Project Propalsal:
A small, low-cost desktop platform that combines short DNA synthesis with cell-free expression. Users (students, community labs, small clinics) design short DNA sequences through a web interface, send them to a benchtop “DNA printer,” and immediately test them in a cell-free system. This pushes “personal fabrication” into biology and could support education and grassroots innovation, but raises serious questions about biosecurity, safety, and equity when DNA writing becomes cheap and widely accessible.
Homework: Waters Part I — Molecular Weight 1. Predicted molecular weight of eGFP The eGFP sequence contains a C-terminal His purification tag (HHHHHH) and a short linker (LE) before the tag.
Using the full provided amino acid sequence:
Length: 247 amino acids Predicted average molecular weight before chromophore maturation: 28,006.60 Da GFP chromophore maturation causes an approximate mass loss of 20.03 Da Corrected theoretical molecular weight: 27,986.58 Da, or 27.987 kDa So the calculated molecular weight is approximately:
Homework — Due by Start of Apr 28 Lecture Part A: The 1,536 Pixel Artwork Canvas | Collective Artwork I contributed one pixel to the global artwork experiment. My contribution was intentionally small, but I liked that the project made each person’s local decision become part of a larger shared image. The final artwork depended on many tiny choices accumulating together rather than on one centralized author.
3.1 Choose your protein
I chose Green Fluorescent Protein (GFP) from the jellyfish Aequorea victoria.
Reasons:
-Classic reporter protein in molecular biology and imaging
-Small, monomeric, and widely used as a fusion tag
sp|P42212|GFP_AEQVI Green fluorescent protein OS=Aequorea victoria OX=6100 GN=GFP PE=1 SV=1 MSKGEELFTGVVPILVELDGDVNGHKFSVSGEGEGDATYGKLTLKFICTTGKLPVPWPTL VTTFSYGVQCFSRYPDHMKQHDFFKSAMPEGYVQERTIFFKDDGNYKTRAEVKFEGDTLV NRIELKGIDFKEDGNILGHKLEYNYNSHNVYIMADKQKNGIKVNFKIRHNIEDGSVQLAD HYQQNTPIGDGPVLLPDNHYLSTQSALSKDPNEKRDHMVLLEFVTAAGITHGMDELYK
3.2 Reverse translate (protein → DNA) reverse translation of sp|P42212|GFP_AEQVI Green fluorescent protein OS=Aequorea victoria OX=6100 GN=GFP PE=1 SV=1 to a 714 base sequence of most likely codons. atgagcaaaggcgaagaactgtttaccggcgtggtgccgattctggtggaactggatggc gatgtgaacggccataaatttagcgtgagcggcgaaggcgaaggcgatgcgacctatggc aaactgaccctgaaatttatttgcaccaccggcaaactgccggtgccgtggccgaccctg gtgaccacctttagctatggcgtgcagtgctttagccgctatccggatcatatgaaacag catgatttttttaaaagcgcgatgccggaaggctatgtgcaggaacgcaccatttttttt aaagatgatggcaactataaaacccgcgcggaagtgaaatttgaaggcgataccctggtg aaccgcattgaactgaaaggcattgattttaaagaagatggcaacattctgggccataaa ctggaatataactataacagccataacgtgtatattatggcggataaacagaaaaacggc attaaagtgaactttaaaattcgccataacattgaagatggcagcgtgcagctggcggat cattatcagcagaacaccccgattggcgatggcccggtgctgctgccggataaccattat ctgagcacccagagcgcgctgagcaaagatccgaacgaaaaacgcgatcatatggtgctg ctggaatttgtgaccgcggcgggcattacccatggcatggatgaactgtataaa
Reference paper: HYDRA – hydrogels by robotic automation Citation
Torchia, E. et al. Fabrication of cell culture hydrogels by robotic liquid handling automation for high-throughput drug testing. Communications Engineering, 4, 222 (2025).
What they did
This paper introduces HYDRA (HYDrogels by Robotic liquid-handling Automation), a method to fabricate thin, planar hydrogel films directly inside standard 96- and 384-well plates using liquid-handling robots.
Part A. Conceptual Questions Question: How many molecules of amino acids do you take with a piece of 500 grams of meat?
(On average an amino acid is ~100 Daltons.)
Answer:
5 mol × 6.02 × 10²³ ≈ 3 × 10²⁴ amino acid molecules.
So eating 500 g of meat gives you on the order of 10²⁴ (about three septillion) amino acid molecules.
Part 1 1. Retrieve SOD1 sequence and introduce A4V MATKVCVVLKGDGPVQGIINFEQKESNGPVKVWGSIKGLTEGLHGFHVHEFGDNTAGCTSAGPHFNPLSRKHGGPKDEERHVGDLGNVTADKDGVADVSIEDSVISLSGDHCIIGRTLVVHEKADDLGKGGNEESTKTGNAGSRLACGVIGIAQ
PepMLM-generated 12-aa peptides + known binder (lower = higher model confidence in the binder).
ID Peptide (12 aa) Source Perplexity (PepMLM) P1 WHSPVAAARLKE PepMLM 11.721713 P2 KRYGAAAARHKK PepMLM 11.211369 P3 WRYPVAGLALKE PepMLM 13.068802 P4 WHSPPAAVALGE PepMLM 12.159801 Ref FLYRWLPSRRGG known SOD1 binder N/A Observation (PepMLM confidence by perplexity):
Among generated candidates, P2 (KRYGAAAARHKK) has the lowest perplexity (highest PepMLM confidence), while P3 (WRYPVAGLALKE) has the highest perplexity (lowest confidence among the four).
Answers to Questions About This Week’s Lab Protocol 1. What are some components in the Phusion High-Fidelity PCR Master Mix and what is their purpose? Phusion High-Fidelity PCR Master Mix typically contains several important components: Phusion DNA polymerase, dNTPs, reaction buffer, and MgCl2. The polymerase synthesizes new DNA strands and has proofreading activity, which lowers the error rate compared with standard Taq polymerase. The dNTPs provide the nucleotide building blocks needed to make the new DNA strands. The buffer maintains the proper chemical environment, including pH and salt concentration, for the enzyme to work efficiently. MgCl2 is an essential cofactor that allows the polymerase to function properly. In this lab, the master mix is provided as a 2X mix, so only template DNA, primers, and water are added separately. (neb.com)
Part 1: IANNs What advantages do IANNs have over traditional genetic circuits? Traditional genetic circuits usually work like Boolean logic gates. They treat inputs and outputs as mostly ON or OFF.
IANNs are more flexible because they can handle graded, continuous biological signals. Instead of only asking whether an input exists, they can respond to the strength of each input and combine multiple inputs together.
Homework Part A: General and Lecturer-Specific Questions Assignees for This Section Group Status MIT/Harvard students Required Committed Listeners Required General Homework Questions Explain the main advantages of cell-free protein synthesis over traditional in vivo methods, specifically in terms of flexibility and control over experimental variables. Name at least two cases where cell-free expression is more beneficial than cell production.
Subsections of Homework
Week 1 HW: Principles and Practices
Project Propalsal:
A small, low-cost desktop platform that combines short DNA synthesis with cell-free expression. Users (students, community labs, small clinics) design short DNA sequences through a web interface, send them to a benchtop “DNA printer,” and immediately test them in a cell-free system. This pushes “personal fabrication” into biology and could support education and grassroots innovation, but raises serious questions about biosecurity, safety, and equity when DNA writing becomes cheap and widely accessible.
Option 1: Mandatory sequence screening and basic customer vetting for all DNA synthesis providers (including cartridge vendors), coordinated through national / international standards.
Option 2: Built-in technical safeguards in desktop devices (on-device sequence screening, hard limits on sequence length and volume, whitelist mode for education deployments).
Option 3: Community lab / school codes of conduct, safety & security training, and an incident-report network co-developed with public agencies and DIYbio / professional societies.
Does the option:
Option 1
Option 2
Option 3
Enhance Biosecurity
• By preventing incidents
1
2
2
• By helping respond
2
3
1
Foster Lab Safety
• By preventing incident
2
2
1
• By helping respond
3
3
1
Protect the environment
• By preventing incidents
2
2
1
• By helping respond
3
3
1
Other considerations
• Minimizing costs and burdens to stakeholders
3
2
1
• Feasibility?
2
3
1
• Not impede research
2
3
1
• Promote constructive applications
2
2
1
Based on this scoring, I would prioritize a combination of Option 1 and Option 3, with Option 2 as a complementary, medium-term measure.
Option 1 scores best on preventing high-consequence biosecurity incidents, especially if screening standards are coordinated internationally and made affordable for smaller providers. However, it is costly and risks concentrating DNA synthesis capacity in a few large actors. Option 3 scores best on lab safety, environmental protection, and promoting constructive applications in community labs and schools, but it is weaker for deterring sophisticated malicious actors. Option 2 could add an important technical layer of protection, yet it faces feasibility and “jailbreaking” challenges and could more easily impede legitimate research if designed too rigidly.
For a national science policy audience or major funders, I would recommend:
Supporting shared, affordable sequence-screening tools and minimum standards (Option 1).
Investing in training, codes of conduct, and incident-report networks for community labs and schools (Option 3).
Encouraging research and early deployment of built-in safeguards, while monitoring how they affect usability and innovation (Option 2).
Key uncertainties include how quickly desktop DNA platforms will diffuse, how easy it will be to circumvent safeguards, and how governance choices in one country will shift risks and opportunities globally.
Reflecting on this week’s class, one ethical concern that became more salient to me is how routine DNA writing already is in modern biology. It no longer feels like a rare, “sci-fi” capability but a basic infrastructure, which makes dual-use risks more mundane and distributed. Another concern is equity: if governance relies only on heavy regulation and expensive compliance, advanced tools may become concentrated in a few wealthy institutions, while informal or under-resourced spaces are pushed into a gray zone with less support and oversight.
In the local context of MIT and Harvard, I think appropriate governance actions include: brief, practical training on DNA synthesis ethics for people who can place synthesis orders; centrally provided sequence-screening tools so individual labs do not each have to solve the problem; and safe channels to ask questions about “borderline” projects and to report concerns. These measures align with Option 1 and Option 3, and feel tractable at the institutional level.
Homework Questions:
Nature’s machinery for copying DNA is called polymerase. What is the error rate of polymerase? How does this compare to the length of the human genome. How does biology deal with that discrepancy?
A:Polymerase is ~1 error per 10⁶ bases, which would mean thousands of errors across the 3.2×10⁹-bp human genome, so cells rely on proofreading plus mismatch repair to bring the effective error rate way down.
How many different ways are there to code (DNA nucleotide code) for an average human protein? In practice what are some of the reasons that all of these different codes don’t work to code for the protein of interest?
A:Because many amino acids have multiple synonymous codons, an average-length protein can be encoded by an astronomically large number of DNA sequences, but many fail in practice due to codon bias/rare tRNAs, harmful mRNA structures, and unintended regulatory or splicing signals that reduce or disrupt expression.
What’s the most commonly used method for oligo synthesis currently?
A:
Oligonucleotide synthesis
Why is it difficult to make oligos longer than 200nt via direct synthesis?
A:Its gonna have errors.
Why can’t you make a 2000bp gene via direct oligo synthesis?
A:Its gonna have a lots of errors.
What are the 10 essential amino acids in all animals and how does this affect your view of the “Lysine Contingency”?
A:The “10 essential amino acids” mnemonic often used for animals is PVT TIM HALL: Phenylalanine, Valine, Threonine, Tryptophan, Isoleucine, Methionine, Histidine, Arginine, Leucine, Lysine.
Since lysine is already an essential amino acid (animals generally can’t synthesize it and must get it from diet), “making an animal lysine-dependent” is basically making it normal, so as a containment strategy it’s weak unless you also control lysine access or engineer dependence on something non-natural (a synthetic nutrient) rather than a widely available dietary essential.
Week 10 HW
Homework: Waters Part I — Molecular Weight
1. Predicted molecular weight of eGFP
The eGFP sequence contains a C-terminal His purification tag (HHHHHH) and a short linker (LE) before the tag.
Using the full provided amino acid sequence:
Length: 247 amino acids
Predicted average molecular weight before chromophore maturation: 28,006.60 Da
GFP chromophore maturation causes an approximate mass loss of 20.03 Da
Corrected theoretical molecular weight: 27,986.58 Da, or 27.987 kDa
So the calculated molecular weight is approximately:
MW_theory = 27.987 kDa
2. Molecular weight from adjacent charge states
I selected two adjacent charge-state peaks from the intact LC-MS spectrum:
Peak
m/z
Assigned charge
Peak 1
933.739
30+
Peak 2
966.039
29+
The charge state can be estimated from the adjacent peak spacing:
[
z \approx \frac{m/z_{higher}}{m/z_{higher} - m/z_{lower}}
]
Yes, the charge state can be observed from the isotope spacing in the zoomed-in peak near m/z 1473.7.
The isotope peaks are separated by about 0.053 m/z. Since isotope spacing is approximately:
[
\Delta(m/z) = \frac{1}{z}
]
then:
[
z \approx \frac{1}{0.053} \approx 19
]
So the zoomed-in peak is approximately:
19+ charge state
Homework: Waters Part II — Secondary/Tertiary Structure
1. Native versus denatured protein conformations
A native protein is folded into its functional three-dimensional structure. A denatured protein is unfolded, so more of its amino acid residues are exposed to solvent.
In mass spectrometry, this changes the charge-state distribution. A denatured protein usually takes on more protons because more basic and polar groups are exposed. This produces higher charge states and therefore peaks at lower m/z values.
In the denatured eGFP spectrum, there are many charge states spread across lower m/z values. In the native eGFP spectrum, the protein remains more compact, so it carries fewer charges. The native spectrum therefore shows lower charge states at higher m/z values, with fewer dominant peaks.
So the main difference is:
Denatured eGFP: more highly charged, lower m/z, broader charge-state distribution. Native eGFP: less highly charged, higher m/z, fewer charge states.
2. Charge state of the native peak near m/z 2800
Yes. The peak near m/z 2800 corresponds to approximately the 10+ charge state.
Reason:
The molecular weight of eGFP is about 28 kDa. A 10+ ion would appear near:
[
\frac{28,000 + 10H^+}{10} \approx 2800
]
The isotope spacing also supports this. The zoomed-in isotope peaks are separated by about 0.1 m/z, and:
[
z = \frac{1}{0.1} = 10
]
Therefore:
The native eGFP peak at ~2800 m/z is 10+.
Homework: Waters Part III — Peptide Mapping / Primary Structure
2. Number of peptides generated by tryptic digestion
Trypsin cleaves after K and R, except when followed by proline.
