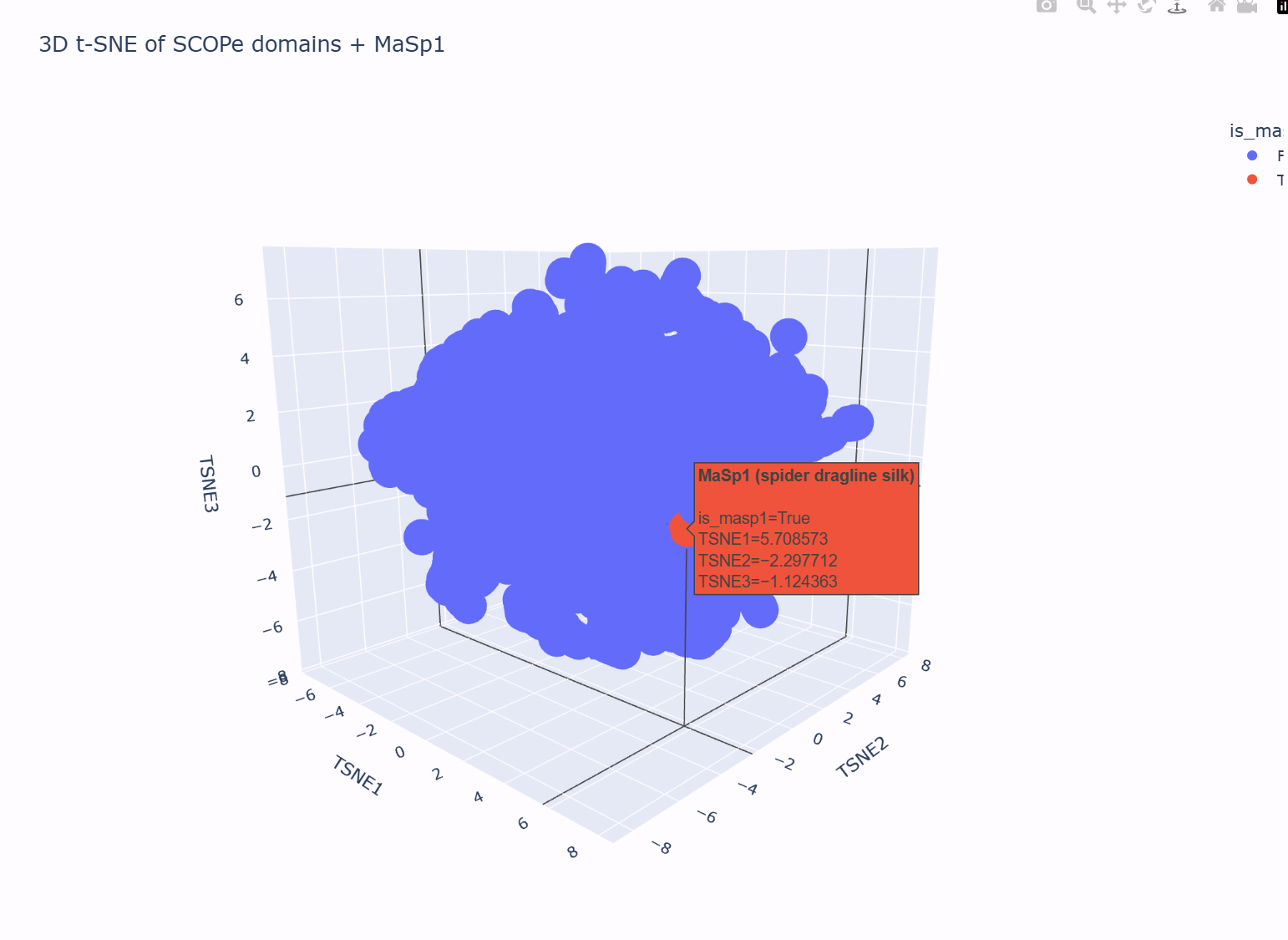
In this 3D map, the SCOPe domains do not form one perfectly uniform “cloud”: there is a dense central region and a somewhat more diffuse shell of points around it. Hovering over different neighborhoods in the plot shows that proteins with related properties tend to live near each other (for example, domains with similar lengths and compositional biases cluster more closely than very different domains). This suggests that the latent space is grouping broadly similar proteins into local neighborhoods.
