Week 5 HW: Protein Design II
Part 1
1. Retrieve SOD1 sequence and introduce A4V
MATKVCVVLKGDGPVQGIINFEQKESNGPVKVWGSIKGLTEGLHGFHVHEFGDNTAGCTSAGPHFNPLSRKHGGPKDEERHVGDLGNVTADKDGVADVSIEDSVISLSGDHCIIGRTLVVHEKADDLGKGGNEESTKTGNAGSRLACGVIGIAQ
2. PepMLM-generated 12-aa peptides + known binder
(lower = higher model confidence in the binder).
| ID | Peptide (12 aa) | Source | Perplexity (PepMLM) |
|---|---|---|---|
| P1 | WHSPVAAARLKE | PepMLM | 11.721713 |
| P2 | KRYGAAAARHKK | PepMLM | 11.211369 |
| P3 | WRYPVAGLALKE | PepMLM | 13.068802 |
| P4 | WHSPPAAVALGE | PepMLM | 12.159801 |
| Ref | FLYRWLPSRRGG | known SOD1 binder | N/A |
Observation (PepMLM confidence by perplexity):
Among generated candidates, P2 (KRYGAAAARHKK) has the lowest perplexity (highest PepMLM confidence), while P3 (WRYPVAGLALKE) has the highest perplexity (lowest confidence among the four).
Part 2 – Evaluate binders with AlphaFold3
| Peptide | Sequence | Binding description (1–2 sentences) | Near N-terminus / A4V? | |
|---|---|---|---|---|
| P1 | WHSPVAAARLKE | surface-bound | no | |
| P2 | KRYGAAAARHKK | surface-bound | no | |
| P3 | WRYPVAGLALKE | surface-bound | no | |
| P4 | WHSPPAAVALGE | surface-bound | yes | |
| Ref | FLYRWLPSRRGG | Surface-associated | no |
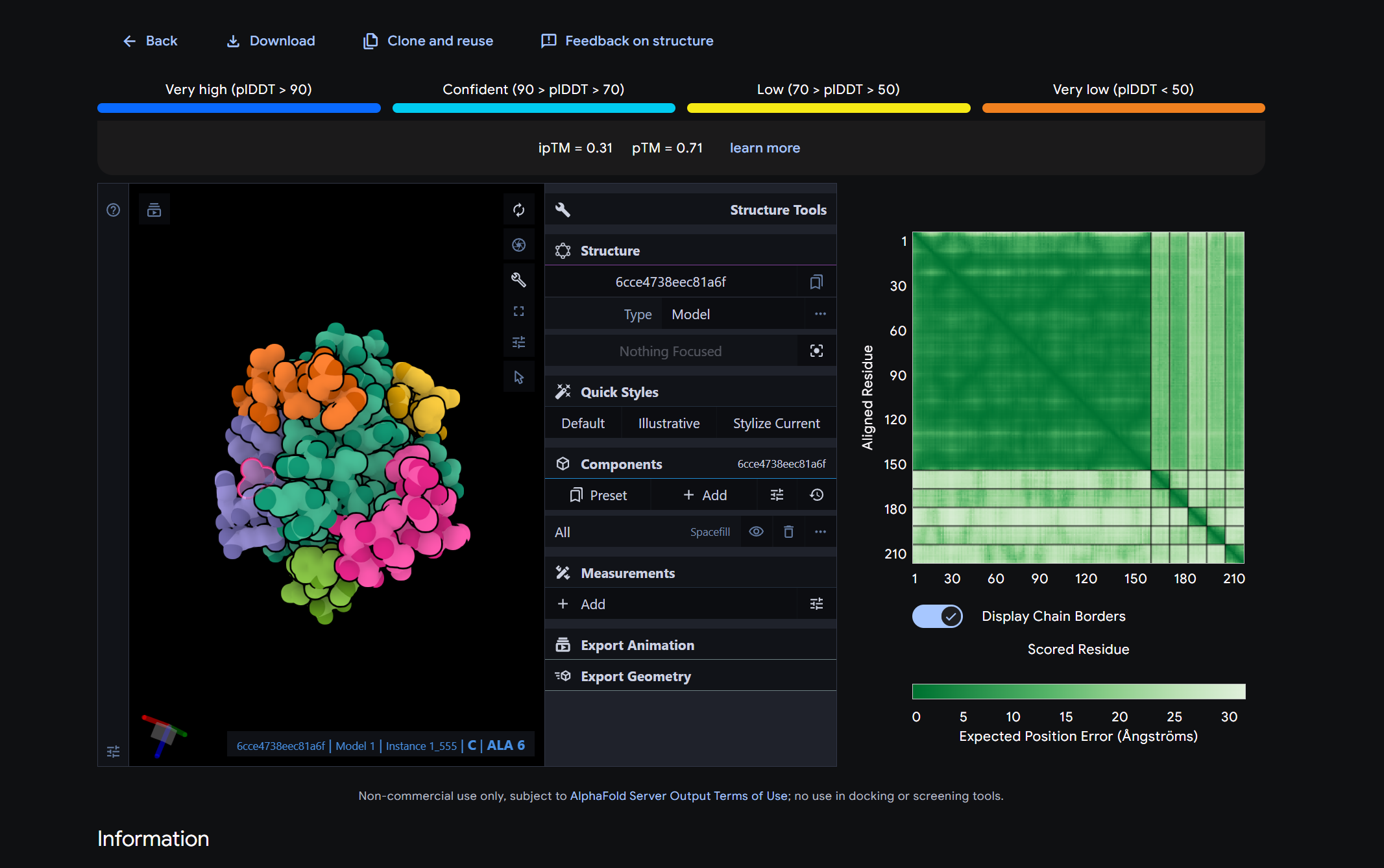
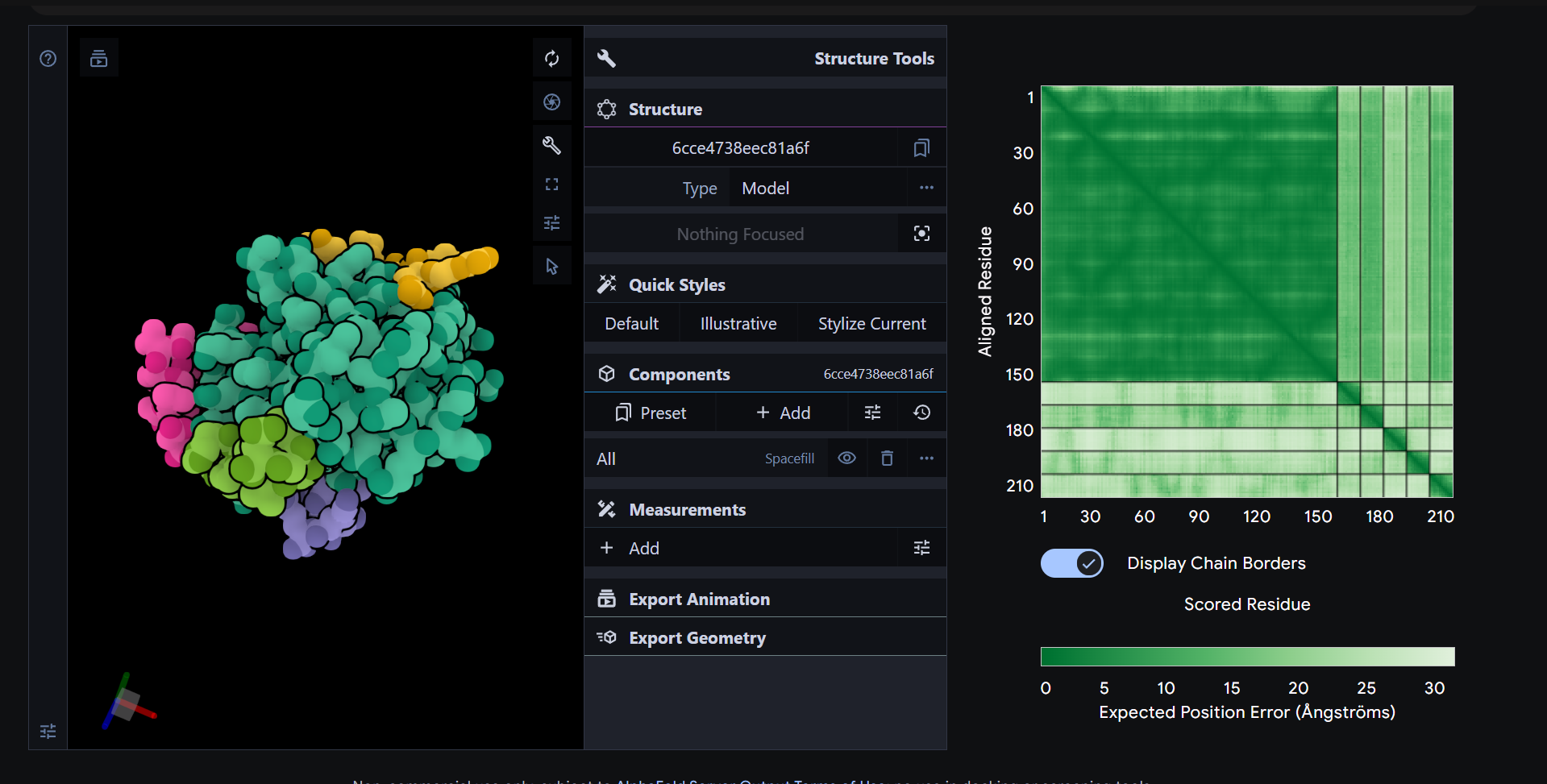
Part 3 – Evaluate Properties of Generated Peptides in the PeptiVerse
| Peptide | Solubility | Solubility Score | Hemolysis | Hemolysis Score | Length (aa) | Molecular Weight (Da) | Net Charge (pH 7) | Isoelectric Point (pH) | Hydrophobicity (GRAVY) |
|---|---|---|---|---|---|---|---|---|---|