Using the PeptideMass-style settings shown in the homework figure:
Enzyme: Trypsin
Missed cleavages: 0
Mass mode: monoisotopic
Output: peptides larger than 500 Da
The digest gives:
27 total tryptic fragments if all fragments are counted. 19 reported peptides if the PeptideMass cutoff of >500 Da is applied.
For the homework answer, I would report:
19 peptides above 500 Da.
3. Number of chromatographic peaks between 0.5 and 6 minutes
From the peptide map TIC, counting peaks above roughly 10% relative abundance between 0.5 and 6 minutes, I see approximately:
18 chromatographic peaks
This is a visual count from the figure, so I would describe it as approximate.
4. Does the number of chromatographic peaks match the predicted peptide number?
The chromatogram shows slightly fewer peaks than the 19 predicted peptides above 500 Da, and far fewer than the 27 total theoretical tryptic fragments.
This does not necessarily mean the digest failed. Several things can happen:
Some peptides may co-elute in the same chromatographic peak.
Some peptides may be too small, too low abundance, or ionize poorly.
Some peptides may not be detected under the LC-MS conditions.
Some chromatographic peaks may include adducts, modified peptides, or background signals.
So the experimental chromatogram is close to the predicted peptide count, but it does not produce a perfect one-peak-per-peptide match.
5. m/z, charge state, and singly charged mass of the peptide in Figure 5b
The most abundant peptide peak in Figure 5b is at approximately:
m/z = 525.767
The isotope spacing is about 0.5 m/z, which indicates:
[
z = \frac{1}{0.5} = 2
]
So the most abundant charge state is:
2+
To calculate the singly charged form ([M+H]^+):
[
[M+H]+ = z(m/z) - (z-1)H+
]
[
[M+H]^+ = 2(525.76712) - 1.0073
]
[
[M+H]^+ \approx 1050.527 \text{ Da}
]
Therefore:
m/z = 525.767, z = 2+, [M+H]+ ≈ 1050.527 Da
6. Peptide identity and mass accuracy
The peptide that best matches this mass in the predicted tryptic digest is:
FEGDTLVNR
The theoretical singly charged mass of this peptide is approximately:
7. Percentage of sequence confirmed by peptide mapping
From the amino acid coverage map:
88% sequence coverage
Bonus 8. Peptide sequence from the fragmentation spectrum
The peptide sequence that best matches the mass and fragmentation spectrum is:
FEGDTLVNR
Bonus 9. Does the peptide map indicate the protein is eGFP?
Yes. The peptide map supports that the sample is the eGFP standard.
The evidence is:
The intact protein mass is close to the theoretical eGFP mass.
The tryptic peptide mass at m/z 525.767, 2+ matches a predicted eGFP peptide.
The fragmentation spectrum supports the sequence FEGDTLVNR.
The peptide mapping coverage is high, at 88%.
Together, these results strongly indicate that the protein is eGFP.
Homework: Waters Part IV — Oligomers
KLH oligomer masses can be estimated by multiplying the subunit mass by the number of subunits in the oligomer.
Oligomeric species
Calculation
Expected mass
Peak position in spectrum
7FU Decamer
10 × 340 kDa
3,400 kDa
~3.4 MDa
8FU Didecamer
20 × 400 kDa
8,000 kDa
~8.3 MDa
8FU 3-Decamer
30 × 400 kDa
12,000 kDa
~12.7 MDa
8FU 4-Decamer
40 × 400 kDa
16,000 kDa
~16 MDa, weak/low abundance
The strongest visible assignments are approximately:
3.4 MDa = 7FU decamer
8.33 MDa = 8FU didecamer
12.67 MDa = 8FU 3-decamer
~16 MDa = expected 8FU 4-decamer, but it appears weak or not clearly resolved in the shown spectrum
Homework: Waters Part V — Did I make GFP?
Measurement
Theoretical
Observed / measured on intact LC-MS
PPM mass error
Molecular weight
27.987 kDa
27.984 kDa
~94 ppm
Based on the intact protein mass, peptide map, and 88% sequence coverage, the sample is consistent with eGFP.
Week 11 HW
Homework — Due by Start of Apr 28 Lecture
Part A: The 1,536 Pixel Artwork Canvas | Collective Artwork
I contributed one pixel to the global artwork experiment. My contribution was intentionally small, but I liked that the project made each person’s local decision become part of a larger shared image. The final artwork depended on many tiny choices accumulating together rather than on one centralized author.
What I liked most was the relationship between scale and authorship. A single pixel is almost invisible on its own, but within the full canvas it becomes part of a collective biological image. That felt appropriate for a bioart project because the artwork behaved almost like a living system: many small inputs produced an emergent pattern that no individual participant could fully control.
For next year, the project could be improved by making the collaboration process more visible. A time-lapse showing how the artwork changed over time would help viewers understand the collective process behind the final image. It could also be interesting to let contributors leave short notes about their choices, so the project records both the final visual outcome and the distributed decisions that produced it.
Part B: Cell-Free Protein Synthesis | Cell-Free Reagents
Roles of each component
E. coli Lysate
BL21(DE3) Star Lysate, including T7 RNA Polymerase The lysate provides the core biological machinery for transcription and translation, including ribosomes, tRNAs, aminoacyl-tRNA synthetases, translation factors, metabolic enzymes, and other cellular components. Because it includes T7 RNA polymerase, it can efficiently transcribe DNA templates under a T7 promoter.
Salts / Buffer
Potassium Glutamate Potassium glutamate helps recreate the ionic environment of the bacterial cytoplasm. Potassium supports protein synthesis, while glutamate is a biologically compatible counterion that helps maintain reaction performance.
HEPES-KOH, pH 7.5 HEPES-KOH buffers the reaction near physiological pH. This is important because transcription, translation, enzyme activity, and fluorescent protein stability are all sensitive to pH changes.
Magnesium Glutamate Magnesium is essential for ribosome structure, tRNA function, nucleotide chemistry, and many enzymes in transcription and translation. Magnesium concentration must be tuned carefully because too little reduces translation, while too much can inhibit or destabilize the system.
Potassium Phosphate Monobasic Potassium phosphate monobasic contributes phosphate and buffering capacity. Together with the dibasic form, it helps maintain pH and supports phosphate-dependent energy metabolism.
Potassium Phosphate Dibasic Potassium phosphate dibasic pairs with the monobasic phosphate to create a phosphate buffer system. It also contributes potassium ions and phosphate needed for metabolic and energy-regeneration reactions.
Energy / Nucleotide System
Ribose Ribose provides a sugar backbone that can support nucleotide regeneration and metabolic activity. In long-duration reactions, it helps the system rebuild nucleotide triphosphates rather than relying only on pre-supplied NTPs.
Glucose Glucose is an energy substrate that can be metabolized by enzymes in the lysate. It supports ATP regeneration over longer reactions.
AMP AMP is a nucleotide precursor that can be converted into ATP through phosphorylation pathways. It supports a lower-cost, regenerating energy system.
CMP CMP is a precursor for CTP regeneration. It supports RNA synthesis once converted into the triphosphate form.
GMP GMP is a precursor for GTP regeneration. GTP is needed for transcription and also plays a major role in translation.
UMP UMP is a precursor for UTP regeneration. UTP is required for RNA synthesis during transcription.
Guanine Guanine is a nucleobase that can enter nucleotide salvage pathways. It can be converted into guanine nucleotides and eventually support GTP production.
Translation Mix / Amino Acids
17 Amino Acid Mix The amino acid mix provides most of the building blocks needed to synthesize the target protein. Without sufficient amino acids, translation stops or produces low yield.
Tyrosine Tyrosine is supplied separately because it has solubility and stability constraints compared with many other amino acids. It is also especially relevant for fluorescent proteins, since aromatic residues help form the chromophore environment.
Cysteine Cysteine is often handled separately because it is chemically reactive and can oxidize. Maintaining cysteine availability helps prevent amino acid limitation during protein synthesis.
Additives
Nicotinamide Nicotinamide supports metabolic cofactor balance, especially pathways related to NAD/NADH chemistry. In a cell-free system, maintaining cofactor availability helps sustain energy metabolism and enzyme activity.
Backfill
Nuclease-Free Water Nuclease-free water is used to bring the reaction to the final volume without introducing nucleases that could degrade DNA or RNA. It is the neutral backfill component for controlling final reaction concentration.
Difference between the 1-hour optimized PEP-NTP master mix and the 20-hour NMP-Ribose-Glucose master mix
The 1-hour optimized PEP-NTP master mix is designed for fast, high-intensity expression by directly supplying NTPs and using phosphoenolpyruvate (PEP) as a strong energy-regeneration substrate. It is useful when the goal is rapid protein production over a short time window.
The 20-hour NMP-Ribose-Glucose master mix is designed for longer reactions by using nucleotide monophosphates, ribose, and glucose to regenerate nucleotides and ATP over time. It is slower but more sustained, making it better for long incubations such as overnight or 36-hour fluorescence development.
Bonus: How can transcription occur if GMP is not included but Guanine is?
Transcription can still occur because guanine can enter nucleotide salvage pathways. The lysate can convert guanine into GMP, then phosphorylate GMP into GDP and GTP. GTP can then be used by RNA polymerase during transcription.
Part C: Planning the Global Experiment | Cell-Free Master Mix Design
Fluorescent protein properties affecting cell-free expression or readout
sfGFP
sfGFP is useful in cell-free systems because it folds and matures quickly and is engineered to tolerate imperfect folding environments. Its main limitation is that fluorescence still requires chromophore maturation, which depends on oxygen and time.
mRFP1
mRFP1 matures faster than the older DsRed protein, but it has lower brightness and photostability than some newer red fluorescent proteins. In a cell-free reaction, this means it may need longer incubation or stronger expression to produce a visible signal.
mKO2
mKO2 is an orange fluorescent protein with moderate acid sensitivity. This means the readout may be affected if the cell-free reaction becomes acidic during long incubation.
mTurquoise2
mTurquoise2 is a cyan fluorescent protein known for strong performance in imaging, including relatively good photostability. In cell-free systems, the main concern is making sure the excitation/emission settings match cyan fluorescence and that the protein has enough time and oxygen to mature.
mScarlet_I
mScarlet_I is a bright monomeric red fluorescent protein with accelerated maturation compared with mScarlet. However, red fluorescent proteins can still be sensitive to maturation time, oxygen availability, and pH during long cell-free incubation.
Electra2
Electra2 is a blue fluorescent protein with low pKa, which suggests relatively strong resistance to acid quenching. Its readout may still be harder to detect than green or red proteins because blue fluorescence can be more sensitive to instrument settings, background, and excitation conditions.
Hypothesis for improving fluorescence over a 36-hour incubation
Protein: mScarlet_I
Reagents to adjust: HEPES-KOH, potassium phosphate buffer, magnesium glutamate, amino acid mix, tyrosine, cysteine, and glucose/ribose energy system.
Hypothesis: For mScarlet_I, increasing buffer capacity with HEPES-KOH and phosphate while maintaining an optimized magnesium glutamate concentration will improve fluorescence over 36 hours by stabilizing pH and supporting ribosome activity. Adding extra amino acid mix, especially tyrosine and cysteine, could prevent amino acid depletion during long translation. Using the NMP-Ribose-Glucose energy system should support sustained ATP and NTP regeneration, allowing protein production and chromophore maturation to continue over a longer time window.
Expected effect: The reaction should produce a stronger final red fluorescence signal after 36 hours because the system remains active for longer, avoids pH-related fluorescence loss, and maintains enough amino acid and energy supply for continued protein synthesis.
Part D: Build-A-Cloud-Lab | Optional Bonus Assignment
For the optional cloud lab design, I would create a circular, modular “biofoundry island” made of Ginkgo Reconfigurable Automation Carts. The center would contain shared analysis equipment, while the outer ring would contain specialized carts for liquid handling, incubation, imaging, and sample storage.
This layout would make the lab feel less like a linear factory and more like a flexible organism. Samples could move around the ring depending on the workflow, and individual carts could be swapped or reconfigured without changing the whole system. The design would be especially useful for experiments like cell-free synthesis, where many small reactions need to be assembled, incubated, imaged, and compared in parallel.
I chose Green Fluorescent Protein (GFP) from the jellyfish Aequorea victoria.
Reasons:
-Classic reporter protein in molecular biology and imaging
-Small, monomeric, and widely used as a fusion tag
sp|P42212|GFP_AEQVI Green fluorescent protein OS=Aequorea victoria OX=6100 GN=GFP PE=1 SV=1
MSKGEELFTGVVPILVELDGDVNGHKFSVSGEGEGDATYGKLTLKFICTTGKLPVPWPTL
VTTFSYGVQCFSRYPDHMKQHDFFKSAMPEGYVQERTIFFKDDGNYKTRAEVKFEGDTLV
NRIELKGIDFKEDGNILGHKLEYNYNSHNVYIMADKQKNGIKVNFKIRHNIEDGSVQLAD
HYQQNTPIGDGPVLLPDNHYLSTQSALSKDPNEKRDHMVLLEFVTAAGITHGMDELYK
3.2 Reverse translate (protein → DNA)
reverse translation of sp|P42212|GFP_AEQVI Green fluorescent protein OS=Aequorea victoria OX=6100 GN=GFP PE=1 SV=1 to a 714 base sequence of most likely codons.
atgagcaaaggcgaagaactgtttaccggcgtggtgccgattctggtggaactggatggc
gatgtgaacggccataaatttagcgtgagcggcgaaggcgaaggcgatgcgacctatggc
aaactgaccctgaaatttatttgcaccaccggcaaactgccggtgccgtggccgaccctg
gtgaccacctttagctatggcgtgcagtgctttagccgctatccggatcatatgaaacag
catgatttttttaaaagcgcgatgccggaaggctatgtgcaggaacgcaccatttttttt
aaagatgatggcaactataaaacccgcgcggaagtgaaatttgaaggcgataccctggtg
aaccgcattgaactgaaaggcattgattttaaagaagatggcaacattctgggccataaa
ctggaatataactataacagccataacgtgtatattatggcggataaacagaaaaacggc
attaaagtgaactttaaaattcgccataacattgaagatggcagcgtgcagctggcggat
cattatcagcagaacaccccgattggcgatggcccggtgctgctgccggataaccattat
ctgagcacccagagcgcgctgagcaaagatccgaacgaaaaacgcgatcatatggtgctg
ctggaatttgtgaccgcggcgggcattacccatggcatggatgaactgtataaa
3.3 Organism chosen and why:
I optimized the sequence for Escherichia coli (e.g. K-12 lab strain).
E. coli is cheap, grows fast, and is a standard workhorse for expressing GFP. There are many well-characterized plasmids and promoters for high-level GFP expression in E. coli.
There are two main ways to produce my GFP protein from this DNA: cell-dependent and cell-free expression.