I then added my own protein sequence — a fragment of the spider dragline silk protein MaSp1 — as an extra point in the embedding. In the t-SNE plot, this MaSp1 point is highlighted in a different color (red in the figure). It does not appear as an isolated outlier: instead, it lies on the outer part of the main cloud, close to other domains that, based on their descriptions, also have biased compositions and/or repetitive, low-complexity features.
This placement is consistent with what we know about MaSp1. The sequence is very long and strongly enriched in glycine and alanine repeats, designed to form structural silk fibers rather than a compact catalytic enzyme. In the latent space, the model therefore locates MaSp1 in a neighborhood of other non-enzymatic, compositionally biased domains, rather than in the core region where many typical enzyme-like domains cluster. In other words, the latent space neighborhoods do approximate “similar” proteins, and MaSp1’s position in the map matches its role as a G/A-rich structural material protein.
C2. Protein Folding (ESMFold)
C2.1 Folding your protein
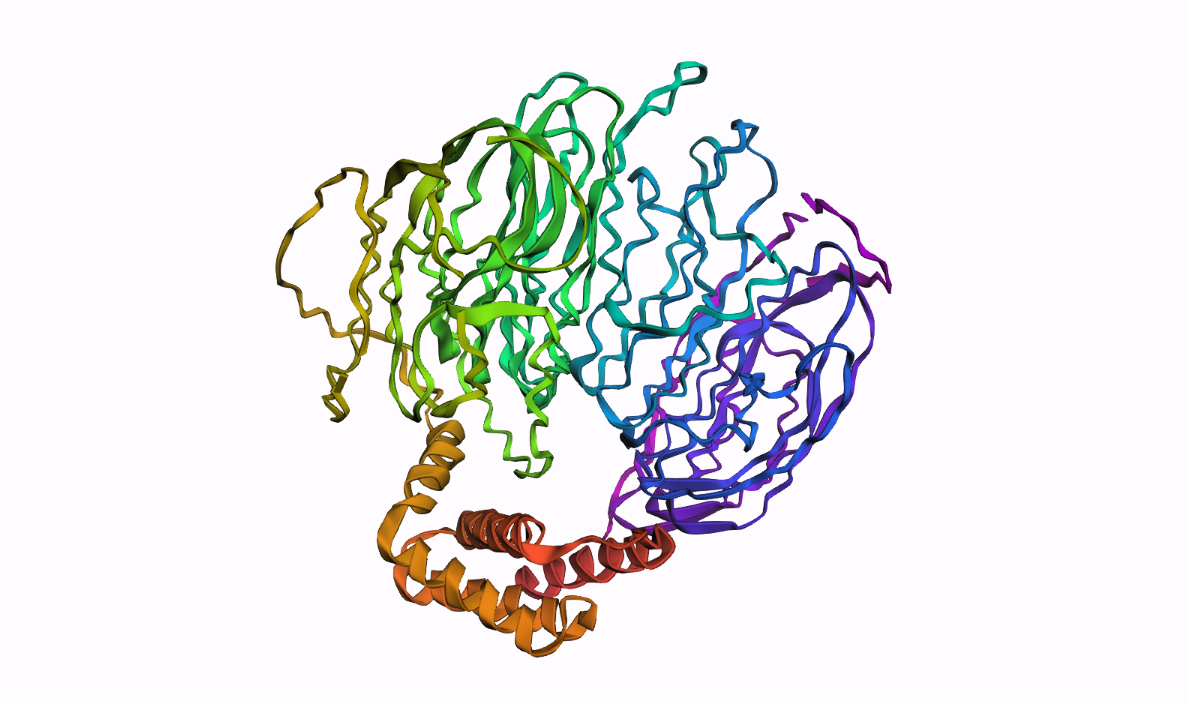
Fold your protein with ESMFold.