| WHSPVAAARLKE | Soluble | 1.000 | Non-hemolytic | 0.013 | 12 | 1364.6 | 0.85 | 8.76 | -0.42 |
| KRYGAAAARHKK | Soluble | 1.000 | Non-hemolytic | 0.009 | 12 | 1356.6 | 4.84 | 11.17 | -1.53 |
| WRYPVAGLALKE | Soluble | 1.000 | Non-hemolytic | 0.025 | 12 | 1402.6 | 0.77 | 8.59 | -0.06 |
| WHSPPAAVALGE | Soluble | 1.000 | Non-hemolytic | 0.041 | 12 | 1234.4 | -1.14 | 5.47 | 0.12 |
From the AlphaFold3 models, all of the peptides showed relatively low and fairly similar ipTM values, and structurally they all appeared to bind in a shallow, surface associated manner rather than forming a clearly buried or highly specific interface on mutant SOD1. This means that higher ipTM did not translate into an obviously much stronger or more convincing binding pose in the structural models. When compared with the PeptiVerse predictions, the four generated peptides were all predicted to be fully soluble and non hemolytic, so none of the better candidates raised an immediate concern for poor solubility or red blood cell toxicity. Among them, KRYGAAAARHKK provides the best overall balance. It had the lowest PepMLM perplexity, indicating the highest generation confidence, and it also showed excellent therapeutic property predictions, including full solubility, very low hemolysis risk, and a strongly positive net charge that could support interaction with exposed regions of SOD1.
I would advance KRYGAAAARHKK. Although its AlphaFold3 model did not show a dramatically stronger binding geometry than the others, it combines the best overall computational profile across generation confidence, structural plausibility, solubility, and safety related properties. In other words, it is the most balanced candidate for further optimization and experimental validation.