Cell-dependent method (E. coli expression) I can clone my codon-optimized GFP sequence into an expression plasmid under a strong promoter (for example a T7 or lac promoter) with a ribosome binding site and terminator. The plasmid is transformed into E. coli. Inside the cells, bacterial RNA polymerase transcribes the GFP gene into mRNA, and ribosomes translate this mRNA into the GFP polypeptide, reading it codon by codon. The peptide folds into the GFP β-barrel and forms its chromophore, so the cells become fluorescent under blue/UV light. This is a classic, cell-dependent way to produce GFP.
Cell-free method (in vitro transcription–translation) Alternatively, I can add the same GFP DNA template to a cell-free transcription–translation system made from E. coli lysate. The lysate contains RNA polymerase, ribosomes, tRNAs, amino acids, NTPs, and energy regeneration components. In the tube, the DNA is transcribed into mRNA and then translated into GFP, again following the central dogma (DNA → RNA → protein), but without living cells. After incubation, the reaction mixture will glow green if GFP is correctly produced and folded.
Week 3 HW: LabAutomation
1. Reference paper: HYDRA – hydrogels by robotic automation
Citation
Torchia, E. et al. Fabrication of cell culture hydrogels by robotic liquid handling automation for high-throughput drug testing. Communications Engineering, 4, 222 (2025).
What they did
This paper introduces HYDRA (HYDrogels by Robotic liquid-handling Automation), a method to fabricate thin, planar hydrogel films directly inside standard 96- and 384-well plates using liquid-handling robots.
Normally, when you cast hydrogels into small wells, capillary forces at the sidewalls create a curved meniscus, which:
makes hydrogel thickness non-uniform across the well
disturbs cell seeding density and imaging focus
reduces the reliability of high-throughput drug screening
HYDRA solves this by:
Robotically dispensing a sub-contact volume of hydrogel precursor (fish gelatin + microbial transglutaminase) into each well, carefully avoiding contact with the sidewalls.
Immediately re-aspirating most of that volume with precisely controlled height and flow rate.
Using contact angle hysteresis so that a thin, meniscus-free layer (about 10–50 μm) remains at the bottom.
The authors show that these hydrogels support drug dose–response assays on engineered epithelial cells and allow long-term imaging on soft, biomimetic substrates. They also demonstrate that HYDRA can be implemented on an open-source Opentrons OT-2 robot, effectively turning a liquid-handling platform into a simple, programmable soft-materials fabrication tool.
They combine this with plate-scale quality control and show that the hydrogels support:
Drug dose–response assays with engineered epithelial cells
Long-term holographic and fluorescence microscopy on soft, biomimetic substrates
How the automation is implemented (and why it’s relevant)
They explicitly use an Opentrons OT-2 as an open-source, low-cost platform to implement HYDRA. The pipeline is built with Opentrons Protocol Designer and custom Python to control dispense/aspirate heights, volumes, and speeds. :contentReference[oaicite:4]{index=4}
The OT-2 mixes gelatin and transglutaminase stocks, casts the precursor into plates, and re-aspirates to leave a controlled film.
Conceptually, this turns a “liquid-handling robot” into a materials fabrication tool: it is designing not only concentrations but also geometry (flat films with controlled thickness) and mechanical properties (tunable stiffness ~1.5–6 kPa). :contentReference[oaicite:5]{index=5}
For my interests (digital fabrication, soft metamaterials, auxetics), this is very close to “2.5D soft material printing”:
Process parameters: volumes, pipette height, flow rate
Output behavior: thickness, flatness, stiffness and cell response
This is exactly the kind of workflow I want to adapt: using Opentrons not just as a biology helper, but as a programmable fabrication device for soft, structured materials.
Opentrons-printed auxetic hydrogel tiles for programmable mechanics
Core idea
Use the Opentrons OT-2 as a “dot-matrix printer” for soft materials: it will deposit small droplets of hydrogel precursor with different formulations onto a thin flexible substrate, forming a 2D auxetic (negative Poisson’s ratio) pattern.
By controlling which beams/tiles are soft vs stiff (or swell more vs less), the overall structure exhibits programmable shape change or auxetic behavior when stretched or stimulated.
This combines:
HYDRA’s idea of robotic hydrogel fabrication
My background in digital fabrication and mechanical metamaterials
A design space of structure × material formulation × process
Biological / material system (high-level)
I will use a crosslinkable hydrogel system compatible with HTGAA and the Opentrons:
Option A: fish gelatin + microbial transglutaminase (following HYDRA)
Option B: a photo-crosslinkable gelatin (e.g., GelMA) if lab infrastructure favors photogels
Each “unit” in the auxetic pattern is defined by two main parameters:
Base stiffness – controlled by gelatin concentration (e.g., 5%, 10%, 20%)
Crosslink density – controlled by enzyme concentration or light exposure time
These combinations create:
Soft segments that deform easily
Stiff segments that act as constraints
Arranged in an auxetic geometry (e.g., re-entrant honeycomb, rotating squares), the global behavior becomes a tunable mechanical metamaterial.
Cells are optional at this stage; the primary goal is to demonstrate programmable mechanical behavior. Cells could later be seeded to test how different stiffness regions affect attachment and morphology.
What will be automated
Automated formulation library
The OT-2 prepares a small library of hydrogel precursor formulations in a 96-well plate:
“Soft”: 5% gelatin + 0.5% crosslinker
“Medium”: 10% gelatin + 1% crosslinker
“Stiff”: 20% gelatin + 2% crosslinker
Using standard pipetting commands, the robot mixes stock solutions to create these recipes in defined wells.
This step establishes a recipe space that can be expanded later (different polymer, different additives, etc.).
Geometric pattern → robot coordinates
I design an auxetic pattern in Rhino/Grasshopper or Python (e.g., 6 × 6 re-entrant lattice).
Each structural element (“beam”) is discretized into a small number of print points.
Each point is annotated with a recipe ID (“soft”, “medium”, “stiff”).
I then map this point set into the OT-2 deck coordinate system by:
3D-printing a simple holder that clamps a thin transparent membrane (PDMS or plastic) on the deck
Calibrating its four corners relative to the robot origin
Converting design coordinates (x, y) into deck positions via a linear transform + offsets
The result: a table of points like (x, y, recipe_id) that the robot can iterate through.
Printing and curing
The robot:
Aspirates small volumes (e.g., 2–5 µL) of each formulation from the 96-well “recipe plate”
Moves to specified (x, y) positions over the substrate
Deposits droplets in the order dictated by the auxetic pattern
After printing:
The membrane is transferred to an incubator (37 °C) or UV station for crosslinking
Once cured, the membrane can be mounted in a simple tensile frame and mechanically tested (even by hand with phone video) to observe auxetic deformation.
This is a direct analog of HYDRA (controlling meniscus and layer thickness), but applied to patterned, multi-formulation structures instead of uniform coatings.
Experimental workflow (high-level)
Preparation (manual)
Prepare gelatin and crosslinker stock solutions.
Design and 3D-print a membrane holder compatible with a specific Opentrons deck slot.
Attach a thin transparent membrane on top and calibrate four reference points on the OT-2.
Deck layout
Slot 1: Eppendorf rack with stock solutions (gelatin, crosslinker, buffer).
Slot 2: 96-well “recipe plate” where the robot generates soft/medium/stiff formulations.
Slot 3: custom membrane holder (“printing bed”).
Slot 4–5: tip racks (P20).
Slot 6: waste reservoir.
Opentrons protocol (concept)
Step 1 – Formulation generation
Robot mixes gelatin + crosslinker into the recipe plate:
Use P300/P20 to combine stocks in wells A1–A3 to produce “soft”, “medium”, “stiff”.
Optional: generate more variants across the plate to explore stiffness/swell space.
Step 2 – Auxetic pattern printing
Load auxetic point list (x, y, recipe) from a CSV or embedded list.
For each point:
Pick up a tip
Aspirate 2 µL from the well corresponding to recipe
Move to the transformed (x, y, z) above the membrane
Dispense the droplet
Drop the tip
Repeat until all beams in the pattern are printed.
Step 3 – Curing and testing
Move the printed membrane to an incubator / UV lamp for crosslinking.
After curing, mount on a simple frame, apply tension, and record deformation.
Optional: overlay grid or markers to track local strain.
My Final Project – Possible Directions
1. Opentrons-printed auxetic hydrogel tiles
One-liner Use an Opentrons robot as a “2.5D printer” to deposit hydrogel droplets in an auxetic pattern, and study how composition plus geometry shape the mechanical behavior.
What I would automate
Opentrons mixes a small library of hydrogel formulations in a 96-well plate (for example, soft / medium / stiff).
It then “prints” droplets onto a thin flexible film in a precomputed auxetic pattern (re-entrant squares, rotating squares, etc.).
Each beam or node of the pattern can use a different formulation, so the auxetic response is not only geometric but also rheology-informed.
Why it is interesting
Treats the OT-2 as a soft-material fabrication machine instead of just a pipetting tool.
Links soft-matter rheology, mechanical metamaterials, and lab automation: geometry, composition, and process are all programmable.
Can start as a purely mechanical experiment and later add biological layers if there is time.
2. High-throughput hydrogel rheology map in a plate
One-liner Use lab automation to build a small materials map of hydrogel rheology in a 96-well plate, linking formulation to mechanical properties as a design tool for later printing.
What I would automate
Opentrons prepares a combinatorial grid of hydrogel recipes across the plate (for example, rows = polymer concentration, columns = crosslinker concentration).
For each well, I run a simple, automatable mechanical proxy (for example, indentation depth under a fixed weight, or image-based deformation).
Collect all measurements into a composition–property map that I can use to choose formulations for the auxetic printing in project 1.
Why it is interesting
Turns subjective “this gel feels soft or stiff” into structured data driven by automation.
Shows a clear automation loop: the robot explores a soft-material design space, not just prepares biological assays.
Directly supports and informs the first project idea.
One-liner Use automation to assemble and place small cell-free reactions that behave like simple logic gates, so that a 2D fluorescent pattern encodes a truth table in space.
What I would automate
Opentrons prepares cell-free reactions with different DNA constructs that approximate AND / OR / NOT behavior (or simpler ON / OFF variants).
Each well corresponds to an input combination (00, 01, 10, 11), arranged in the plate as a visual truth table, or optionally printed as droplets onto a flat substrate.
After incubation, the pattern of fluorescence across wells or positions becomes a spatial representation of the logic.
Why it is interesting
Uses automation to build and arrange many small reactions that would be tedious by hand.
Connects synthetic biology, simple computation, and digital fabrication: logic is expressed both in biochemical reactions and in spatial layout.
Offers a complementary direction where the “printed pattern” carries information and function, not just mechanical behavior.
Week 4 HW: ProteinDesign
Part A. Conceptual Questions
Question: How many molecules of amino acids do you take with a piece of 500 grams of meat? (On average an amino acid is ~100 Daltons.)
So eating 500 g of meat gives you on the order of 10²⁴ (about three septillion) amino acid molecules.
Question: Why do humans eat beef but do not become a cow, eat fish but do not become fish?
Answer:
These free amino acids are absorbed and reused as generic building blocks to make human proteins, under the instructions of human DNA and our own gene expression programs.
Part B. Protein Analysis and Visualization
In this part, pick any protein (with a known 3D structure) and answer the following.
B1. Protein choice
Briefly describe the protein you selected and why you selected it.
Answer:
I chose the spider dragline silk protein, specifically a major ampullate spidroin (MaSp1). This is one of the main structural proteins that spiders use to spin their dragline silk, the “lifeline” they hang from and use as a safety rope. Dragline silk is famous for combining very high tensile strength (comparable to steel) with remarkable toughness and elasticity, and these mechanical properties come directly from its unusual amino acid sequence: long, repetitive blocks that form a mix of crystalline β-sheet nanodomains and more disordered, elastic regions.
I selected MaSp1 because it sits at the intersection of protein sequence, hierarchical structure, and macroscopic material behavior. It’s a natural example of how repeating sequence motifs can encode a programmable mechanical material, which fits nicely with the theme of protein design and with thinking about proteins as engineerable structural materials, not just enzymes or receptors.
This sequence is heavily enriched in glycine (G) and alanine (A), which together make up the majority of residues. Glycine is extremely small and flexible, while alanine is small and slightly hydrophobic. In spider silk, repeated (Gly–Ala)n motifs are known to stack into tightly packed β-sheet nanocrystals that provide the fiber’s high stiffness and tensile strength.
B4. Sequence homologs
How many protein sequence homologs are there for your protein?
Answer:
UniProt BLAST returns hundreds of homologous sequences, mostly other spider silk proteins from related species, showing that MaSp1 belongs to a large and diversified silk protein family.
B5. Protein family
Does your protein belong to any protein family?
Answer:
Belongs to the silk fibroin family
B6. Structure identification (RCSB PDB)
Identify the structure page of your protein in RCSB.
When was the structure solved? Is it a good quality structure?
Answer:
Year solved: 2016
Resolution: 2.02A
Comment on quality: Good
B8. Other molecules in the structure
Are there any other molecules in the solved structure apart from the protein?
Answer:
Ligands: None – there are no bound small-molecule ligands or cofactors reported.
Ions: None – no metal ions are annotated in this structure.
Water / other components: Only crystallographic water molecules (HOH) are present as part of the solvent.
Comments: The 5IZ2 structure is essentially just the N-terminal domain of MaSp1A plus bulk water, with no extra cofactors or metal ions. This makes it a relatively “clean” system for analyzing the intrinsic protein–protein interface and packing.
B9. Structural classification
Does your protein belong to any structure classification family? (e.g., SCOP, CATH)
This shows that the N-terminal domain of MaSp1 belongs to a specific spidroin N-terminal structural family in SCOP.
