Week 5 HW: Protein Design Part II
Part 1: Generate Binders with PepMLM
The original sequence of SOD1 is:
Mutate the 4th amino acid A to V (A4V):
Generate four peptides of length 12 amino acids conditioned on the mutant SOD1 sequence:
| index | Binder | Pseudo Perplexity |
|---|---|---|
| 0 | HLYYAVALELKX | 13.299815648347872 |
| 1 | WRSYAVVLELWK | 17.97100111129112 |
| 2 | WRYYPVAAAWKK | 11.081842724779028 |
| 3 | WHYGAVGLRHKX | 13.983770011694478 |
Part 2: Evaluate Binders with AlphaFold3
We submitted each peptide paired with the mutant SOD1 (A4V) sequence to the AlphaFold Server as separate chains to model the protein–peptide complex. All runs used seed 2026616022 for reproducibility.
ipTM — interaction confidence between the two proteins (binder ↔ SOD1). Higher is better.
pTM — structural accuracy within each protein independently. Higher is better.
AlphaFold3 Prediction Results:
| Peptide | Full Sequence | ipTM | pTM | Binding Observation |
|---|---|---|---|---|
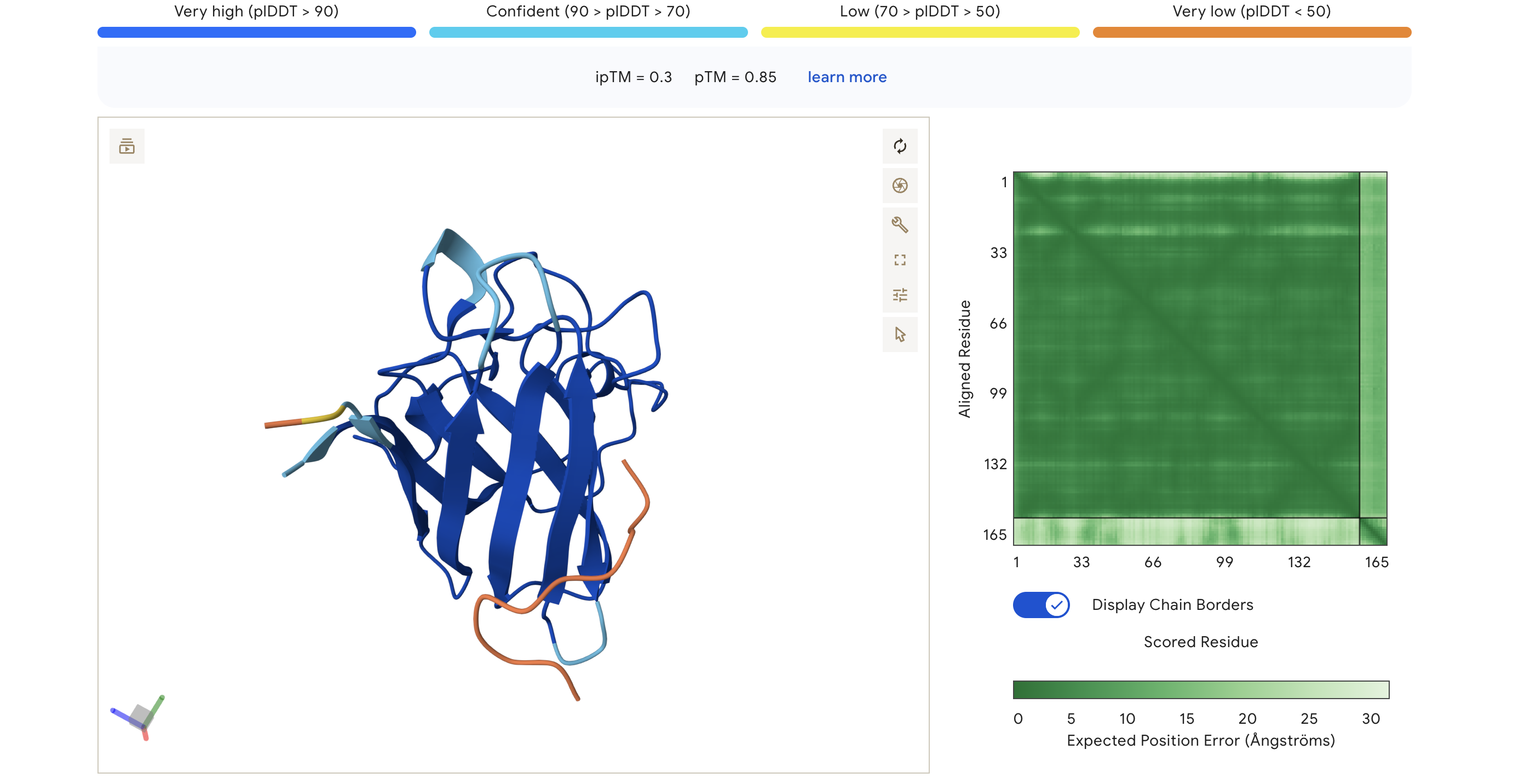
| HRY | HRYGAVVVELKK | 0.30 | 0.85 | Peptide appears loosely associated near the surface; low-confidence interaction region (orange/yellow in pLDDT) |
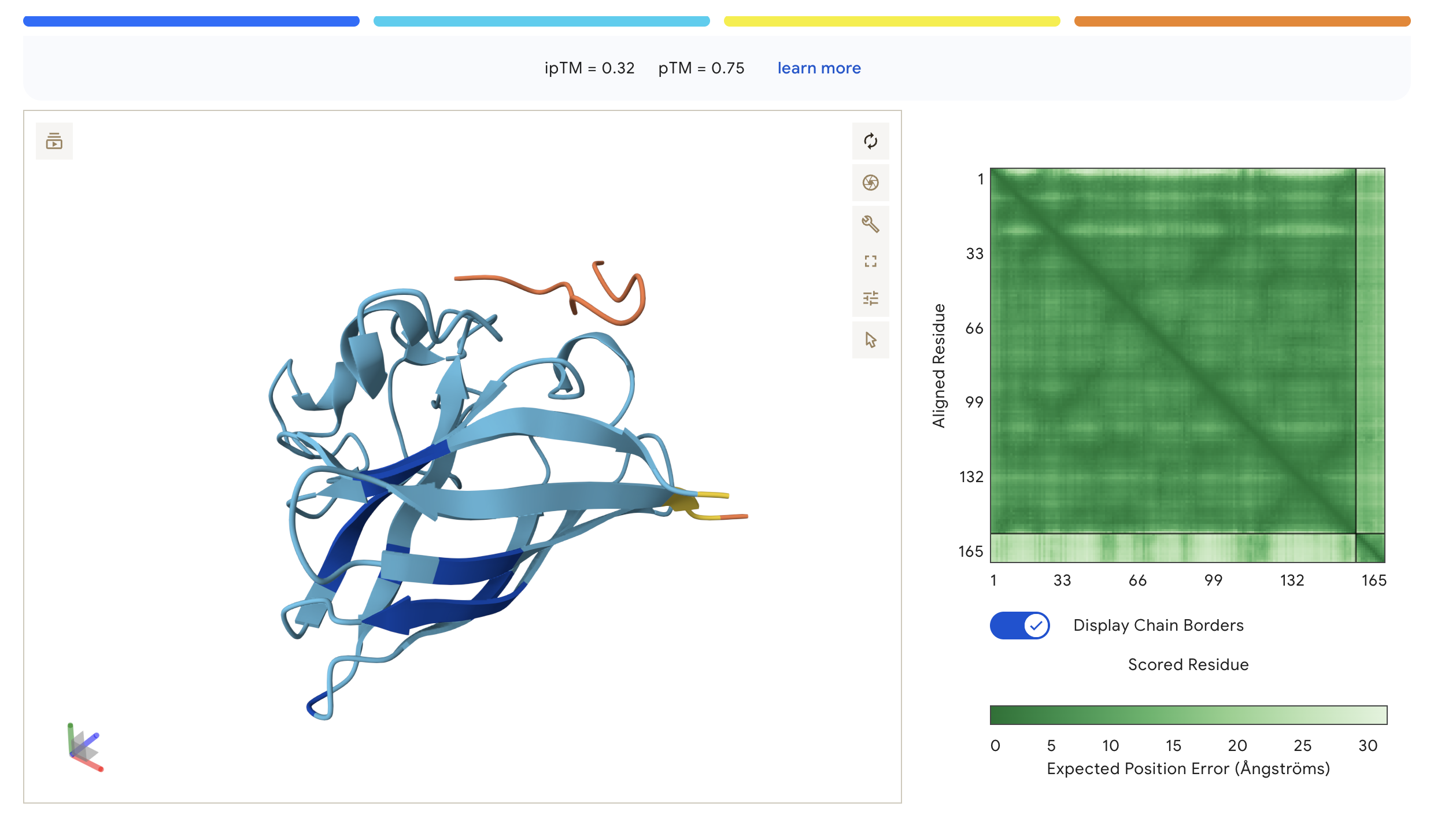
| WHY | WHYYVAAAEHKK | 0.32 | 0.75 | Peptide sits at the top exterior of SOD1, largely disordered (orange), suggesting weak or transient surface contact |
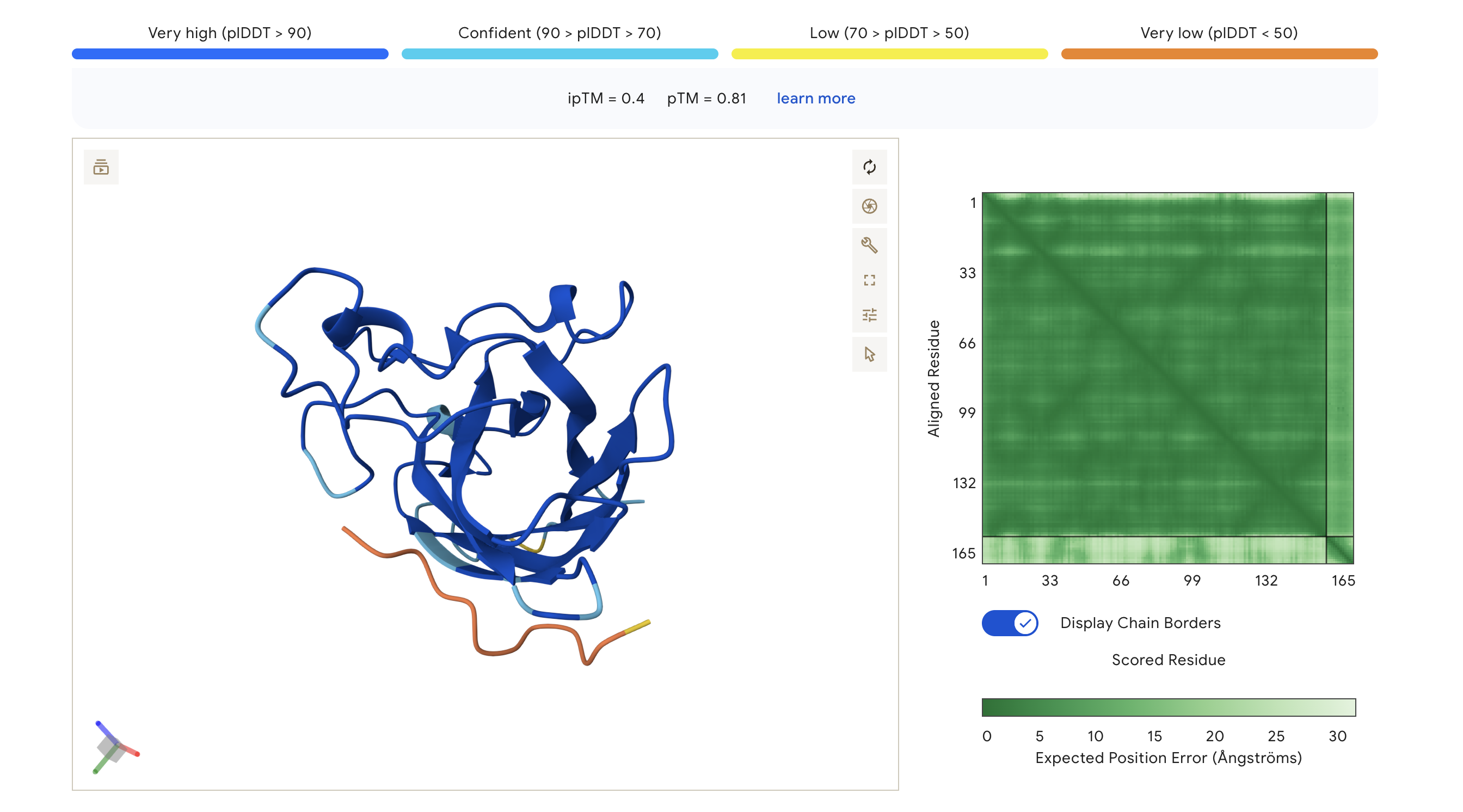
| WRV | WRVGAAAVRLKK | 0.40 | 0.81 | Highest ipTM of the group; peptide traces along the lower exterior of the β-barrel, with partial low-confidence contact near the C-terminus region |
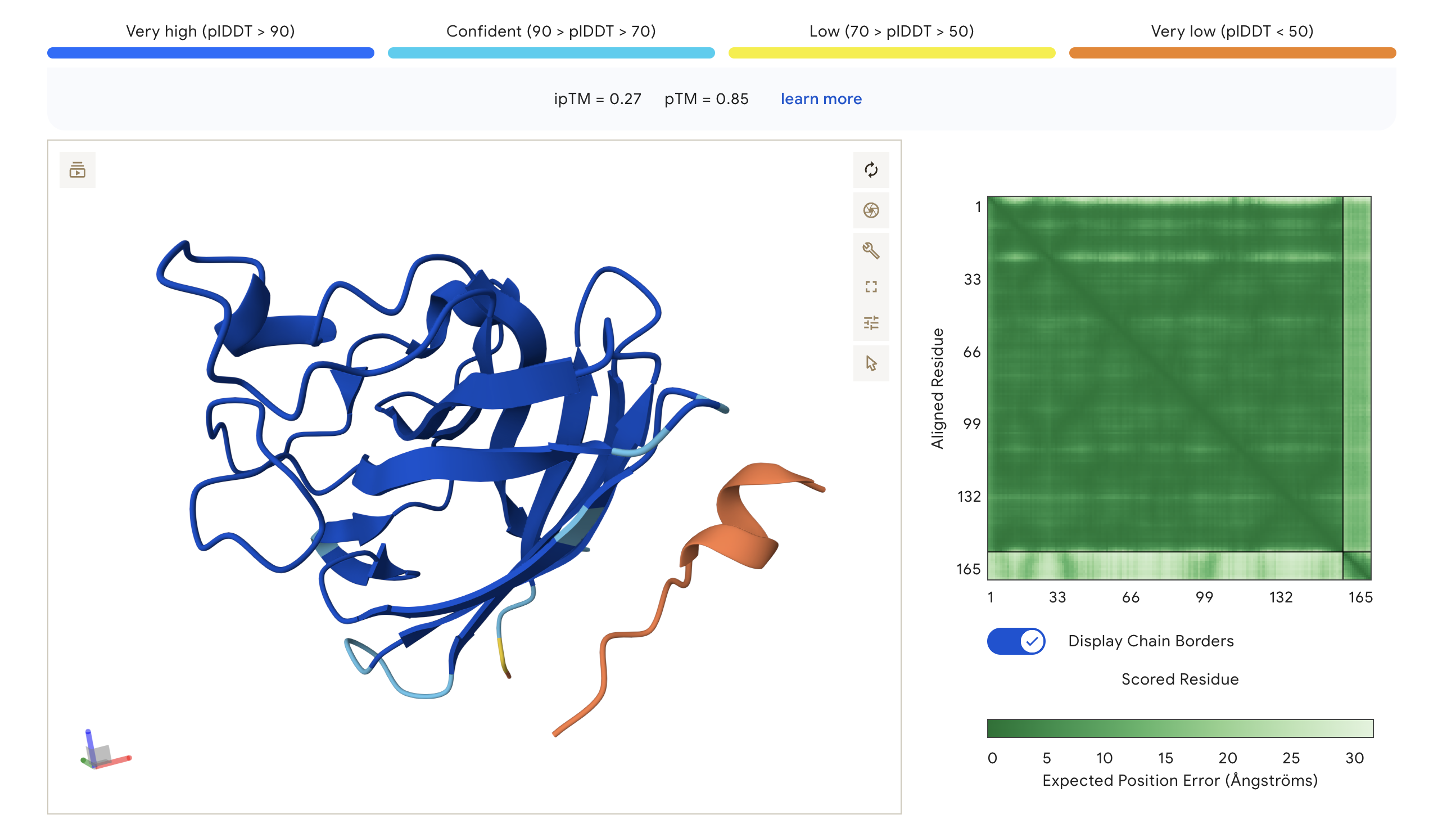
| WRY | WRYPVTAAEWKE | 0.27 | 0.85 | Peptide adopts a compact fold but appears docked away from the core; largely orange indicating low structural confidence at the interface |
