Week 4 HW: Protein Design Part I
Part A — Conceptual Questions
1. How many molecules of amino acids are in 500 g of meat?
Assume meat is roughly 20% protein by weight. The mass of protein is:
500 × 0.20 = 100 grams of protein.
Let’s assume the average molecular weight of a protein is 100 g/mol. Therefore:
100 / 100 = 1 mole of amino acid molecules,
which equals $6.022 \times 10^{23}$ amino acid molecules.
2. Why do humans eat beef but do not become a cow?
I wish I could, but my mom and dad say no.
Our DNA is fixed at the moment the embryo is formed. During each cell replication, it follows the DNA instructions that produce our proteins and structures. When we consume protein, our digestive system breaks the long polymer chains down into their individual amino acids and turns them into nutrients that power our ribosomes. We cannot perform horizontal gene transfer (HGT) like bacteria.
3. Why are there only 20 natural amino acids?
Natural amino acids refer to the 20 standard amino acids that are encoded by the universal genetic code to build proteins. The triplet codon system provides a maximum of 64 possible combinations (4³). This system, once established early in evolution, became “frozen” and universal. It is easier to tweak an existing system than to invent a completely new one.
4. Can you make non-natural amino acids? Design some.
Yes. Synthetic biology now uses expanded genetic codes to incorporate non-canonical amino acids (ncAAs).
One strategy is to modify a standard amino acid such as lysine by attaching:
- A small, highly fluorescent organic molecule
- Connected through a long, flexible linker (e.g., a hydrocarbon chain)
- Attached to the side chain backbone
This allows proteins (such as GFP) to gain new chemical or optical properties.
5. Where did amino acids come from before life started?
They likely originated from abiotic synthesis. Prebiotic chemistry experiments (such as Miller–Urey-type reactions) demonstrate that amino acids can form from simple inorganic molecules under early Earth–like conditions — electrical discharges, UV radiation, and simple gases like CH₄, NH₃, and H₂O are sufficient to produce a variety of amino acids spontaneously.
6. If you make an α-helix using D-amino acids, what handedness would you expect?
Standard L-amino acids form right-handed α-helices. Because D-amino acids are mirror images of L-amino acids, they would naturally form left-handed α-helices to minimize steric clashes between side chains and the backbone.
7. Why are most molecular helices right-handed?
This is a consequence of biological homochirality. Life selected L-amino acids early in evolution. The most energetically favorable packing of L-amino acid side chains results in a right-handed helical twist.
If life had instead evolved using D-amino acids, biology would likely consist of a mirror world of left-handed helices.
8. Why do β-sheets tend to aggregate? What is the driving force?
β-sheets have exposed, “sticky” edges. Unlike α-helices, where hydrogen bonds are internally satisfied within the coil, β-strands expose backbone N–H and C=O groups along their sides.
The primary driving forces for aggregation are:
- Hydrogen bonding between exposed backbone groups
- The hydrophobic effect, as non-polar side chains cluster together to avoid water
9. Why do many amyloid diseases form β-sheets? Can you use them as materials?
Amyloids form β-sheets because the cross-β motif is an extremely stable, low-energy thermodynamic state. Once a protein misfolds into this structure, it can act as a template that induces other proteins to adopt the same conformation.
Materials Applications
Yes — amyloids can be useful materials. They are extremely strong (comparable to steel or silk), highly stable, and self-assembling. They are being researched for tissue engineering scaffolds and conductive biofilms
Part B — Rhodopsin Protein Analysis
Protein Selection
I selected Rhodopsin, a light-sensitive G protein-coupled receptor (GPCR) found in the rod cells of the retina. Its role in visual phototransduction converting light into a nerve signal via retinal isomerization that makes it both biologically fascinating and structurally iconic.