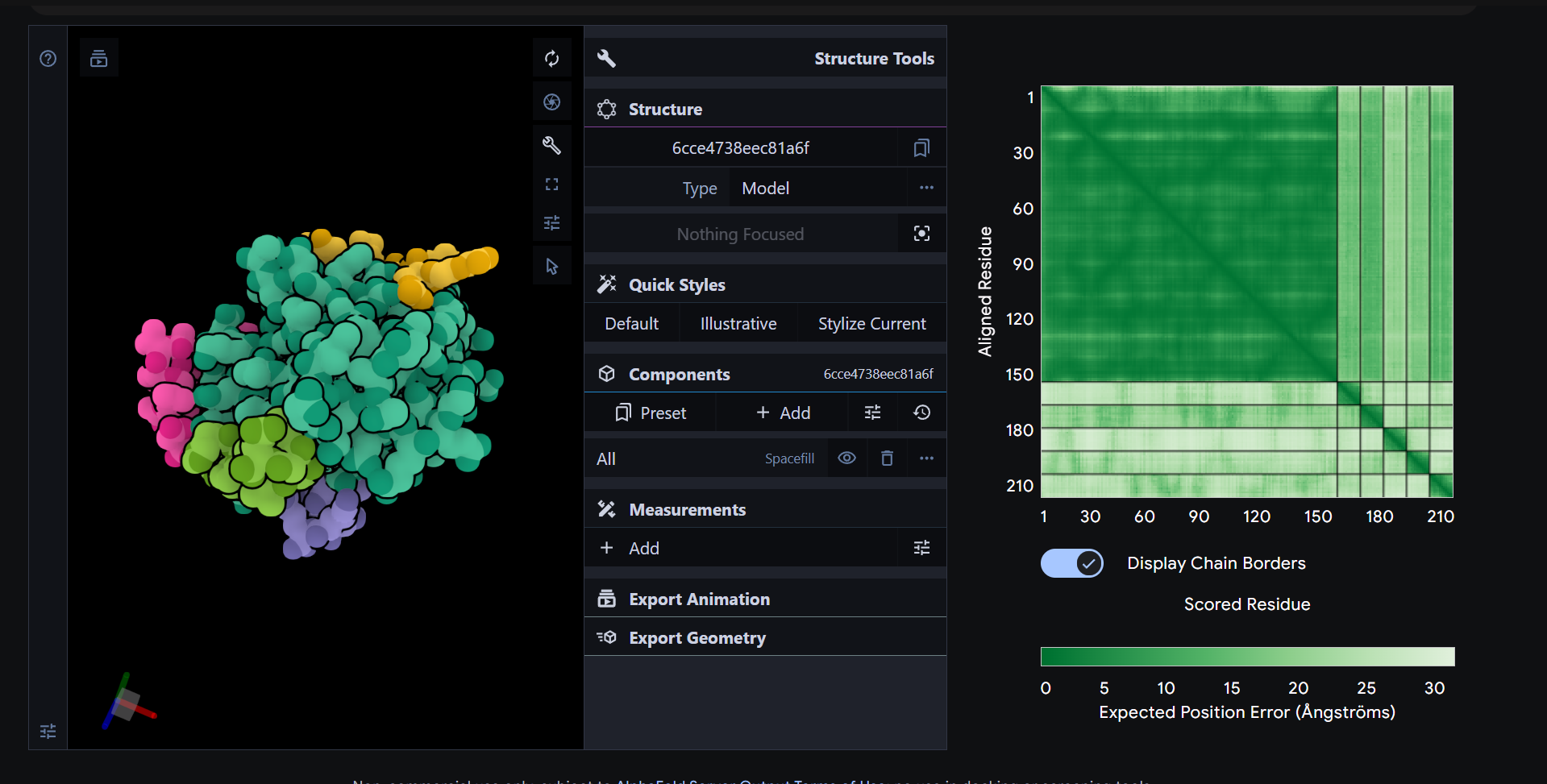
B10. 3D visualization
B10.1 Cartoon / ribbon / ball-and-stick views
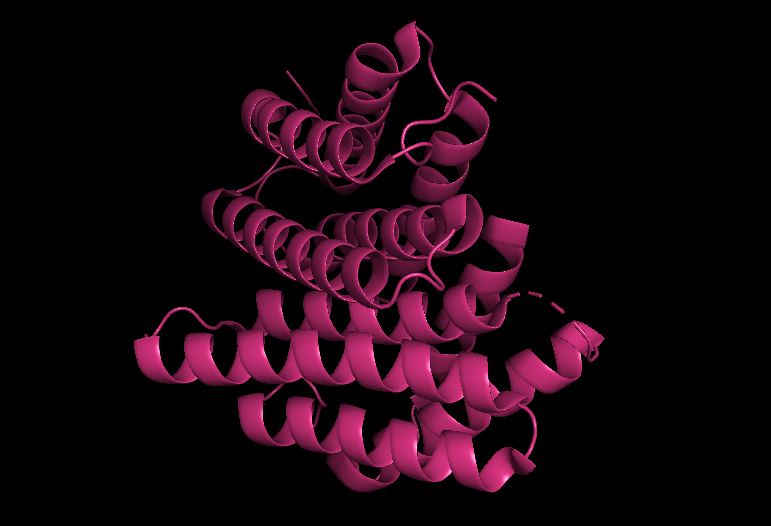
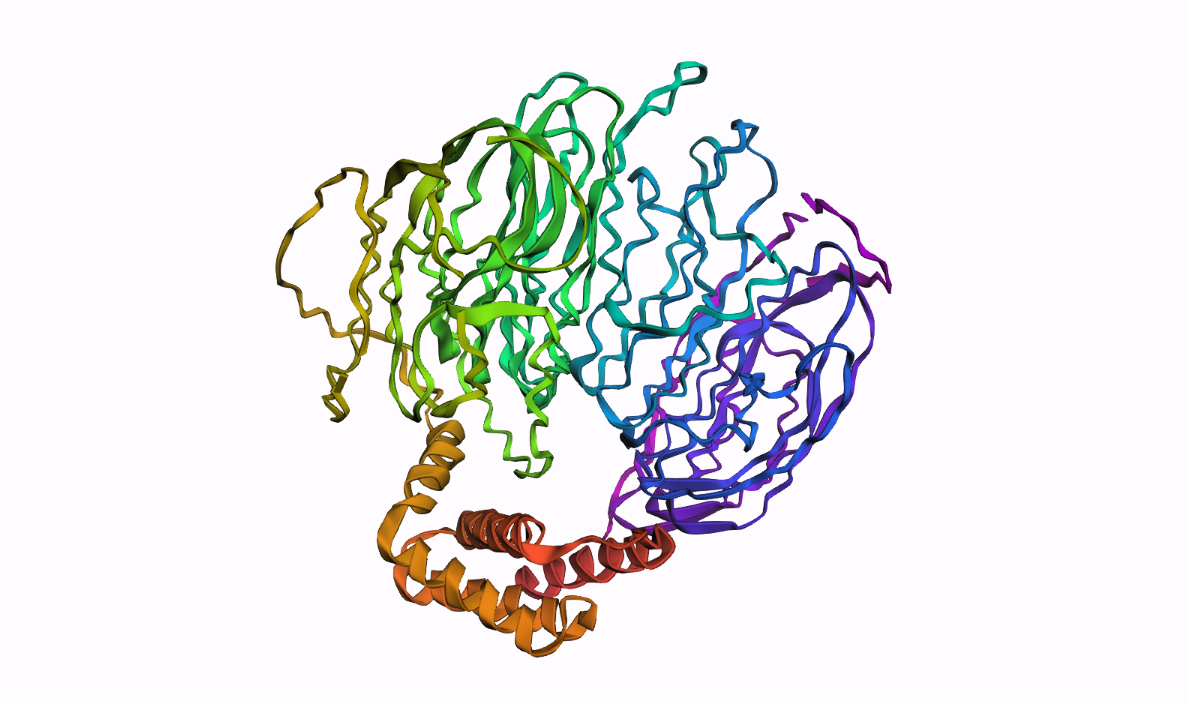
In PyMOL, I loaded the N-terminal domain of MaSp1 (PDB 5IZ2) and visualized it with different representations:
Cartoon view (secondary structure):
Ribbon view:
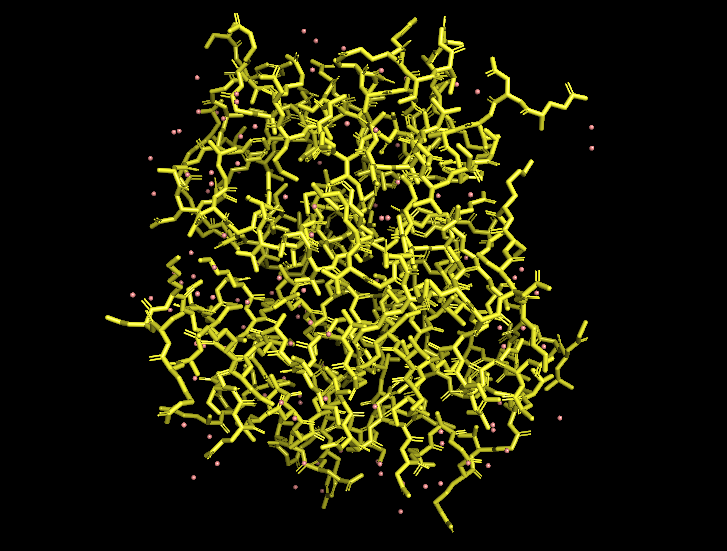
Ball-and-stick view (side chains):
B10.2 Hydrophobic vs hydrophilic residue distribution
When I color the protein by residue type / hydrophobicity, I observe a typical hydrophobic-core, hydrophilic-surface pattern:
Many hydrophobic residues (e.g., Leu, Val, Ile, Phe) are buried in the interior of each helical bundle and at the dimer interface.
Polar and charged residues (e.g., Gln, Glu, Lys, Arg, Ser) are mostly exposed on the outside, where they can interact with solvent and potentially mediate pH-dependent behavior.
This distribution is consistent with the idea that the MaSp1 N-terminal domain is a soluble, dimerizing helical bundle, with its hydrophobic core shielded from water and its polar residues decorating the surface.
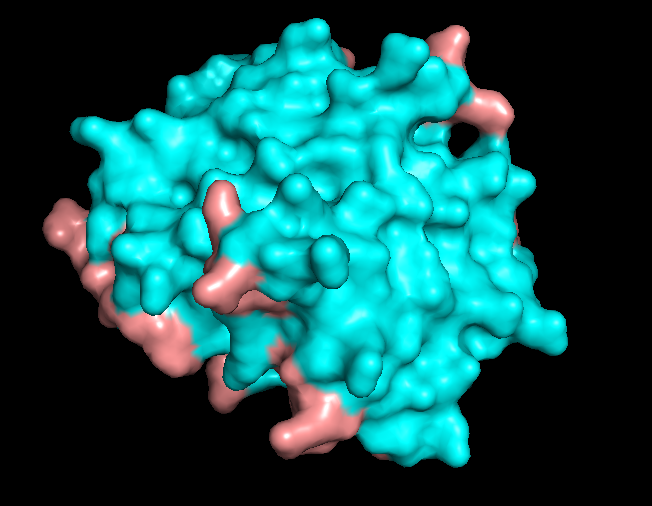
B10.3 Surface and potential “holes” (pockets)
Finally, I visualized the molecular surface of the dimer:
The overall shape is compact and smooth, but there are some shallow grooves and one noticeable depression on the surface. This depression looks like a small, shallow pocket rather than a deep enzyme-like active site or tunnel. It is more likely to be a protein–protein interaction groove (or simply a consequence of how the helices pack) than a dedicated small-molecule binding pocket.
Overall, the surface of 5IZ2 matches its role as a structural N-terminal dimerization domain, not a classical ligand-binding enzyme.
Part C. Using ML-Based Protein Design Tools
C1. Protein Language Modeling
C1.1 Deep mutational scan with ESM2
Use ESM2 to generate an unsupervised deep mutational scan of your protein. Can you explain any particular pattern? (Choose a residue and a mutation that stands out.)
Answer:
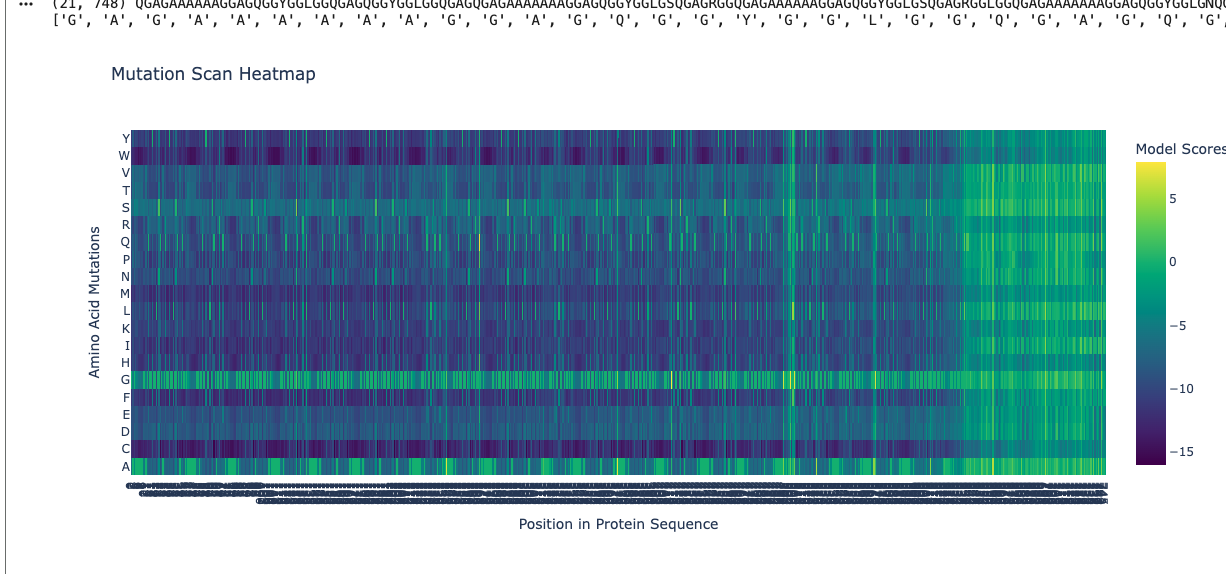
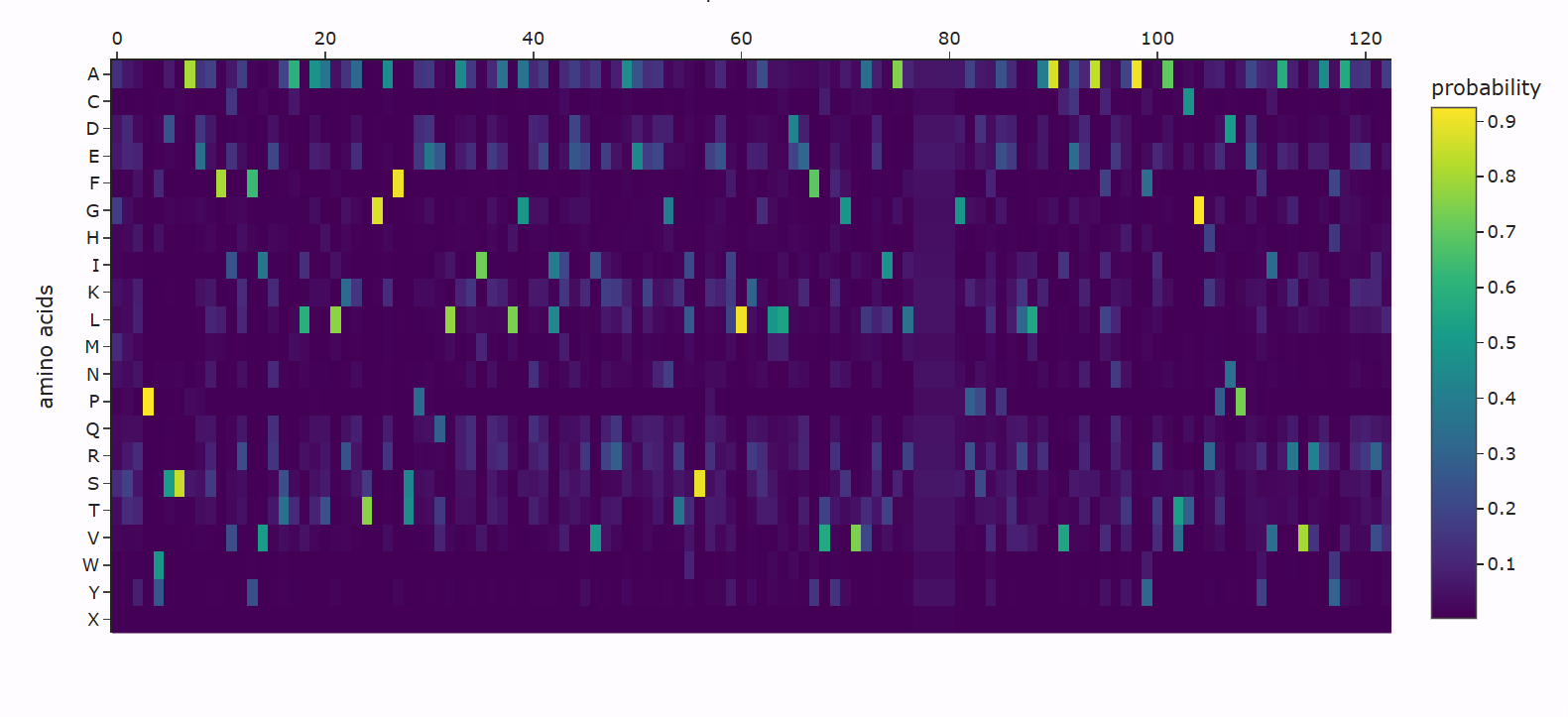
Using ESM2, I generated a deep mutational scan for the full MaSp1 sequence. A clear pattern is that the model strongly prefers small residues like glycine (G) and alanine (A) in the long repetitive core of the silk protein, and strongly dislikes large or strongly charged residues at those positions.