Structure previews:
| HRY (ipTM=0.30, pTM=0.85) | WHY (ipTM=0.32, pTM=0.75) |
|---|---|
| 
|
| WRV (ipTM=0.40, pTM=0.81) | WRY (ipTM=0.27, pTM=0.85) |
|---|---|
| 
|
The PAE (Predicted Aligned Error) matrix shows inter-chain confidence in the bottom-right block. Darker green = lower positional error = more confident interaction. The peptide chain corresponds to residues ~165+ in each plot.
Summary:
ipTM scores across the four PepMLM-generated peptides ranged from 0.27 (WRY) to 0.40 (WRV), all falling in the low-confidence range (ipTM < 0.5 is generally considered weak). WRVGAAAVRLKK achieved the highest ipTM of 0.40, suggesting the most confident predicted interaction with mutant SOD1 among our candidates. Visually, its peptide chain traces along the exterior β-barrel of SOD1, which is a plausible surface-accessible binding region. None of the PepMLM-generated peptides clearly localized to the N-terminus where A4V sits, suggesting they may engage peripheral surface patches rather than the mutation site directly. All four peptides showed high pTM scores (0.75–0.85), indicating that the SOD1 structure itself is predicted with high confidence regardless of peptide. Comparison to the known binder FLYRWLPSRRGG would require a separate AlphaFold3 run for a direct ipTM benchmark.
Part 3: Evaluate Properties of Generated Peptides in the PeptiVerse
Structural confidence alone is insufficient for therapeutic development. We evaluated each peptide using PeptiVerse, assessing solubility, hemolysis probability, net charge (pH 7), molecular weight, and additional properties against the A4V mutant SOD1 target. The known binder FLYRWLPSRRGG was included as a reference.