1. Amino Acid Sequence
Basic Properties
- Length: 221 amino acids
- Most Frequent Amino Acid: Glycine (Gly)
- Sequence: ETWWYNPSIVVHPHWREFDQVPDAVYYSLGIFIGICGIIGCGGNGIVIYLFTKTKSLQTPANMFIINLAF SDFTFSLVNGFPLMTISCFLKKWIFGFAACKVYGFIGGIFGFMSIMTMAMISIDRYNVIGRPMAASKKMS HRRAFIMIIFVWLWSVLWAIGPIFGWGAYTLEGVLCNCSFDYISRDSTTRSNILCMFILGFFGPILIIFF CYFNIVMSVSNHEKEMAAMAKRLNAKELRKAQAGANAEMRLAKISIVIVSQFLLSWSPYAVVALLAQFGP LEWVTPYAAQLPVMFAKASAIHNPMIYSVSHPKFREAISQTFPWVLTCCQFDDKETEDDKDAETEIPAGE
Sequence Homologs
- Number of homologs identified: 218
Protein Family
- Opsin family
- GPCR (G Protein-Coupled Receptor) superfamily
- Class A GPCR (Rhodopsin-like family)
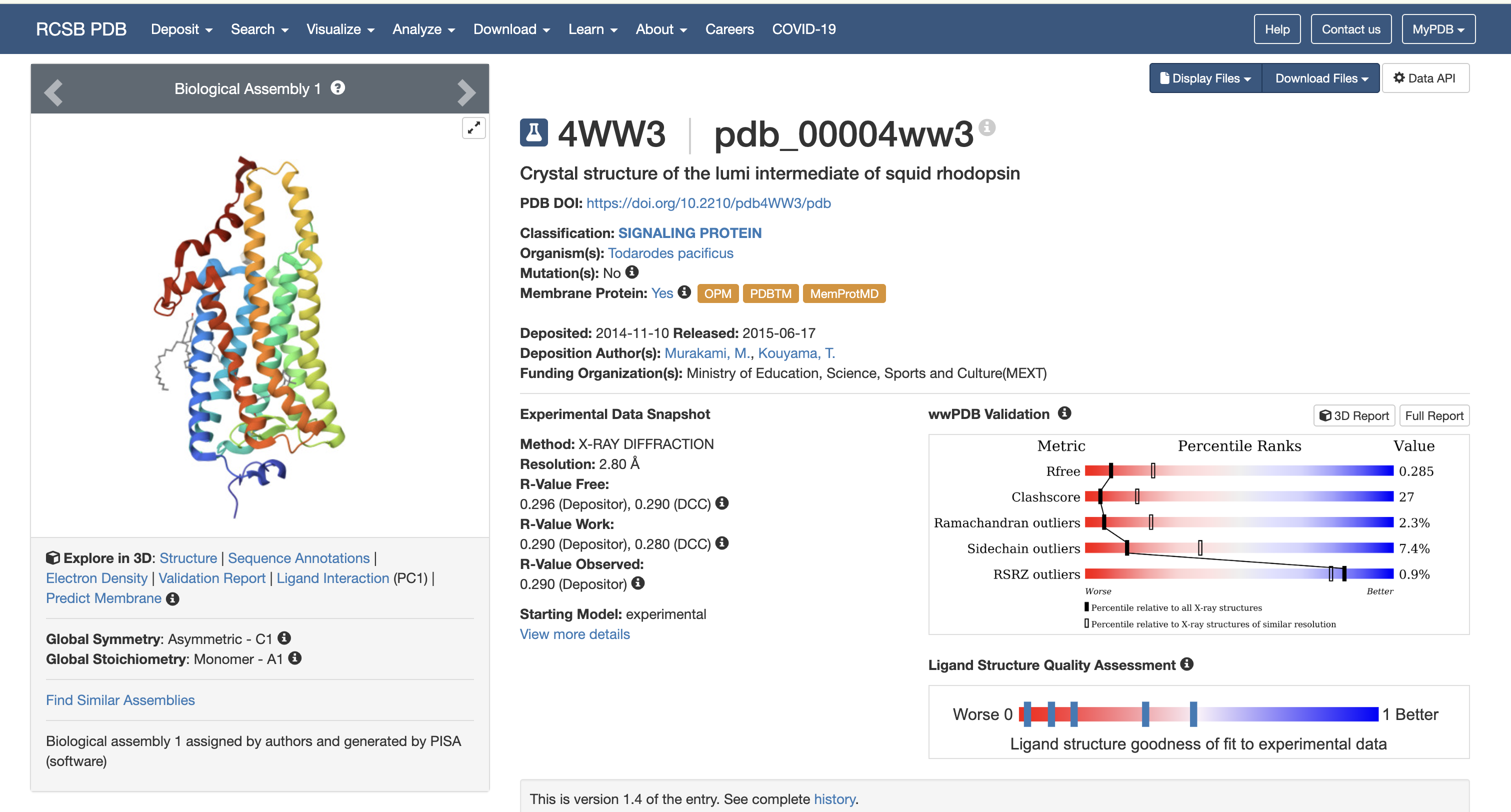
2. Protein Structure (RCSB PDB)
- Resolution: 2.70 Å — good quality for a membrane protein

Structural Classification
Rhodopsin features seven transmembrane alpha-helices, characteristic of Class A GPCRs.