Do the predicted coordinates match your original structure?
Answer:
C3. Protein Generation
C3.1 Inverse folding with ProteinMPNN
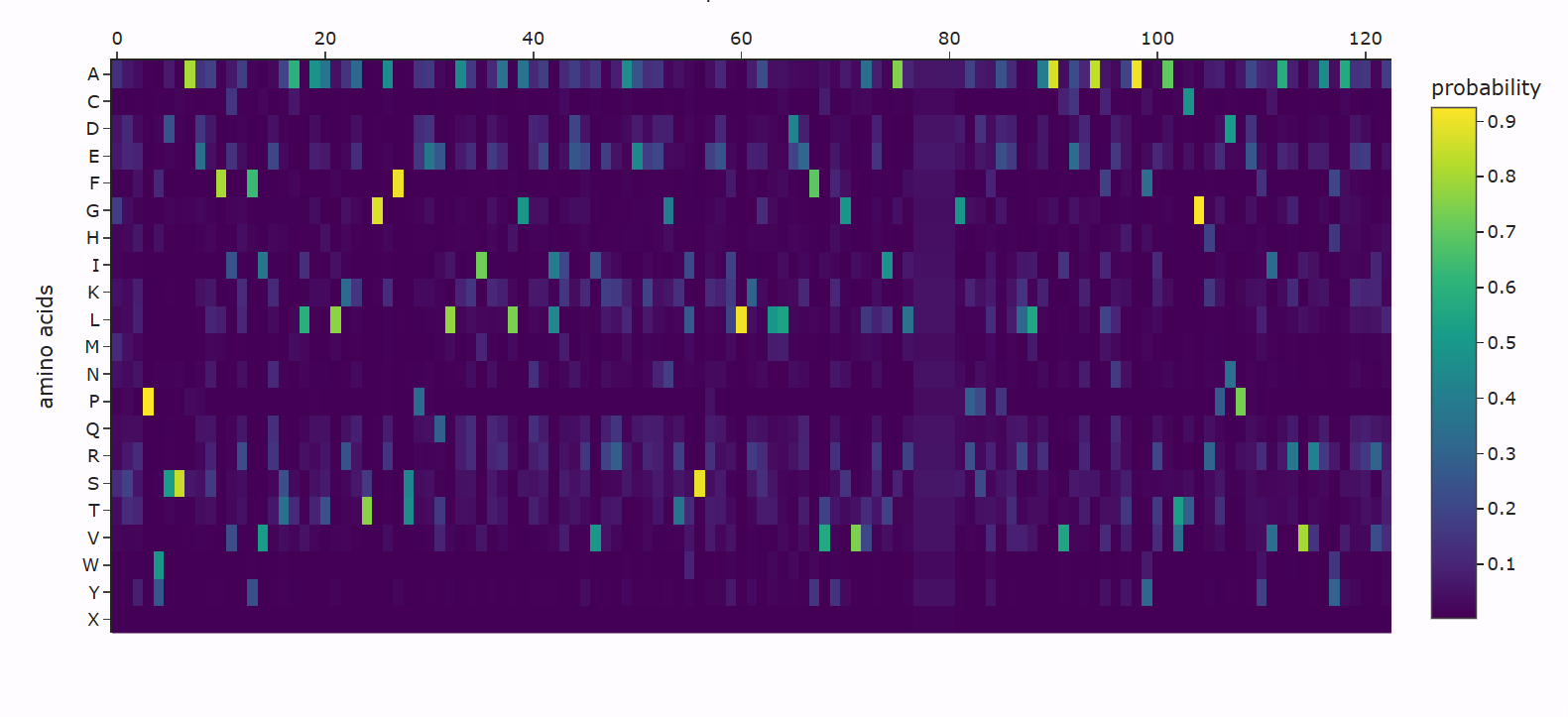
Use the backbone of your chosen PDB to propose sequence candidates via ProteinMPNN.
Analyze the predicted sequence probabilities and compare the predicted sequence vs the original one.