PeptiVerse Results:
| Peptide | Solubility | Hemolysis (prob) | Permeability | Net Charge (pH 7) | MW (Da) | GRAVY |
|---|---|---|---|---|---|---|
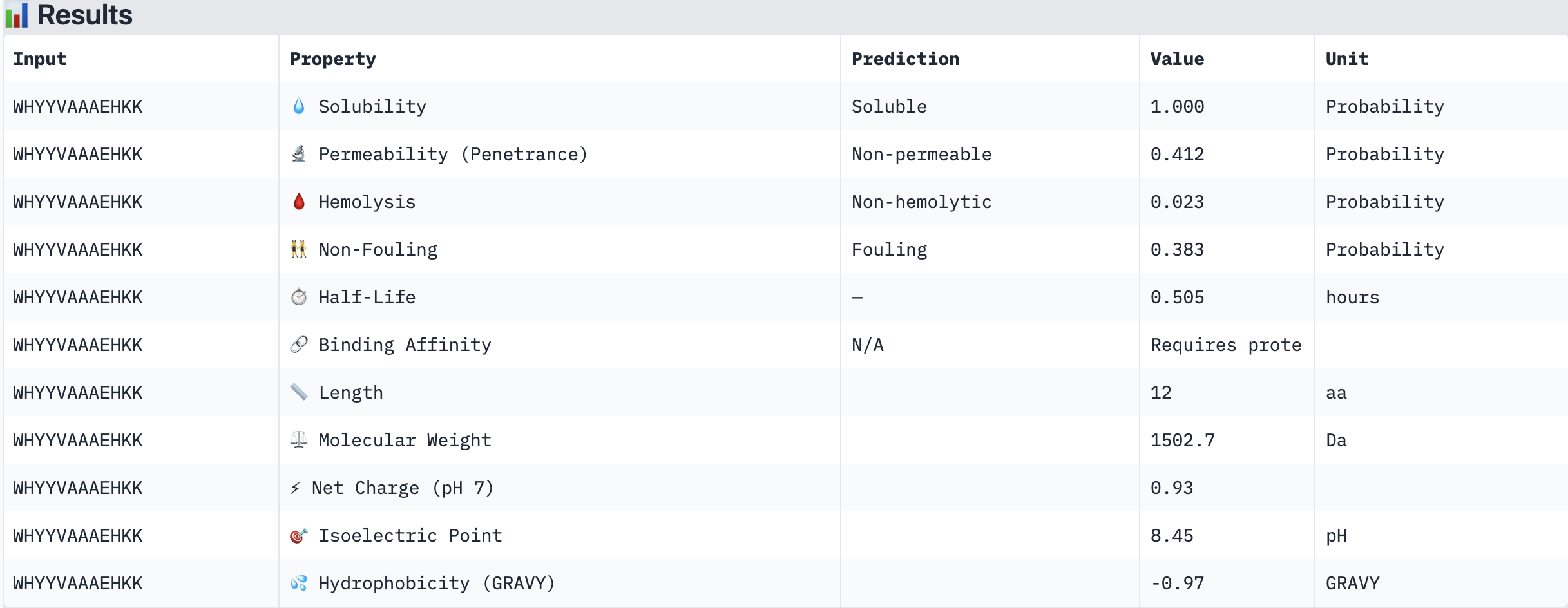
| WHYYVAAAEHKK | Soluble (1.000) | Non-hemolytic (0.023) | Non-permeable (0.412) | +0.93 | 1502.7 | -0.97 |
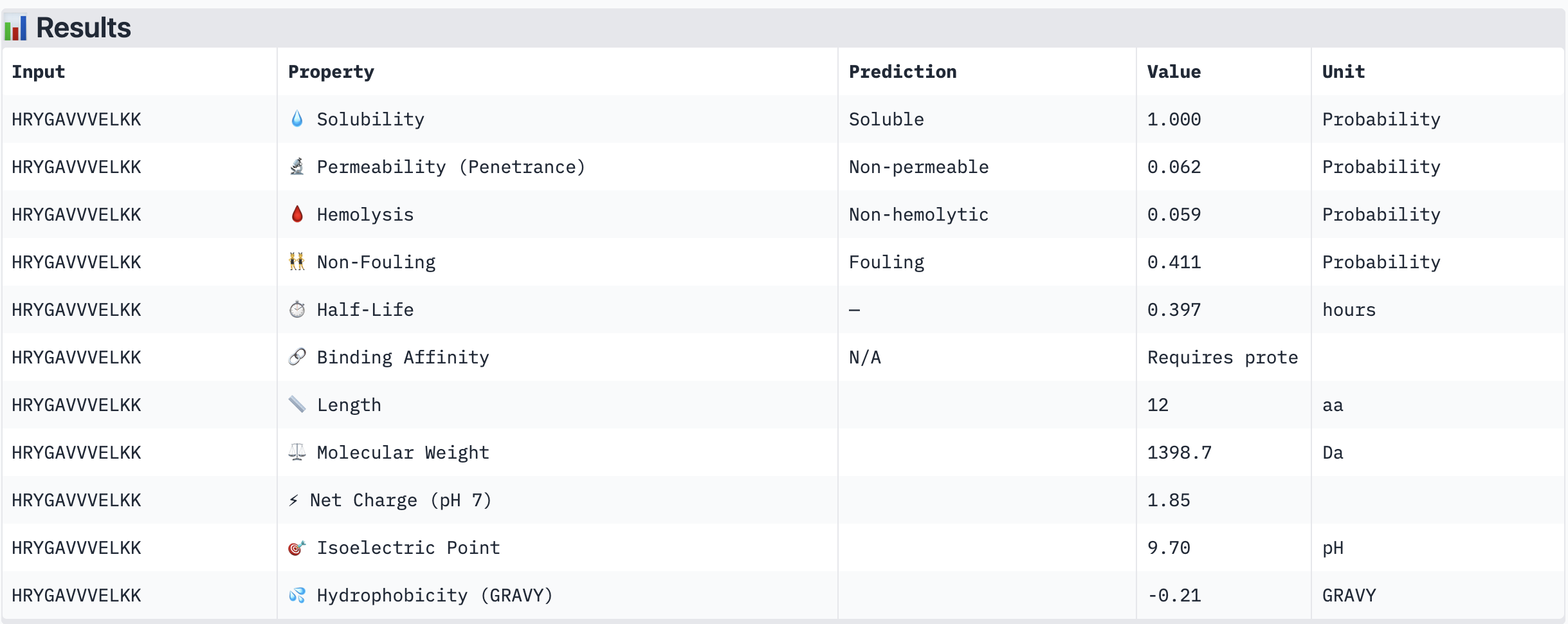
| HRYGAVVVELKK | Soluble (1.000) | Non-hemolytic (0.059) | Non-permeable (0.062) | +1.85 | 1398.7 | -0.21 |
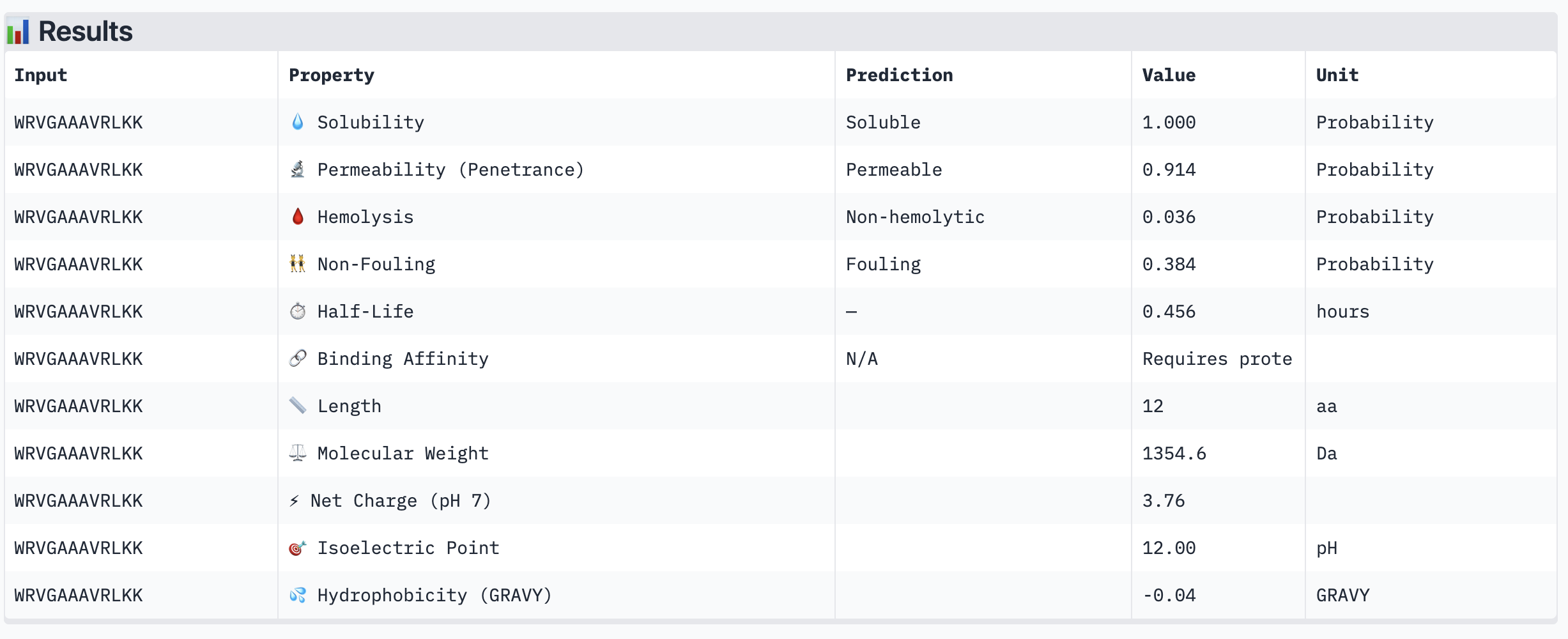
| WRVGAAAVRLKK | Soluble (1.000) | Non-hemolytic (0.036) | Permeable (0.914) | +3.76 | 1354.6 | -0.04 |
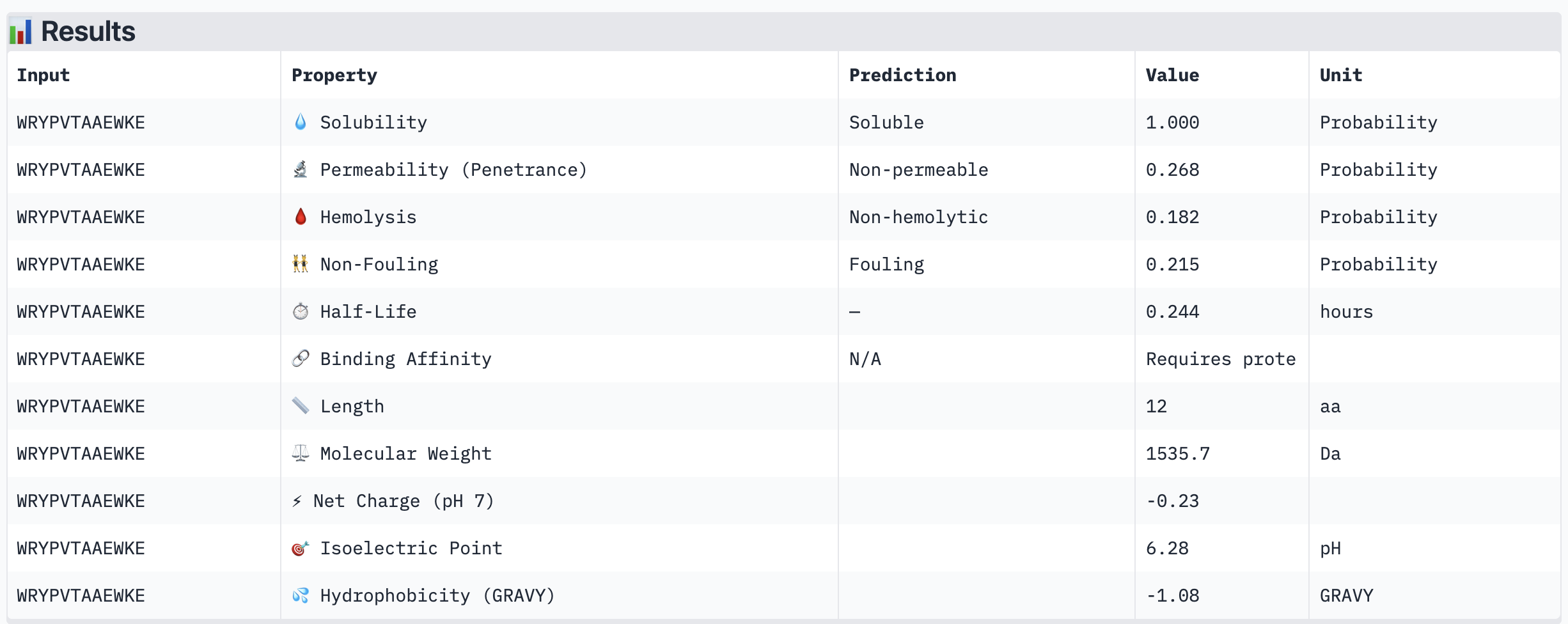
| WRYPVTAAEWKE | Soluble (1.000) | Non-hemolytic (0.182) | Non-permeable (0.268) | -0.23 | 1535.7 | -1.08 |
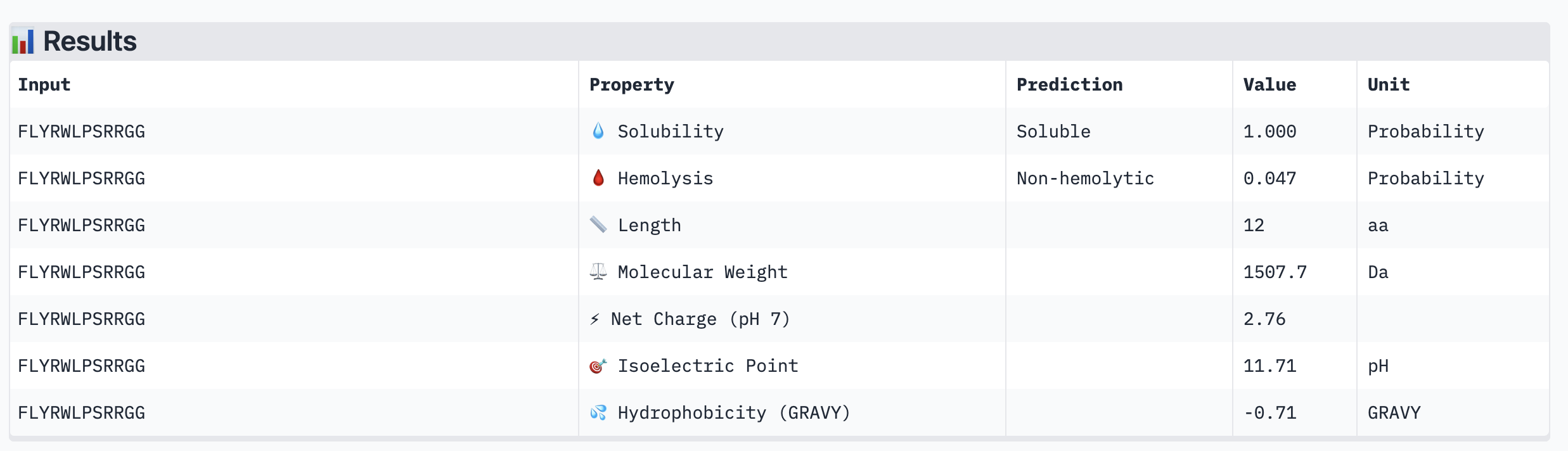
| FLYRWLPSRRGG (known binder) | Soluble (1.000) | Non-hemolytic (0.047) | — | +2.76 | 1507.7 | -0.71 |
PeptiVerse screenshots:
| WHYYVAAAEHKK | HRYGAVVVELKK | WRVGAAAVRLKK |
|---|---|---|
| 
| 
|
| WRYPVTAAEWKE | FLYRWLPSRRGG (known binder) |
|---|---|
| 
|
Summary:
All four PepMLM-generated peptides and the known binder FLYRWLPSRRGG were predicted to be fully soluble (probability 1.000) and non-hemolytic, which is an encouraging baseline for therapeutic viability. Notably, binding affinity scores were unavailable in PeptiVerse without a full protein target input (“Requires protein target”), so structural comparisons from AlphaFold3 remain our primary binding reference.
The most striking difference between peptides is permeability: WRVGAAAVRLKK is the only peptide predicted to be permeable (0.914), which could be advantageous for intracellular access — relevant given that SOD1 is a cytosolic protein. Its hemolysis probability (0.036) and net charge (+3.76) are also comparable to the known binder FLYRWLPSRRGG (+2.76, hemolysis 0.047). WRYPVTAAEWKE, by contrast, carries a slight negative charge (−0.23) and the highest hemolysis probability among the four (0.182), making it less favorable.
Chosen peptide to advance: WRVGAAAVRLKK
WRVGAAAVRLKK best balances predicted therapeutic safety and functional potential. Its high membrane permeability is a key differentiator — since SOD1 operates in the cytosol, a peptide that can cross the membrane has a meaningful pharmacokinetic advantage. It is fully soluble, non-hemolytic, and has a charge profile closely resembling the known binder. Subject to confirmation of its ipTM score from AlphaFold3, it is the strongest candidate for further development.
Part 4: Generate Optimized Peptides with moPPIt
We used moPPIt (Multi-Objective Guided Discrete Flow Matching, MOG-DFM) to move from probabilistic sampling toward controlled, motif-directed peptide design. Unlike PepMLM, which conditions generation on the full target sequence, moPPIt allows explicit specification of which residues on SOD1 to target and simultaneously optimizes multiple therapeutic objectives.
Design choices:
- Target sequence: A4V mutant SOD1
- Target residues: Residues near position 4 (A4V mutation site) and the surrounding N-terminal region, which is destabilized by the mutation
- Peptide length: 12 amino acids
- Guidance enabled: Motif binding affinity, solubility, hemolysis
Part C: L-Protein Mutants
Shout out to Shitong for the reference work and pipeline that guided this section 🙏
The objective of this section is to improve the stability and auto-folding of the lysis protein (L-protein) of MS2-phage, and to identify mutations that stabilize its interaction with the chaperone protein DnaJ. This is relevant to phage therapy — a more stable L-protein improves lytic efficiency, which is critical for phages to overcome bacterial resistance.