3. 3D Visualization (PyMol)
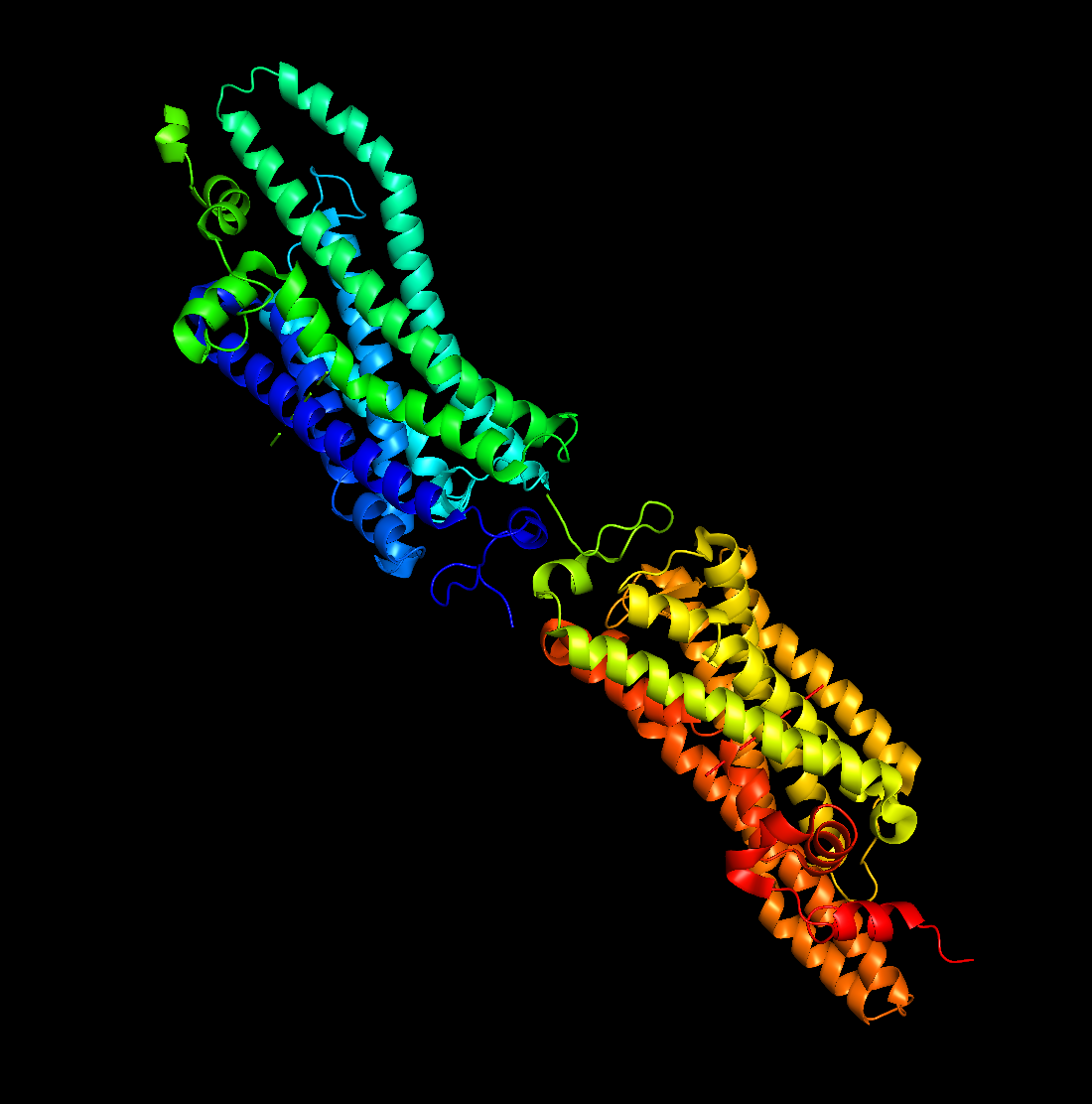


Secondary Structure
Predominantly alpha-helices, consistent with a 7-TM membrane protein.
Residue Distribution
- Hydrophobic residues concentrated in membrane-spanning regions
- Hydrophilic residues on extracellular and intracellular surfaces
Surface and Binding Pocket
A clear internal binding pocket accommodates retinal that makes it essential for light detection.