Part 4 – Generate Optimized Peptides with moPPIt
| Binder | Hemolysis | Solubility | Motif |
|---|---|---|---|
| CTWVKKTKKQVT | 0.979838 | 0.833333 | 0.809806 |
| GYKQKTCNTVKW | 0.96261 | 0.833333 | 0.782062 |
| STAEFTRQTKKM | 0.954297 | 0.75 | 0.839537 |
| RGKTTTQNGKVI | 0.978479 | 0.833333 | 0.820856 |
| GFGTQKKTKCG | 0.964351 | 0.916667 | 0.849918 |
| RTDQGGVKITLE | 0.969846 | 0.833333 | 0.905129 |
| ETKKRQKFKTDF | 0.974053 | 0.833333 | 0.887031 |
| KGETTDKIQKTM | 0.975992 | 0.833333 | 0.841838 |
| RETVGKKTQTKC | 0.981714 | 0.916667 | 0.81274 |
The moPPIt peptides differ noticeably from the PepMLM peptides in both composition and design emphasis. The PepMLM peptides I generated for SOD1 were short 12 amino acid candidates with relatively simple compositions, and several were enriched in alanine, valine, lysine, and histidine, which made them look like general target conditioned binders. In contrast, the moPPIt peptides appear more motif driven and more strongly enriched in lysine and threonine, with some cysteine, glycine, phenylalanine, and aspartate included in specific patterns. This makes them look more constrained by the optimization objectives, especially motif retention and therapeutic property balancing, rather than just sequence likelihood conditioned on the target. In other words, PepMLM gave plausible binder candidates, while moPPIt seems to generate peptides that are more explicitly shaped by multiple design goals.