For example, I looked at one position in the middle of the repetitive region where the wild-type residue is glycine. In the heatmap, mutating this site to alanine (G → A) has a relatively neutral or mildly favorable score (greenish color), but mutating it to tryptophan (G → W) gives a very unfavorable score (dark purple). This makes sense: natural spider dragline silk is built from G/A-rich motifs such as (Gly–Ala)n that pack into tight β-sheet nanocrystals. Replacing a small glycine with a bulky aromatic residue like tryptophan would likely disrupt this packing, so the language model correctly treats this mutation as strongly deleterious.
C1.2 Latent Space Analysis
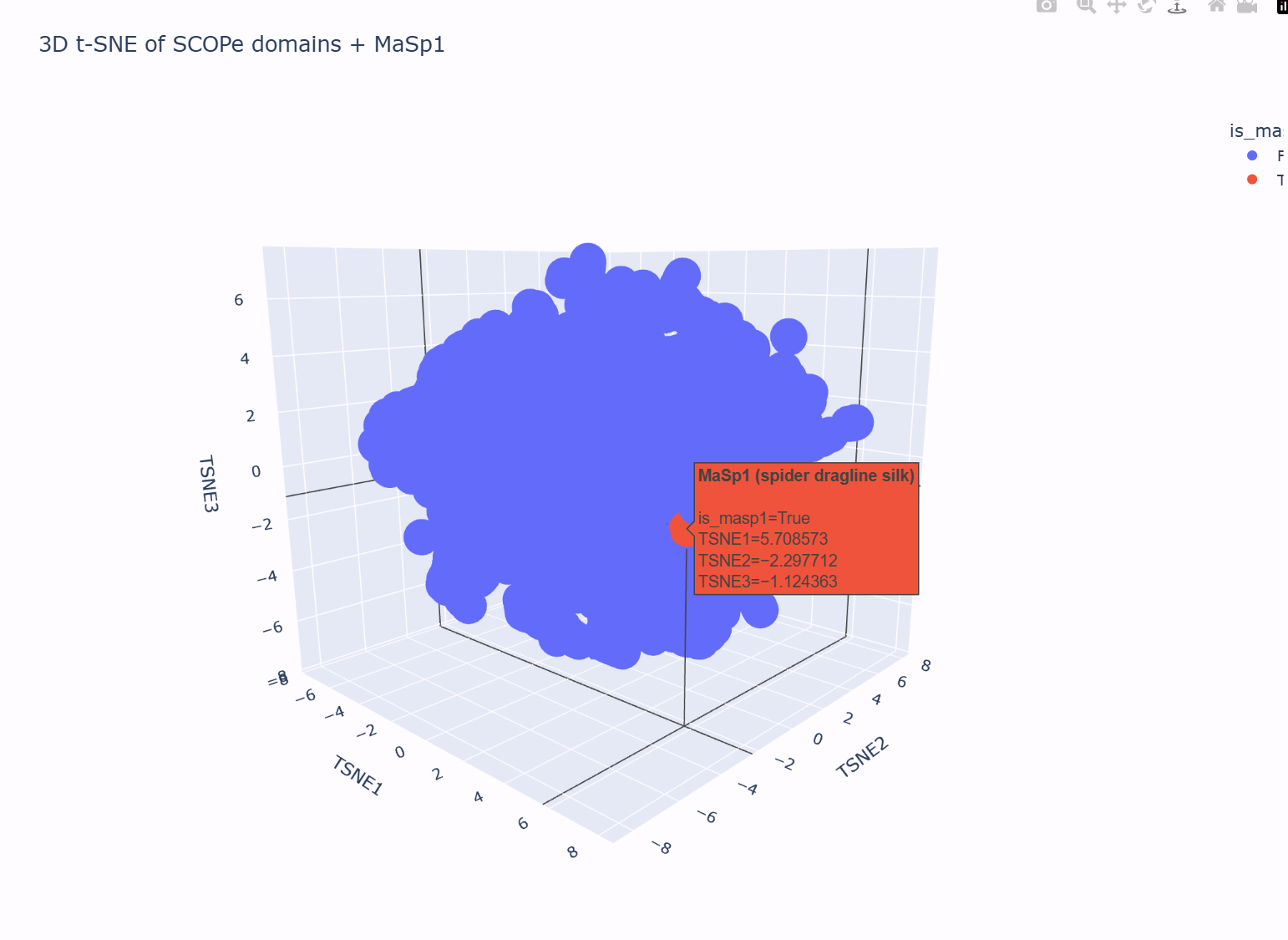
Using the notebook’s SCOPe ASTRAL dataset, I first embedded all protein domains with ESM2 and then reduced the embedding dimensionality to 3D using t-SNE (n_components = 3, perplexity = 30). Each point in the plot below corresponds to one protein domain in the SCOPe dataset; nearby points represent proteins that the language model considers similar in terms of sequence statistics and learned evolutionary patterns.
In this 3D map, the SCOPe domains do not form one perfectly uniform “cloud”: there is a dense central region and a somewhat more diffuse shell of points around it. Hovering over different neighborhoods in the plot shows that proteins with related properties tend to live near each other (for example, domains with similar lengths and compositional biases cluster more closely than very different domains). This suggests that the latent space is grouping broadly similar proteins into local neighborhoods.
I then added my own protein sequence — a fragment of the spider dragline silk protein MaSp1 — as an extra point in the embedding. In the t-SNE plot, this MaSp1 point is highlighted in a different color (red in the figure). It does not appear as an isolated outlier: instead, it lies on the outer part of the main cloud, close to other domains that, based on their descriptions, also have biased compositions and/or repetitive, low-complexity features.
This placement is consistent with what we know about MaSp1. The sequence is very long and strongly enriched in glycine and alanine repeats, designed to form structural silk fibers rather than a compact catalytic enzyme. In the latent space, the model therefore locates MaSp1 in a neighborhood of other non-enzymatic, compositionally biased domains, rather than in the core region where many typical enzyme-like domains cluster. In other words, the latent space neighborhoods do approximate “similar” proteins, and MaSp1’s position in the map matches its role as a G/A-rich structural material protein.
C2. Protein Folding (ESMFold)
C2.1 Folding your protein
Fold your protein with ESMFold. Do the predicted coordinates match your original structure?
Answer:
C3. Protein Generation
C3.1 Inverse folding with ProteinMPNN
Use the backbone of your chosen PDB to propose sequence candidates via ProteinMPNN. Analyze the predicted sequence probabilities and compare the predicted sequence vs the original one.
Observation (PepMLM confidence by perplexity): Among generated candidates, P2 (KRYGAAAARHKK) has the lowest perplexity (highest PepMLM confidence), while P3 (WRYPVAGLALKE) has the highest perplexity (lowest confidence among the four).
Part 2 – Evaluate binders with AlphaFold3
Peptide
Sequence
Binding description (1–2 sentences)
Near N-terminus / A4V?
P1
WHSPVAAARLKE
surface-bound
no
P2
KRYGAAAARHKK
surface-bound
no
P3
WRYPVAGLALKE
surface-bound
no
P4
WHSPPAAVALGE
surface-bound
yes
Ref
FLYRWLPSRRGG
Surface-associated
no
Part 3 – Evaluate Properties of Generated Peptides in the PeptiVerse
Peptide
Solubility
Solubility Score
Hemolysis
Hemolysis Score
Length (aa)
Molecular Weight (Da)
Net Charge (pH 7)
Isoelectric Point (pH)
Hydrophobicity (GRAVY)
WHSPVAAARLKE
Soluble
1.000
Non-hemolytic
0.013
12
1364.6
0.85
8.76
-0.42
KRYGAAAARHKK
Soluble
1.000
Non-hemolytic
0.009
12
1356.6
4.84
11.17
-1.53
WRYPVAGLALKE
Soluble
1.000
Non-hemolytic
0.025
12
1402.6
0.77
8.59
-0.06
WHSPPAAVALGE
Soluble
1.000
Non-hemolytic
0.041
12
1234.4
-1.14
5.47
0.12
From the AlphaFold3 models, all of the peptides showed relatively low and fairly similar ipTM values, and structurally they all appeared to bind in a shallow, surface associated manner rather than forming a clearly buried or highly specific interface on mutant SOD1. This means that higher ipTM did not translate into an obviously much stronger or more convincing binding pose in the structural models. When compared with the PeptiVerse predictions, the four generated peptides were all predicted to be fully soluble and non hemolytic, so none of the better candidates raised an immediate concern for poor solubility or red blood cell toxicity. Among them, KRYGAAAARHKK provides the best overall balance. It had the lowest PepMLM perplexity, indicating the highest generation confidence, and it also showed excellent therapeutic property predictions, including full solubility, very low hemolysis risk, and a strongly positive net charge that could support interaction with exposed regions of SOD1.
I would advance KRYGAAAARHKK. Although its AlphaFold3 model did not show a dramatically stronger binding geometry than the others, it combines the best overall computational profile across generation confidence, structural plausibility, solubility, and safety related properties. In other words, it is the most balanced candidate for further optimization and experimental validation.
Part 4 – Generate Optimized Peptides with moPPIt
Binder
Hemolysis
Solubility
Motif
CTWVKKTKKQVT
0.979838
0.833333
0.809806
GYKQKTCNTVKW
0.96261
0.833333
0.782062
STAEFTRQTKKM
0.954297
0.75
0.839537
RGKTTTQNGKVI
0.978479
0.833333
0.820856
GFGTQKKTKCG
0.964351
0.916667
0.849918
RTDQGGVKITLE
0.969846
0.833333
0.905129
ETKKRQKFKTDF
0.974053
0.833333
0.887031
KGETTDKIQKTM
0.975992
0.833333
0.841838
RETVGKKTQTKC
0.981714
0.916667
0.81274
The moPPIt peptides differ noticeably from the PepMLM peptides in both composition and design emphasis. The PepMLM peptides I generated for SOD1 were short 12 amino acid candidates with relatively simple compositions, and several were enriched in alanine, valine, lysine, and histidine, which made them look like general target conditioned binders. In contrast, the moPPIt peptides appear more motif driven and more strongly enriched in lysine and threonine, with some cysteine, glycine, phenylalanine, and aspartate included in specific patterns. This makes them look more constrained by the optimization objectives, especially motif retention and therapeutic property balancing, rather than just sequence likelihood conditioned on the target. In other words, PepMLM gave plausible binder candidates, while moPPIt seems to generate peptides that are more explicitly shaped by multiple design goals.
Before advancing any of these peptides toward clinical studies, I would evaluate them in several stages. First, I would confirm binding experimentally using assays such as surface plasmon resonance, biolayer interferometry, or microscale thermophoresis to measure affinity and specificity for mutant SOD1 compared with wild type SOD1 and unrelated proteins. Second, I would test whether the peptides actually improve a disease relevant phenotype, for example by reducing SOD1 aggregation, toxicity, or misfolding in cell based ALS models. Third, I would assess safety and developability, including hemolysis, cytotoxicity, serum stability, protease resistance, immunogenicity risk, and off target effects. Fourth, I would evaluate pharmacology, including uptake, biodistribution, half life, and whether the peptide can reach the relevant tissues, especially the central nervous system. Only peptides that show convincing binding, functional benefit, acceptable safety, and realistic delivery potential should move forward into animal studies and later clinical development.
L-Protein Engineering | Option 3: Random Mutagenesis
Goal
In this option, I generated random multi-site L-protein mutants using mutation information from prior mutational analysis experiments, then selected candidates for co-folding with DnaJ using AF2 Multimer. The goal is to explore combinations of tolerated or favorable mutations rather than relying only on single-site manual design. The L protein contains a soluble N-terminal domain followed by a transmembrane region in the last 35 residues, so both structural context and prior mutation evidence were considered when building the mutation pool. :contentReference[oaicite:1]{index=1}
Strategy
Instead of introducing fully random amino acid substitutions, I used a constrained random mutagenesis strategy. I first built a mutation pool from mutations that were supported by prior mutational analysis experiments or appeared tolerated or favorable in the provided mutation data. Then I randomly sampled combinations containing at least 2 mutated positions. This makes the search more biologically grounded than unconstrained random mutagenesis.
A good mutant should not simply contain many mutations. I define an effective mutant as one that satisfies several criteria:
It preserves or improves the foldability and structural plausibility of the L protein.
It does not obviously disrupt the soluble N-terminal region that is implicated in DnaJ interaction.