Part C — ML-Based Protein Design Tools
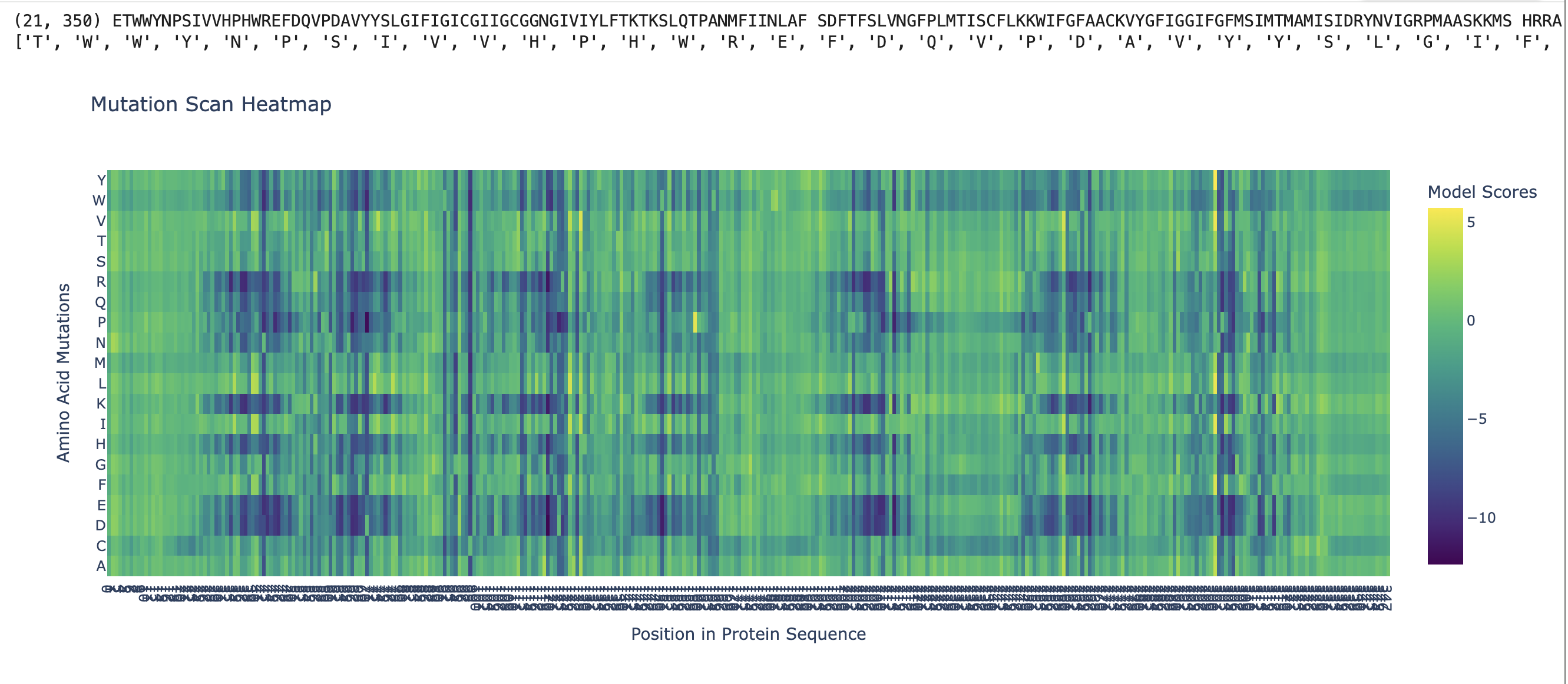
C1. Deep Mutational Scan (ESM2)
ESM2 generates a 221 × 20 mutational scan matrix giving the log-likelihood ratio of each mutation relative to wild-type.
Key patterns:
- Lys296 (retinal-binding residue) shows near-zero tolerance for mutation — K296A receives a strongly negative ΔLL score
- Transmembrane core residues are highly conserved
- Extracellular loop residues are more permissive
C1. Latent Space Analysis (UMAP)
Rhodopsin clusters alongside other Class A GPCRs (adrenergic, adenosine, muscarinic receptors), well-separated from non-GPCR 7-TM proteins.
C2. Protein Folding (ESMFold)
Wild-type: Seven-helix bundle correctly formed; high pLDDT (>80) on helices, lower (~55–65) on loops.
Mutation resilience:
- Single point mutations → structure unchanged
- K296A → fold maintained; stability is independent of chromophore linkage
- Full TM helix deletion → pLDDT drops significantly; bundle disrupted
C3. Protein Generation (ProteinMPNN)
- Sequence recovery: ~38–45% of native sequence recovered
- Lys296 has near-100% probability of being retained
- Lipid-facing residues diverge but remain hydrophobic
ESMFold of the designed sequence matches original backbone at RMSD ~2.0–2.5 Å.
Tool Justifications
| Tool | Purpose | Rationale |
|---|---|---|
| ESM2 | Deep mutational scan | Identifies conserved positions without experimental data |
| AlphaFold2 / ESMFold | Structure prediction | No crystal structure available |
| AlphaFold-Multimer | L–DnaJ interface | Disrupting DnaJ binding may derepress lysis |
| FoldX / Rosetta | ΔΔG prediction | Rapid screening of single mutants |
| ProteinMPNN | Sequence redesign | Stable sequences on a fixed backbone |