Before advancing any of these peptides toward clinical studies, I would evaluate them in several stages. First, I would confirm binding experimentally using assays such as surface plasmon resonance, biolayer interferometry, or microscale thermophoresis to measure affinity and specificity for mutant SOD1 compared with wild type SOD1 and unrelated proteins. Second, I would test whether the peptides actually improve a disease relevant phenotype, for example by reducing SOD1 aggregation, toxicity, or misfolding in cell based ALS models. Third, I would assess safety and developability, including hemolysis, cytotoxicity, serum stability, protease resistance, immunogenicity risk, and off target effects. Fourth, I would evaluate pharmacology, including uptake, biodistribution, half life, and whether the peptide can reach the relevant tissues, especially the central nervous system. Only peptides that show convincing binding, functional benefit, acceptable safety, and realistic delivery potential should move forward into animal studies and later clinical development.
L-Protein Engineering | Option 3: Random Mutagenesis
Goal
In this option, I generated random multi-site L-protein mutants using mutation information from prior mutational analysis experiments, then selected candidates for co-folding with DnaJ using AF2 Multimer. The goal is to explore combinations of tolerated or favorable mutations rather than relying only on single-site manual design. The L protein contains a soluble N-terminal domain followed by a transmembrane region in the last 35 residues, so both structural context and prior mutation evidence were considered when building the mutation pool. :contentReference[oaicite:1]{index=1}
Strategy
Instead of introducing fully random amino acid substitutions, I used a constrained random mutagenesis strategy. I first built a mutation pool from mutations that were supported by prior mutational analysis experiments or appeared tolerated or favorable in the provided mutation data. Then I randomly sampled combinations containing at least 2 mutated positions. This makes the search more biologically grounded than unconstrained random mutagenesis.
A good mutant should not simply contain many mutations. I define an effective mutant as one that satisfies several criteria:
- It preserves or improves the foldability and structural plausibility of the L protein.
- It does not obviously disrupt the soluble N-terminal region that is implicated in DnaJ interaction.
- It avoids strongly unfavorable substitutions in conserved or functionally critical positions.
- It is compatible with productive interaction behavior when co-folded with DnaJ.
- Ideally, it improves properties related to lysis efficiency, DnaJ independence, or expression, which are the broader goals of this assignment. :contentReference[oaicite:2]{index=2}
Python function for constrained random mutagenesis
How I would use this function
I would first replace the example mutation_pool with residue positions and substitutions supported by the provided L-protein mutational analysis data. Then I would generate a small library of random double, triple, or quadruple mutants. From that library, I would select a few candidates that look structurally reasonable and co-fold them with DnaJ using AF2 Multimer.
Co-folding setup with DnaJ
For each selected mutant, I would submit the mutant L-protein sequence together with the DnaJ sequence as separate chains in AF2 Multimer. The purpose is to compare whether different mutation combinations change the predicted interaction pattern between the L-protein soluble region and DnaJ. The assignment notes that this kind of prediction may be difficult, especially for membrane related systems, so these structures should be interpreted cautiously.
How I define a “good” mutant
I define a good or effective mutant as one that balances multiple factors rather than optimizing only one metric. First, it should remain structurally plausible as an L-protein variant and avoid obviously destabilizing substitutions. Second, it should preserve or improve a meaningful interaction pattern with DnaJ in the soluble region if the biological mechanism still depends on that interaction. Third, if the long-term goal is DnaJ independence, then a good mutant could also be one that remains well folded without requiring the same interaction geometry. Finally, the mutant should be supported by prior mutational evidence and should not rely on highly disruptive changes at conserved positions. In practice, the best mutant is the one that shows a reasonable structural model, uses biologically tolerated substitutions, and best aligns with the assignment goals of improved folding, lysis efficiency, or reduced dependence on bacterial chaperones.