It avoids strongly unfavorable substitutions in conserved or functionally critical positions.
It is compatible with productive interaction behavior when co-folded with DnaJ.
Ideally, it improves properties related to lysis efficiency, DnaJ independence, or expression, which are the broader goals of this assignment. :contentReference[oaicite:2]{index=2}
Python function for constrained random mutagenesis
importrandomfromtypingimportDict,List,TupleL_PROTEIN_WT="METRFPQQSQQTPASTNRRRPFKHEDYPCRRQQRSSTLYVLIFLAIFLSKFTNQLLLSLLEAVIRTVTTLQQLLT"# Example mutation pool format:# key = 1-indexed residue position# value = (wildtype_residue, [allowed_mutant_residues])## You should replace these example entries with positions supported by your# mutational analysis sheet or notebook results.mutation_pool:Dict[int,Tuple[str,List[str]]]={8:("Q",["K","R"]),14:("A",["S","T"]),24:("E",["D","Q"]),31:("R",["K"]),35:("S",["T","A"]),41:("L",["I","V"]),44:("A",["V"]),58:("L",["I","V"]),63:("A",["V","L"]),69:("V",["I","L"]),}defapply_mutations(sequence:str,mutations:List[Tuple[int,str]])->str:seq_list=list(sequence)forpos,new_resinmutations:seq_list[pos-1]=new_resreturn"".join(seq_list)defformat_mutation_name(sequence:str,mutations:List[Tuple[int,str]])->str:names=[]forpos,new_resinmutations:wt=sequence[pos-1]names.append(f"{wt}{pos}{new_res}")return",".join(names)defgenerate_random_mutant(sequence:str,pool:Dict[int,Tuple[str,List[str]]],min_sites:int=2,max_sites:int=4):n_sites=random.randint(min_sites,max_sites)chosen_positions=random.sample(list(pool.keys()),n_sites)mutations=[]forposinchosen_positions:wt_expected,allowed=pool[pos]wt_actual=sequence[pos-1]ifwt_actual!=wt_expected:raiseValueError(f"WT mismatch at position {pos}: expected {wt_expected}, found {wt_actual}")new_res=random.choice(allowed)mutations.append((pos,new_res))mutations=sorted(mutations,key=lambdax:x[0])mutant_seq=apply_mutations(sequence,mutations)mutant_name=format_mutation_name(sequence,mutations)return{"mutations":mutant_name,"sequence":mutant_seq,"num_mutations":len(mutations)}defgenerate_unique_mutants(sequence:str,pool:Dict[int,Tuple[str,List[str]]],n_mutants:int=10,min_sites:int=2,max_sites:int=4):seen=set()results=[]whilelen(results)<n_mutants:mutant=generate_random_mutant(sequence,pool,min_sites,max_sites)key=mutant["mutations"]ifkeynotinseen:seen.add(key)results.append(mutant)returnresults# Example usagerandom.seed(42)mutants=generate_unique_mutants(sequence=L_PROTEIN_WT,pool=mutation_pool,n_mutants=10,min_sites=2,max_sites=4)fori,minenumerate(mutants,1):print(f"Mutant {i}")print("Mutations:",m["mutations"])print("Sequence :",m["sequence"])print("Count :",m["num_mutations"])print()
How I would use this function
I would first replace the example mutation_pool with residue positions and substitutions supported by the provided L-protein mutational analysis data. Then I would generate a small library of random double, triple, or quadruple mutants. From that library, I would select a few candidates that look structurally reasonable and co-fold them with DnaJ using AF2 Multimer.
Co-folding setup with DnaJ
For each selected mutant, I would submit the mutant L-protein sequence together with the DnaJ sequence as separate chains in AF2 Multimer. The purpose is to compare whether different mutation combinations change the predicted interaction pattern between the L-protein soluble region and DnaJ. The assignment notes that this kind of prediction may be difficult, especially for membrane related systems, so these structures should be interpreted cautiously.
How I define a “good” mutant
I define a good or effective mutant as one that balances multiple factors rather than optimizing only one metric. First, it should remain structurally plausible as an L-protein variant and avoid obviously destabilizing substitutions. Second, it should preserve or improve a meaningful interaction pattern with DnaJ in the soluble region if the biological mechanism still depends on that interaction. Third, if the long-term goal is DnaJ independence, then a good mutant could also be one that remains well folded without requiring the same interaction geometry. Finally, the mutant should be supported by prior mutational evidence and should not rely on highly disruptive changes at conserved positions. In practice, the best mutant is the one that shows a reasonable structural model, uses biologically tolerated substitutions, and best aligns with the assignment goals of improved folding, lysis efficiency, or reduced dependence on bacterial chaperones.
Week 6 HW: Genetic Circuits Part I: Assembly Technologies
Answers to Questions About This Week’s Lab Protocol
1. What are some components in the Phusion High-Fidelity PCR Master Mix and what is their purpose?
Phusion High-Fidelity PCR Master Mix typically contains several important components: Phusion DNA polymerase, dNTPs, reaction buffer, and MgCl2. The polymerase synthesizes new DNA strands and has proofreading activity, which lowers the error rate compared with standard Taq polymerase. The dNTPs provide the nucleotide building blocks needed to make the new DNA strands. The buffer maintains the proper chemical environment, including pH and salt concentration, for the enzyme to work efficiently. MgCl2 is an essential cofactor that allows the polymerase to function properly. In this lab, the master mix is provided as a 2X mix, so only template DNA, primers, and water are added separately. (neb.com)
2. What are some factors that determine primer annealing temperature during PCR?
Primer annealing temperature is mainly determined by the melting temperature (Tm) of the primer binding region. According to the course page, a good primer binding region is usually about 18–22 base pairs, with a Tm around 52–58 °C, and the Tm values of the two primers should ideally be within about 5 °C of each other. GC content affects annealing temperature because GC base pairs are more stable than AT base pairs, so primers with higher GC content tend to have higher Tm values. A GC clamp at the 3′ end can improve binding stability, while hairpins or primer dimers can reduce efficiency. In this lab, the backbone PCR uses an annealing temperature of 57 °C, while the insert PCR uses 53 °C, showing that different primers may require different annealing conditions. (docs.google.com)
3. There are two methods from this class that create linear fragments of DNA: PCR, and restriction enzyme digests. Compare and contrast these two methods, both in terms of protocol as well as when one may be preferable to use over the other.
PCR creates a linear DNA fragment by amplifying a selected region from a DNA template using primers, polymerase, dNTPs, and a thermocycler. In this week’s lab, PCR is used both to generate the backbone fragment and the color fragment, and it also introduces mutations through the primer sequence. Restriction enzyme digestion, by contrast, cuts DNA at specific recognition sites already present in the sequence. This means restriction digestion is limited by whether suitable restriction sites exist in the right positions, while PCR allows much more flexibility in choosing the exact boundaries of the fragment. (docs.google.com)
PCR is often preferable when you want to customize the fragment, such as adding Gibson overlaps, introducing mutations, or amplifying a region that does not have convenient restriction sites. Restriction enzyme digestion is often preferable when a plasmid already has well-placed restriction sites and you want a straightforward way to cut out a fragment without designing long primers. PCR is more versatile but can introduce amplification errors or require more optimization. Restriction digests are simpler in some cases, but they are less flexible because they depend on the DNA sequence already containing the right enzyme recognition sites. In this lab, PCR is the better choice because the experiment depends on introducing specific mutations into the color sequence and creating Gibson-compatible overlaps at the same time. (docs.google.com)
4. How can you ensure that the DNA sequences that you have digested and PCR-ed will be appropriate for Gibson cloning?
To make DNA fragments appropriate for Gibson cloning, the adjacent fragments must contain the correct overlapping homologous sequences. The course page states that Gibson Assembly usually requires 20–40 bp overlaps between neighboring fragments, and these overlaps are designed directly into the primers. You also need to verify that the fragments are in the correct orientation and that their ends match exactly with the intended assembly junctions. In practice, this means checking the fragment design in a sequence editor before running the experiment, and then confirming fragment size by gel electrophoresis after PCR. (docs.google.com)
Another important step in this lab is DpnI digestion. DpnI digests the original methylated plasmid template but leaves the new unmethylated PCR products intact. This reduces background from the original plasmid and helps ensure that the fragments going into Gibson Assembly are the newly generated ones rather than leftover parental template. After that, the DNA is purified and quantified before assembly, which helps confirm that the samples are clean and present at usable concentrations. (docs.google.com)
5. How does the plasmid DNA enter the E. coli cells during transformation?
In this lab, plasmid DNA enters E. coli cells during transformation after the cells are made permeable by heat shock. The course page explains that heat shock or electroporation causes the bacterial membrane to temporarily open up, allowing plasmid DNA to enter the cell by diffusion. After the DNA enters, the cells are placed in SOC medium to recover and start expressing the antibiotic resistance gene carried by the plasmid. The cells are then plated on selective media containing chloramphenicol, so only cells that successfully took up the plasmid can grow. (docs.google.com)
6. Describe another assembly method in detail (such as Golden Gate Assembly).
Another widely used DNA assembly method is Golden Gate Assembly. Golden Gate uses a Type IIS restriction enzyme such as BsaI or BsmBI together with T4 DNA ligase in a one-pot reaction. Unlike standard restriction enzymes, Type IIS enzymes cut outside of their recognition sequence, which allows the user to design custom sticky ends. These sticky ends determine the order in which fragments assemble, so multiple fragments can be assembled directionally in one reaction. During repeated digestion-ligation cycles, incorrectly assembled products are recut, while correctly assembled products are ligated and preserved because the recognition sites are removed in the final construct. This makes Golden Gate highly efficient for assembling multiple modular parts in a predefined order, often without leaving scars between fragments. (neb.com)
7. Explain the other method in 5–7 sentences plus diagrams.
Golden Gate Assembly is a one-pot DNA assembly method that uses a Type IIS restriction enzyme and DNA ligase. The Type IIS enzyme cuts outside its recognition site, creating user-defined sticky ends instead of fixed ones. Those sticky ends determine which DNA fragments can join to each other, allowing multiple fragments to assemble in a chosen order. During thermal cycling, the enzyme cuts unassembled or misassembled molecules, while ligase seals the correct junctions. Because the recognition sites are typically removed during assembly, the final product is often scarless and is no longer recut in the same reaction. Compared with Gibson Assembly, Golden Gate uses short sticky ends rather than long homologous overlaps. This makes it especially useful for modular cloning systems with many interchangeable parts. (neb.com)
Diagram
Fragment A Fragment B Vector
[ BsaI ][ A overhang ] [ matching ][ BsaI ] [ BsaI ][ matching ][ BsaI ]
Step 1: Type IIS digestion
BsaI cuts outside its recognition sequence
↓ ↓ ↓
sticky end exposed matching sticky end exposed vector ends exposed
Step 2: Ligation
[ A overhang ] + [ matching overhang ] → joined fragment
Step 3: Final product
Correctly assembled product no longer contains the original Type IIS sites at the junction
→ scarless, ordered assembly
Week 7 HW
Part 1: IANNs
What advantages do IANNs have over traditional genetic circuits?
Traditional genetic circuits usually work like Boolean logic gates. They treat inputs and outputs as mostly ON or OFF.
IANNs are more flexible because they can handle graded, continuous biological signals. Instead of only asking whether an input exists, they can respond to the strength of each input and combine multiple inputs together.
IANNs can:
process noisy biological signals
weight different inputs differently
produce gradual output levels instead of only ON/OFF
recognize complex patterns of inputs
represent more complex behaviors than simple Boolean circuits
This makes them useful for biological systems where signals are rarely perfectly binary.
Useful application for an IANN
A useful application would be an engineered diagnostic cell that detects a disease-like state.
For example, the IANN could receive several inputs:
high inflammation signal
low oxygen level
cancer-associated microRNA
abnormal metabolic signal
The output could be a fluorescent protein or a therapeutic molecule.
Input/output behavior:
If only one weak disease signal is present, the output stays low.
If several disease signals are present, the output becomes medium.
If the full disease-like pattern is detected, the output becomes strong.
If the cell looks healthy, the output remains OFF.
This is useful because many diseases are not defined by one marker. They are defined by combinations of signals.
Limitations:
Biological circuits are noisy.
The response may be slow because transcription and translation take time.
Too many components can burden the cell.
The circuit may behave differently in different cell types.
It may mutate or become unstable over time.
It may be hard to tune the correct input weights.
Intracellular multilayer perceptron description
In a multilayer intracellular perceptron, the first layer receives DNA inputs and produces an intermediate molecular output. For example, layer 1 could produce the Csy4 endoribonuclease.
Csy4 then acts as the output of layer 1 and the regulatory input for layer 2. In layer 2, the fluorescent protein gene is transcribed into mRNA that contains a Csy4 recognition site. If enough Csy4 is produced, it cleaves the fluorescent protein mRNA and reduces fluorescence.
So the system works like this:
Input DNA signals → layer 1 gene expression → Csy4 production → regulation of fluorescent protein mRNA → final fluorescence output
The final output is the fluorescence level, which depends on how strongly the upstream inputs activate Csy4.
Part 2: Fungal Materials
Existing fungal materials and uses
Mycelium packaging
Mycelium can be grown through agricultural waste to make packaging materials.
Uses:
protective packaging
foam replacement
disposable molded products
Advantages:
biodegradable
lightweight
made from waste materials
lower environmental impact than plastic foam
Disadvantages:
less water-resistant
slower to produce
mechanical properties can vary
harder to standardize at large scale
Mycelium leather
Fungal mycelium can be processed into leather-like sheets.
Uses:
fashion
bags
shoes
furniture surfaces
Advantages:
animal-free
potentially more sustainable than animal leather
can be grown into sheets
texture and thickness can be tuned
Disadvantages:
may be less durable than traditional leather
often needs coating or finishing
water resistance can be limited
large-scale production is still developing
Mycelium insulation and building panels
Mycelium composites can be made into panels, bricks, acoustic tiles, or insulation.
Uses:
wall panels
acoustic panels
thermal insulation
temporary architecture
Advantages:
lightweight
biodegradable
good insulation potential
made from agricultural waste
can have good fire resistance
Disadvantages:
usually not strong enough for structural loads
sensitive to moisture
long-term durability is uncertain
building code approval can be difficult
What I might genetically engineer fungi to do
I would genetically engineer fungi to make responsive building materials.
For example, a mycelium wall panel could sense humidity, pollution, or damage and respond visibly or materially.
Possible input/output behavior:
Input: high humidity Output: produce water-resistant compounds
Input: pollutant or toxin Output: change color as a warning signal
Input: physical damage Output: produce binding polymers to help self-repair
This would make fungal materials more than passive materials. They could become living or bio-based interfaces that sense and respond to their environment.
