Week 1 HW: Principles and Practices

HTGAA Homework 1
Describe a biological engineering application or tool you want to develop and why:
Bioremediation of heavy metal contamination from soils in former industrial, or military areas to allow for future development.
At present there are an estimated 1,340 superfund sites on the EPA’s National Priority List (NPL) in the United States and according to a 2023 study, 23 million people live within 1 mile of a site.
I come from an architectural background and have a passion for reuse and restoration. I have watched the attempts, successes, and mistakes made during the remediation processes at some former superfund sites that have since gone on to be used as multi-unit housing, and commercial sites. Many of these sites used traditional removal and sequestration practices that disturbed topsoil and introduced contaminants into the surrounding air and water, creating toxic conditions for new residents.
I am interested in the use of engineered bacterial biofilms to remediate as much of the heavy metals present as possible to allow for access to previously uninhabitable land to allow for construction of housing, new community infrastructure, and other resources.
Describe one or more governance/policy goals related to ensuring that this application or tool contributes to an “ethical” future, like ensuring non-malfeasance (preventing harm). Break big goals down into two or more specific sub-goal:
Goal 1: Prevention of harm to flora and fauna
Bioremediation would require the introduction of engineered bacteria, such as B. subtilis, that is capable of not only thriving in the soil but also in the roots of plants, that could result in unintended and harmful consequences.
- Establish testing protocols before introduction into the environment.
- Establishment of a biosafety certification
Goal 2: Accessibility allowing use for diverse development
Describe at least three different potential governance “actions” by considering the four aspects below (Purpose, Design, Assumptions, Risks of Failure & “Success”)
Action 1. Transparency & education for public knowledge
- Purpose: Develop transparent protocols for development and testing that allow for public transparency at all levels.
- Design: Establish oversight and peer review
- Assumptions: Misunderstanding of existing review mechanisms.
- Risks of Failure: Public fear regarding bioengineering
Action 2. Establishment of funding
- Purpose: Attract institutional, or private funding for development of an accessible process
- Design: Work with stakeholders with an interest in earth regeneration or restoration
- Assumptions: Funding exists to allow for this work
- Risks of Failure: Lack of capital. Lack of interest.
Action 3. Establishment of oversight and cooperation with existing governance
- Purpose: Work with existing regulatory organizations
- Design: Create a board of scientists and stakeholders to enforce governance
- Risks of Failure: restrictive regulatory environment.
Score (from 1-3 with, 1 as the best, or n/a) each of your governance actions against your rubric of policy goals. The following is one framework but feel free to make your own:
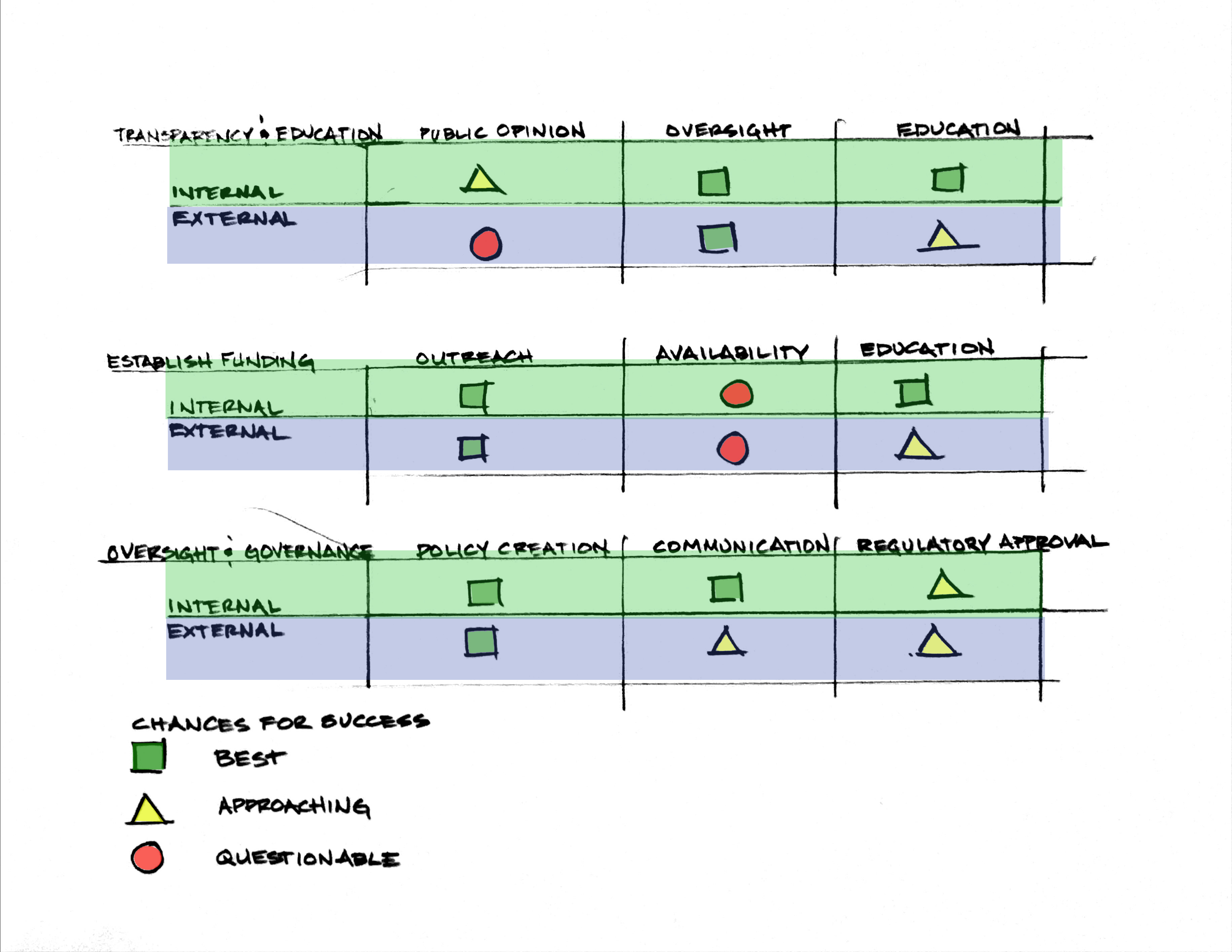
Drawing upon this scoring, describe which governance option, or combination of options, you would prioritize, and why. Outline any trade-offs you considered as well as assumptions and uncertainties.
The best chances of success for this require the prioritization of transparency and oversight, without which the availability of funding, public opinion and approval of outside governance will be possible.