Why use fungi instead of bacteria?
Fungi are useful for synthetic biology because they naturally form material structures. Their hyphae grow into dense networks, which can become sheets, foams, panels, or composites.
Advantages over bacteria:
fungi naturally make large physical networks
they can grow on cheap agricultural waste
they can secrete useful enzymes, pigments, and polymers
they are better suited for macroscopic materials
as eukaryotes, they can process more complex proteins than bacteria
However, fungi are often slower-growing and harder to genetically engineer than bacteria. Their growth can also be harder to control.
Week 9 HW
Homework Part A: General and Lecturer-Specific Questions
Assignees for This Section
Group
Status
MIT/Harvard students
Required
Committed Listeners
Required
General Homework Questions
Explain the main advantages of cell-free protein synthesis over traditional in vivo methods, specifically in terms of flexibility and control over experimental variables. Name at least two cases where cell-free expression is more beneficial than cell production.
Describe the main components of a cell-free expression system and explain the role of each component.
Why is energy provision regeneration critical in cell-free systems? Describe a method you could use to ensure continuous ATP supply in your cell-free experiment.
Compare prokaryotic versus eukaryotic cell-free expression systems. Choose a protein to produce in each system and explain why.
How would you design a cell-free experiment to optimize the expression of a membrane protein? Discuss the challenges and how you would address them in your setup.
Imagine you observe a low yield of your target protein in a cell-free system. Describe three possible reasons for this and suggest a troubleshooting strategy for each.
Homework Question from Kate Adamala
Prompt
Design an example of a useful synthetic minimal cell as follows.
Function
Pick a function and describe it.
What would your synthetic cell do? What is the input and what is the output?
Could this function be realized by cell-free Tx/Tl alone, without encapsulation?
Could this function be realized by a genetically modified natural cell?
Describe the desired outcome of your synthetic cell operation.
Synthetic Cell Components
Design all components that would need to be part of your synthetic cell.
What would the membrane be made of?
What would you encapsulate inside? Enzymes, small molecules.
Which organism will your Tx/Tl system come from? Is bacterial OK, or do you need a mammalian system for some reason?
Hint: For example, if you want to use small-molecule-modulated promoters, like Tet-ON, you need a mammalian system.
How will your synthetic cell communicate with the environment?
Hint: Are substrates permeable, or do you need to express the membrane channel?
Experimental Details
List all lipids and genes.
Bonus: Find the specific genes. For example, instead of just saying “small molecule membrane channel,” pick the actual gene.
How will you measure the function of your system?
Example Solution
Based on: Lentini, R. et al., 2014. Nature Communications, 5, p. 4012.
Pick a function and describe it.
Expand the sensing capacity of bacteria.
Input: theophylline, which is inert to bacteria.
Output of the synthetic minimal cell: IPTG.
Output of the whole system: GFP produced in bacteria.
Theophylline aptamer reference: Martini, L. & Mansy, S. S., 2011. “Cell-like systems with riboswitch controlled gene expression.” Chemical Communications, 47(38), p. 10734.
Could this function be realized by cell-free Tx/Tl alone, without encapsulation?
No. If the IPTG were not encapsulated, it would enter the bacteria without the need for theophylline-induced membrane channel synthesis. In that case, the synthetic cell actuator would not exist.
Could this function be realized by a genetically modified natural cell?
Yes, in this particular case. The theophylline aptamer could be incorporated into a transformed gene. However, this lacks generality. It is easier to make synthetic minimal cells than to modify bacteria, so in this system a single bacterial reporter can be used to detect various small molecules.
Describe the desired outcome of your synthetic cell operation.
In the presence of synthetic minimal cells, bacteria can sense theophylline.
Design all components that would need to be part of your synthetic cell.
Membrane
Phospholipids
Cholesterol
Encapsulated components
Cell-free Tx/Tl system
IPTG
Gene for membrane transporter under the control of the theophylline aptamer
Which organism will your Tx/Tl system come from?
Bacterial, because the system uses the theophylline riboswitch as the synthetic minimal cell input.
How will your synthetic cell communicate with the environment?
The membrane is permeable to the input molecule, theophylline. The output is IPTG, which crosses the membrane through the membrane pore created after theophylline-initiated gene expression.
Experimental details
Lipids
POPC
Cholesterol
Enzymes
Bacterial cell-free Tx/Tl
Genes
α-hemolysin, or aHL, to encapsulate in the synthetic minimal cell
Biological cells
E. coli transformed with GFP under a T7 promoter and a lac operator
How will you measure the function of your system?
Measure GFP output of the cells using flow cytometry. Alternatively, use an enzymatic reporter, such as luciferase, and measure the bulk output of the enzyme.
Figure caption
Artificial cells translate chemical signals for E. coli.
In the absence of artificial cells, E. coli cannot sense theophylline.
Artificial cells can be engineered to detect theophylline and release IPTG in response.
IPTG then induces a response in E. coli.
Homework Question from Peter Nguyen
Freeze-dried cell-free systems can be incorporated into many kinds of materials as biological sensors or as inducible enzymes that modify the material itself or the surrounding environment.
Choose one application field:
Architecture
Textiles/Fashion
Robotics
Then propose an application using cell-free systems that are functionally integrated into the material.
Answer each of these key questions for your proposal pitch:
Write a one-sentence summary pitch describing your concept.
How will the idea work, in more detail? Write 3–4 sentences or more.
What societal challenge or market need will this address?
How do you envision addressing the limitations of cell-free reactions, such as activation with water, stability, or one-time use?
Homework Question from Ally Huang
Freeze-dried cell-free reactions have great potential in space, where resources are constrained. As described in the lecture, the Genes in Space competition challenges students to consider how biotechnology, including cell-free reactions, can be used to solve biological problems encountered in space.
While the competition is limited to high school students, this assignment asks you to develop your own mock Genes in Space proposal to practice thinking about biotech applications in space.
For this assignment, your proposal must incorporate the BioBits® cell-free protein expression system. You may also use other tools in the Genes in Space toolkit, including:
Provide background information that describes the space biology question or challenge you propose to address. Explain why this topic is significant for humanity, relevant for space exploration, and scientifically interesting. Maximum: 100 words.
Name the molecular or genetic target that you propose to study. Examples of molecular targets include individual genes and proteins, DNA and RNA sequences, or broader -omics approaches. Maximum: 30 words.
Describe how your molecular or genetic target relates to the space biology question or challenge your proposal addresses. Maximum: 100 words.
Clearly state your hypothesis or research goal and explain the reasoning behind it. Maximum: 150 words.
Outline your experimental plan. Identify the sample or samples you will test in your experiment, including any necessary controls, the type of data or measurements that will be collected, and any relevant workflow details. Maximum: 100 words.
Concept What if you could write a line of genetic code and have it determine how a surface curls in the rain? This project treats DNA sequence as a material programming language. Rather than engineering biology to produce a molecule or report a signal, I use genetically encoded elastin-like proteins (ELPs) to ask a more fundamental question: can editing the sequence of a repetitive protein produce predictable, distinct material behaviors at the macroscale?
Sequence-Programmed Hygromorphic Coatings from Engineered Elastin-Like Proteins
DNA Sequence as a Material Design Variable for Programming Environmental Response
Yao Wang | HTGAA 2026
1. Concept
What if you could write a line of genetic code and have it determine how a surface curls in the rain? This project treats DNA sequence as a material programming language. Rather than engineering biology to produce a molecule or report a signal, I use genetically encoded elastin-like proteins (ELPs) to ask a more fundamental question: can editing the sequence of a repetitive protein produce predictable, distinct material behaviors at the macroscale?
The core proposition is that DNA is the only existing system where a discrete, editable, reproducible instruction set compiles into a physical material with tunable macroscopic behavior. Unlike conventional responsive materials, where achieving different behavior requires reformulating chemistry, adjusting processing, or introducing new components, sequence-encoded materials allow variation to live in the code rather than the process. The fabrication pipeline—same vector, same bacteria, same induction, same coating method—stays identical. What changes is the material output.
ELP is the right chassis for this proof of concept because it is one of the few protein systems where the sequence-property relationship is unusually direct. ELPs are intrinsically disordered: they do not need to fold correctly to function, which means expression almost always works and the pipeline does not break when the sequence changes. Their lower critical solution temperature (LCST) transition—soluble and extended below the transition temperature, collapsed and aggregated above it—provides environmentally responsive behavior intrinsic to the material, without requiring any engineered sensor or switch. And because the transition temperature shifts predictably with sequence composition, editing the DNA directly tunes the trigger point. The distance between a sequence edit and a material behavior change is shorter, more predictable, and less failure-prone in ELPs than in virtually any other protein system.
Most responsive materials today rely on synthetic polymers, complex fabrication, or external mechanical actuation. Biologically derived responsive systems exist—whole-cell bio-actuators, for instance—but they are difficult to control and deploy. This project occupies a third space: genetically encoded material behavior without living cells in the final product. The material is dead but programmed. That framing opens a path toward adaptive biomaterials, environmentally responsive interfaces, and biofabrication systems tunable for different deployment conditions.
2. Project Aims
Aim 1: Experimental Aim — This Project
Test whether DNA-encoded ELP sequence variants produce distinct humidity-responsive material behaviors using Benchling-based DNA design, molecular cloning, E. coli expression, basic protein purification, and small-scale film/bilayer fabrication on PET substrates.
I will construct three ELP variants that differ at the guest residue position of the canonical (VPGXG) pentapeptide repeat, producing a hydrophilicity gradient: (VPGSG)₂₀ hydrophilic/serine, (VPGAG)₂₀ balanced/alanine, and (VPGVG)₂₀ hydrophobic/valine. Each construct includes an N-terminal 6xHis tag for detection and optional purification. The three variants share an identical backbone architecture and differ by a single residue per repeat unit, isolating amino acid identity as the sole design variable.
I will compare their water uptake, swelling, bending amplitude, recovery speed, and structural stability as one-sided coatings on PET film strips under controlled humidity changes. Key metrics include maximum bending angle, time to peak deformation, recovery during drying, and coating integrity over repeated wet/dry cycles.
If the three single-composition variants produce distinguishable behaviors, I will additionally test diblock ELP constructs that combine two guest residues in a single chain—for example, (VPGSG)₁₀-(VPGAG)₁₀ or (VPGSG)₁₀-(VPGVG)₁₀. These diblocks test whether sequence organization, not just composition, changes film behavior, which is a stronger claim for the “sequence as design language” framework. Diblocks may exhibit phase separation, domain-specific swelling, or asymmetric film morphology that single-composition variants cannot produce. These constructs are included in the design files for feasibility assessment; synthesis and testing will proceed only if timeline and Twist compatibility allow.
Aim 2: Development Aim — Multi-Axis Sequence Programming
Aim 1 demonstrates that sequence edits along one axis, guest residue hydrophilicity, produce measurable differences in one class of material behavior: humidity response. Aim 2 would expand the system to a second, qualitatively different behavioral axis by introducing a charged guest residue.
The proposed construct is (VPGKG)₂₀, with lysine at the guest position. Lysine introduces a positively charged side chain at physiological pH, making this variant not just humidity-responsive but also pH-responsive and salt-responsive. Under Aim 2, I would test the same coating-on-PET pipeline but expose samples to acidic vs. basic conditions and varying ionic strength, comparing whether the same sequence framework can be tuned to respond to fundamentally different environmental inputs. This would transform the project from “I varied one parameter” to “I showed that sequence edits can switch which environmental variable the material responds to.”
Aim 2 would also include expanding environmental inputs for the Aim 1 variants, such as temperature and salinity, and improving film robustness, bilayer control, and material processing methods.
In the long term, I envision this project becoming a platform for environmentally responsive biomaterials in which DNA sequence serves as a programmable design language. The endpoint is a searchable, combinatorial design space: given a target environmental condition and a desired material response, a designer could consult a sequence–environment–behavior map to select or computationally predict the right ELP construct. This would bring materials design closer to the logic of software—version-controlled, editable, reproducible—and enable new approaches to adaptive interfaces, climate-specific materials, and sustainable biofabrication.
3. Background
Why ELP?
Elastin-like proteins are among the few protein systems where structure is optional. Most proteins require correct folding to function; ELPs are intrinsically disordered below their transition temperature and do not depend on tertiary structure for their material properties. This means expression almost always succeeds, the pipeline tolerates sequence variation without catastrophic failure, and multiple variants can be produced in parallel with confidence. The LCST phase transition is intrinsic to the material—no engineered sensor or switch is needed—and shifts predictably with guest residue identity. ELPs also film well: the existing literature supports that they can be processed into coatings, films, fibers, and hydrogels, providing reasonable confidence that expressed protein will form usable coatings on PET.
Literature Context
Srokowski et al., “Surface and adsorption characteristics of three elastin-like polypeptides with sequence length and guest residue variations.” This study demonstrates that ELP sequence length and guest residue composition significantly alter thin film and coating surface properties, directly supporting the logic that DNA sequence variation translates into material behavior differences.
Välisalmi et al., “Highly Hydrophobic Films of Engineered Silk Proteins by a Simple Fusion Protein Concept.” This work shows that genetically engineered spider silk proteins can be processed into thin films with strongly hydrophobic surface properties, demonstrating that recombinant protein design can directly program film behavior.
Zhu et al., “Humidity-responsive self-assembly of short peptides with super-flexibility.” This study reports that short peptide materials undergo reversible structural transformation under changing humidity, driven by hydrogen bonding and conformational changes. It provides direct precedent for biomolecular humidity-responsive actuators.
Liu et al., “Spider dragline silk as torsional actuator driven by humidity.” This research demonstrates that natural protein materials can convert humidity input into large-amplitude mechanical deformation, establishing a strong precedent for using biological molecules as humidity-driven actuators.
Novelty
This project is novel because it treats DNA sequence as a design variable for programming material behavior, not merely as biological information for producing a molecule. Instead of engineering biology to express a protein and then asking what material it makes, I propose a controlled comparison: a small family of variants that differ by a single residue per repeat unit, tested through an identical pipeline, to isolate the effect of sequence on macroscale response.
The project also reframes bio-inspired responsive design. Rather than replicating whole-cell bio-actuators or mimicking biological morphology, it focuses on a minimal and testable platform where DNA-encoded peptide structure directly shapes macroscale material response. The inclusion of diblock architecture variants, Aim 1b, and a charged variant, Aim 2, extends this beyond parametric tuning toward a genuine demonstration that sequence can control qualitatively different dimensions of material behavior.
Significance
Most responsive materials rely on synthetic polymers, complex fabrication, or external mechanical components. These systems are difficult to customize at the molecular level, and variations in behavior typically require reformulating chemistry or processing. This project addresses the problem by demonstrating that variation can live in the genetic code itself: the fabrication pipeline stays identical while the material output changes.
If successful, this work establishes that even minimal sequence edits—a single amino acid substitution per repeat unit—produce measurably different humidity-responsive behaviors. This is the minimum viable proof that DNA can function as a material programming language. The broader implication is a path toward biomaterials whose behavior is version-controlled, computationally searchable, and tunable for specific deployment conditions, with applications in adaptive interfaces, environmental sensing, and sustainable biofabrication.
4. Ethical Considerations
This project raises ethical questions around biosafety, environmental responsibility, and scientific framing. Although the work uses standard laboratory expression systems and is limited to small-scale peptide/protein materials, it involves synthetic biology methods that should be guided by non-maleficence and responsibility. A key ethical concern is avoiding exaggerated claims: a sequence-programmed biomaterial is not automatically sustainable, safe, or deployment-ready. Observed material behavior may depend not only on DNA sequence but also on processing conditions and experimental context, and these limitations must be stated clearly.
All work will be conducted in contained laboratory conditions using non-pathogenic systems, with no environmental release or real-world deployment. The project will be described as a proof of concept, not an immediate solution. Uncertainties and limitations will be explicitly stated in interpreting results. Potential unintended consequences include overstating sustainability, overgeneralizing from a small number of variants, or assuming biomaterials are inherently beneficial. Alternatives include first testing similar responsive behaviors with non-engineered biological polymers or fully synthetic materials before scaling toward more complex engineered systems.
5. Experimental Design
Central Hypothesis
Changing the guest residue in DNA-encoded repetitive elastin-like proteins will alter their water uptake, swelling behavior, coating morphology, and bilayer mismatch on a passive PET substrate. As a result, different ELP sequence variants will generate distinct humidity-responsive material behaviors such as bending, curling, wrinkling, or changes in surface texture.
Sequence Design
Three ELP variants, differing only at the guest position X of the (VPGXG) pentapeptide repeat:
Variant
Sequence
Guest Residue
Expected Tendency
Hydrophilic
6xHis-(VPGSG)₂₀
Serine (S)
High water uptake, strong swelling
Balanced
6xHis-(VPGAG)₂₀
Alanine (A)
Moderate swelling, baseline behavior
Hydrophobic
6xHis-(VPGVG)₂₀
Valine (V)
Low swelling, possible aggregation
Stretch goal constructs, Aim 1b: Diblock variants combining two guest residues in a single chain, such as (VPGSG)₁₀-(VPGAG)₁₀ and (VPGSG)₁₀-(VPGVG)₁₀. These test whether sequence organization—not just composition—affects film behavior. Feasibility of Twist synthesis for these constructs will be assessed before ordering.
Future construct, Aim 2: (VPGKG)₂₀, a charged variant with lysine at the guest position. This variant is designed but not tested in the current project. It would introduce pH-sensitivity and salt-sensitivity, demonstrating that the same ELP scaffold can be tuned to respond to qualitatively different environmental inputs.
Detailed Protocol
Define the sequence design strategy and project scope, Day 1. Finalize three ELP variants with a clear sequence gradient. Expected outcome: a set of three protein variants with a strong conceptual link between sequence composition and expected material response.
Design three DNA-encoded ELP variants in Benchling, Days 1–2. Design constructs with N-terminal 6xHis tag, annotate inserts, confirm reading frames, check restriction sites, verify repeat organization. Expected result: three fully annotated DNA constructs ready for synthesis.
Prepare cloning strategy compatible with Twist Bioscience synthesis, Days 2–3. Prepare insert sequences with flanking regions for cloning into an IPTG-inducible E. coli expression vector. Select target vector backbone and ensure synthesis compatibility. This avoids cloning errors associated with repetitive DNA.
Submit Twist gene fragment order and finalize vector design, Day 3. Order each variant as a single Gene Fragment from Twist Bioscience, guaranteed 100–1000 ng per fragment. Three variants = three fragments, approximately $63 total. This quantity is sufficient for Gibson Assembly into pET-28a; protein yield depends on bacterial amplification, not DNA input quantity. If budget allows, order two copies of each fragment as backup. Alternatively, ordering Clonal Genes, where Twist clones and sequence-verifies for you, costs more but saves 3–5 days of cloning time. In parallel, finalize the expression vector design including promoter, RBS, His tag, antibiotic resistance, and cloning junctions.
Clone synthesized fragments into expression vector, 2–3 days after DNA arrival. Use Gibson Assembly, restriction enzyme cloning, or another standard method. Transform into DH5α or TOP10, plate on selective agar, pick colonies.
Validate constructs by colony PCR, MiniPrep, and sequencing, 1–2 days. Colony PCR, liquid culture, MiniPrep, diagnostic digestion and/or sequencing. Sequence confirmation is especially important for repetitive regions.
Transform verified plasmids into expression strain, 1 day. Transform into BL21(DE3) or equivalent for inducible production.
Perform small-scale expression tests, 2 days. Pilot expression under IPTG induction. Assess by SDS-PAGE comparing induced and uninduced samples. Goal: determine whether each construct expresses detectably and whether the protein is soluble enough for coating experiments.
Optimize expression conditions if needed, 1–2 days. Test induction timing and IPTG concentration. Critical: expression temperature must be 18–25°C with overnight induction, and all protein solutions must be kept at 4°C throughout handling and storage. This is not an optional optimization—it is a prerequisite for the entire downstream pipeline. ELPs expressed at 37°C are far more likely to aggregate irreversibly, and protein left at room temperature will degrade or phase-separate before coating. The project requires only small-scale material, so the yield optimization threshold is modest, but temperature discipline is non-negotiable.
Extract and optionally purify recombinant proteins, 2 days. Lyse cells. Use Ni-NTA affinity purification for His-tagged constructs if feasible; otherwise proceed with partially purified fractions and state this limitation. Quantify by Nanodrop or BCA assay.
Prepare standardized PET substrates, 1 day. Use 6 µm Mylar/PET film as the primary substrate, Premier Lab Supply, approximately $12–60 per roll. Cut into uniform strips or small rectangles. 6 µm is thin enough to respond visibly to coating-induced stress; if it proves too flimsy to handle reliably, 12 µm Mylar is the fallback. Before coating, treat the PET surface with oxygen plasma, available at MIT.nano after registration and training or by arranging brief access through a nearby microfluidics group. Plasma treatment is critical: it activates the PET surface for better protein adhesion. The coating must be applied within 30 minutes of plasma treatment, as the hydrophilic surface will revert.
Create protein coatings on one side of PET strips, 1–2 days. Apply each protein sample onto one side of plasma-treated PET using repeated drop-casting: deposit one layer, allow to dry completely, then deposit the next layer, repeating 3–5 times to build up coating thickness. Confirm each layer is fully dry before applying the next. This multi-pass approach produces a thicker and more continuous active layer than a single application. The result is a bilayer system: PET as passive layer, ELP coating as humidity-responsive active layer.
Document initial coating quality and morphology, 1 day. Photograph and image coated samples. Note differences in smoothness, cracking, opacity, aggregation, thickness uniformity, and adhesion across variants.
Expose coated strips to controlled humidity changes, 1–2 days. Use a sealed chamber with moisture source or standardized mist exposure. Introduce a reproducible humidity change and observe how coated PET strips respond. Different variants should swell differently and generate different bilayer mismatch.
Record macroscopic responses with video and images, same day. Capture time-lapse images or short videos during humidity exposure and drying. Observable readouts: bending angle, curling, lifting, wrinkling, surface texture change.
Quantify deformation and response kinetics, 1–2 days. Measure maximum bending angle, time to peak deformation, recovery time, curl radius, or extent of wrinkling. If bending is minimal, quantify alternative outputs such as end displacement or surface-area distortion.
Perform repeat-cycle testing for reversibility, 1 day. Expose samples to at least three dry/wet cycles. Record whether response is reversible, degrades, or delaminates—degradation patterns are themselves sequence-dependent outcomes.
Include controls, 1 day. Uncoated PET, PET with blank buffer/lysate, and if possible PET with a non-repetitive control protein. Controls distinguish sequence effects from substrate or solvent artifacts.
Interpret results through biological and materials frameworks, 1–2 days. Biologically: does DNA sequence function as a design variable for protein material properties? From materials: does sequence-dependent water uptake and bilayer mismatch produce distinguishable macroscale behaviors?
Plan for technical risk and fallback outcomes. Two-variant comparison is sufficient if one fails to express. Partially purified samples can support preliminary coating tests. Wrinkling, swelling, or coating integrity differences are meaningful even without large bending.
Assemble final figures, workflow diagrams, and documentation, final 2 days. Organize all design materials, cloning maps, expression data, coating images, response videos, and quantitative plots into a coherent final presentation.
Condensed Timeline
Phase
Activities
Days 1–3
Define scope, design ELP variants in Benchling, prepare Twist-compatible constructs, finalize vector strategy, submit Twist order
Post-arrival 1–3
Clone inserts into expression vector, transform into cloning strain, validate constructs
Post-arrival 4–6
Transform into expression strain, small-scale expression tests
Post-arrival 6–8
Optimize expression, prepare protein samples
Post-arrival 8–10
Prepare PET substrates, fabricate one-sided coatings
Post-arrival 10–12
Humidity-response experiments, video recording, quantify deformation
Post-arrival 12–14
Repeat-cycle testing, controls, data interpretation, final figures
Critical Experimental Notes
Expression temperature is non-negotiable. Induce at 18–25°C overnight. Keep all protein solutions at 4°C from lysis through coating. ELPs expressed at 37°C are prone to irreversible aggregation, and protein stored at room temperature will phase-separate or degrade before it can be used. This is not an optimization variable—it is a prerequisite for the entire downstream pipeline.
Plasma treatment timing. After oxygen plasma treatment of PET, the activated hydrophilic surface begins reverting within 30 minutes. All coating must be applied within this window. Plan the workflow so that plasma treatment and drop-casting happen in immediate sequence.
Multi-layer coating protocol. A single drop-cast layer is unlikely to produce sufficient thickness for visible bilayer mismatch on 6 µm PET. Build up the coating through 3–5 repeated cycles of drop-cast → dry → drop-cast. Confirm each layer is fully dry before the next application. This is especially important for the more hydrophobic variants, which may form thinner or less continuous films per layer.
Substrate thickness selection. Start with 6 µm Mylar: thinner substrates amplify bilayer bending response. If 6 µm is too fragile to handle or cut cleanly, switch to 12 µm as the fallback. Document which thickness was used for each sample, as substrate thickness directly affects the magnitude of observable deformation.
6. Techniques
Relevant Techniques Checklist
Pipetting
Lab safety
Bioethical considerations
DNA sequencing
DNA construct design
Restriction enzyme digestion
Gel electrophoresis
DNA purification from gel
Databases: GenBank, NCBI
Designing a Twist order
Protein design
Use of Benchling
Chassis selection: BL21(DE3)
Plasmid preparation
Bacterial culturing
Bacterial processing: centrifugation, lysis, DNA purification
Protein purification
Gibson Assembly
Primer design or selection
PCR reactions
Two Key Techniques in Detail
Benchling-based DNA construct design. I will use Benchling to design, annotate, and validate all three ELP constructs before synthesis. This includes confirming the reading frame across all 20 repeat units, checking for internal restriction sites that could interfere with downstream cloning, verifying the 6xHis tag is in-frame with the start codon, and ensuring the flanking regions are compatible with the chosen cloning method and expression vector. Because repetitive sequences are prone to recombination and deletion, careful construct design in Benchling is critical to catching problems before synthesis rather than after. Benchling will also serve as the central documentation and version-control platform for the project’s DNA design files.
SDS-PAGE for expression validation. After IPTG induction, I will use SDS-PAGE to compare induced and uninduced cell lysates for each ELP variant. ELPs are repetitive and often migrate anomalously on gels—appearing at a higher apparent molecular weight than predicted—so I will calculate expected molecular weights in advance and look for bands in the appropriate range. SDS-PAGE will also allow me to assess relative expression levels across the three variants and to determine whether each protein is primarily in the soluble or insoluble fraction, which directly affects whether the downstream coating experiments are feasible. This technique is the gatekeeper between the cloning/expression phase and the materials phase of the project.
Associated Industry Council Companies
Twist Biosciences, gene synthesis; Addgene, vectors; New England Biolabs, cloning enzymes and Gibson Assembly; Opentrons, potential automation; Thermo Fisher Scientific, expression and purification reagents; Benchling, construct design platform.
7. Expected Results and Quantitative Expectations
Validation Approach
I will validate the project by designing and ordering the three ELP DNA constructs via Twist Bioscience, confirming successful cloning by colony PCR and sequencing, and verifying protein expression by SDS-PAGE. This validates the core pipeline from sequence design to protein production.
Expected Outcomes
Humidity response gradient. The serine variant (VPGSG)₂₀ is expected to show the highest water uptake and strongest swelling-driven deformation. The alanine variant (VPGAG)₂₀ should show moderate response. The valine variant (VPGVG)₂₀ is expected to show the weakest swelling, potentially with more brittle or opaque coatings due to increased hydrophobic aggregation.
Coating morphology. Different guest residues may produce visible differences in film smoothness, cracking patterns, opacity, and adhesion to PET, even before humidity testing.
Reversibility. Some variants may retain reversible actuation over multiple cycles while others degrade, crack, or delaminate—degradation patterns are themselves sequence-dependent and meaningful.
Minimum success threshold. The project is designed to be meaningful even with partial success. Two expressing variants with distinguishable humidity response is sufficient to support the core hypothesis. If dramatic bending does not occur, differences in wrinkling, swelling, coating integrity, or surface texture still constitute evidence that sequence programs material behavior.
Technical Risks and Fallbacks
Expression failure. If one variant does not express, a two-variant comparison is still sufficient. ELPs are well-established expression targets; complete failure of all three is unlikely.
Purification difficulty. Partially purified fractions can support preliminary coating tests. This limitation will be clearly stated.
Twist synthesis issues with repeats. ELP repeats are short, 15 bp per pentapeptide, and within Twist’s capabilities, but synthesis of highly repetitive regions can occasionally fail. Fallback: adjust codon usage to reduce exact repeat identity while preserving the same amino acid sequence.
Diblock synthesis feasibility. The Aim 1b diblock constructs may be more challenging for Twist due to longer repetitive regions. These are explicitly marked as stretch goals pending feasibility confirmation.
8. References
Srokowski et al., “Surface and adsorption characteristics of three elastin-like polypeptides with sequence length and guest residue variations.” Journal of Biomaterials Science, Polymer Edition.https://pmc.ncbi.nlm.nih.gov/articles/PMC3540362/
Estimated total: $377–$838 with the Gene Fragments route; up to ~$1,175 if using Clonal Genes. Costs depend on lab availability of shared reagents and equipment.