Assuming that any public communication or work with larger regulatory bodies will take time and multiple iterations towards approval, this process could take some time before it becomes available.
This work was done without an extensive review of available in-situ engineered bioremediation strategies, and as such can assume that there are a number of existing or in research solutions that could achieve the same ends, but that might not meet the criteria of transparency and availability to all socio-economic classes.
Reflecting on what you learned and did in class this week, outline any ethical concerns that arose, especially any that were new to you. Then propose any governance actions you think might be appropriate to address those issues. This should be included on your class page for this week.
The majority of ethical concerns within this process were the possible effects of the introduction of an engineered bacteria into an uncontrolled and future publicly accessible area. Without foreknowledge of the mechanisms of the synthesis, and release of this, I was and largely still am in the dark. I was pleased to learn about the existence of regulatory bodies that review these concerns in various capacities and was able to find a number of papers outlining the creation of bioengineered bacteria that met the criteria for treatment of contaminated soils. I would without a doubt work with the existing regulatory bodies and establish a group dedicated to public transparency and education to encourage the use of safe and effective solutions for soil treatments.
Week 2 Homework Questions
- Nature’s machinery for copying DNA is called polymerase. What is the error rate of polymerase? How does this compare to the length of the human genome. How does biology deal with that discrepancy?
- 1:10e6 . The human genome is cited as 3.2:10e9 . Biology relies on the MutS repair system to identify and repair mismatched base pairs.
- How many different ways are there to code (DNA nucleotide code) for an average human protein? In practice what are some of the reasons that all of these different codes don’t work to code for the protein of interest?
Slide 6 references 1036bp for average human protein. I’m uncertain about this question, but if this refers to human proteins, I can see that there are error correction issues at longer lengths, and that existing human protein sequences contain encoding that would contribute to them not folding correctly for another application.
Dr. LeProust
- What’s the most commonly used method for oligo synthesis currently?
- Phosphoramidite chemistry
- Why is it difficult to make oligos longer than 200nt via direct synthesis?
- Though Phosphoramidite chemistry has a high degree of accuracy (`>99% per coupling), the small errors accumulate and approaching 200nt become disproportionate to the whole.
- Why can’t you make a 2000bp gene via direct oligo synthesis?
- Using Google & Prof. Church’s slide #4 What are the 10 essential amino acids in all animals and how does this affect your view of the “Lysine Contingency”?
- Arginine, Histidine, Isoleucine, Leucine, Lysine, Methionine, Phenylalanine, Threonine, Tryptophan, and Valine
- OK. If we’re talking Jurassic Park here.. Lysine is readily available in nature (and required by all animals to survive) from animal meat, so carnivore/omnivores would have no issues, and herbivores could find it readily in vegetation in varying amounts. You’re gonna need a bigger boat for that BS, Dr. Arnold.
Week 2 HW: Read, Write & Edit

HTGAA Homework 2
Part 1: Benchling & In-silico Gel Art
See the Gel Art: Restriction Digests and Gel Electrophoresis protocol for details. Overview:
Make a free account at benchling.com
Import the Lambda DNA.
Simulate Restriction Enzyme Digestion with the following Enzymes:
- EcoRI
- HindIII
- BamHI
- KpnI
- EcoRV
- SacI
- SalI
Create a pattern/image in the style of Paul Vanouse’s Latent Figure Protocol artworks.
Part 2: Gel Art - Restriction Digests and Gel Electrophoresis
Part 3: DNA Design Challenge
3.1. Choose your protein.
- In recitation, we discussed that you will pick a protein for your homework that you find interesting. Which protein have you chosen and why? Using one of the tools described in recitation (NCBI, UniProt, google), obtain the protein sequence for the protein you chose.
Example from our group homework, you may notice the particular format — The example below came from UniProt
sp|P03609|LYS_BPMS2 Lysis protein OS=Escherichia phage MS2 OX=12022 PE=2 SV=1 METRFPQQSQQTPASTNRRRPFKHEDYPCRRQQRSSTLYVLIFLAIFLSKFTNQLLLSLL EAVIRTVTTLQQLLT
3.2. Reverse Translate: Protein (amino acid) sequence to DNA (nucleotide) sequence.
- The Central Dogma discussed in class and recitation describes the process in which DNA sequence becomes transcribed and translated into protein. The Central Dogma gives us the framework to work backwards from a given protein sequence and infer the DNA sequence that the protein is derived from. Using one of the tools discussed in class, NCBI or online tools (google “reverse translation tools”), determine the nucleotide sequence that corresponds to the protein sequence you chose above.
Example: Get to the original sequence of phage MS2 L-protein from its genome phage MS2 genome - Nucleotide - NCBI
- Lysis protein DNA sequence
- atggaaacccgattccctcagcaatcgcagcaaactccggcatctactaatagacgccggccattcaaacatgaggattacccatgtcgaagacaacaaagaagttcaactctttatgtattgatcttcctcgcgatctttctctcgaaatttaccaatcaattgcttctgtcgctactggaagcggtgatccgcacagtgacgactttacagcaattgcttacttaa
3.3. Codon optimization.
Once a nucleotide sequence of your protein is determined, you need to codon optimize your sequence. You may, once again, utilize google for a “codon optimization tool”. In your own words, describe why you need to optimize codon usage. Which organism have you chosen to optimize the codon sequence for and why?
Example from Codon Optimization Tool | Twist Bioscience while avoiding Type IIs enzyme recognition sites BsaI, BsmBI, and BbsI
- Lysis protein DNA sequence with Codon-Optimization
- ATGGAAACCCGCTTTCCGCAGCAGAGCCAGCAGACCCCGGCGAGCACCAACCGCCGCCGCCCGTTCAAACATGAAGATTATCCGTGCCGTCGTCAGCAGCGCAGCAGCACCCTGTATGTGCTGATTTTTCTGGCGATTTTTCTGAGCAAATTCACCAACCAGCTGCTGCTGAGCCTGCTGGAAGCGGTGATTCGCACAGTGACGACCCTGCAGCAGCTGCTGACCTAA
3.4. You have a sequence! Now what?
What technologies could be used to produce this protein from your DNA? Describe in your words the DNA sequence can be transcribed and translated into your protein. You may describe either cell-dependent or cell-free methods, or both.
3.5. [Optional] How does it work in nature/biological systems?
- Describe how a single gene codes for multiple proteins at the transcriptional level.
- Try aligning the DNA sequence, the transcribed RNA, and also the resulting translated Protein!!! See example below.
- Example shows the biomolecular flow in central dogma from DNA to RNA to Protein] Special note that all “T” were transcribed into “U” and that the 3-nt codon represents 1-AA.
Part 4: Prepare a Twist DNA Synthesis Order
4.1. Create a Twist account, and Benchling account
4.2. Build Your DNA Insert Sequence
- For example, let’s make a sequence that will make E. coli glow fluorescent green under UV light by constitutively (always) expressing sfGFP (a green fluorescent protein):
4.3. Select “Clonal Genes” option
For this demonstration, we’ll choose Clonal Genes. You’ll select clonal genes or gene fragments depending on your final project.
Historically, HTGAA projects using clonal genes (circular DNA) have reached experimental results 1-2 weeks quicker because they can be transformed directly into E. coli without additional assembly.
Gene fragments (linear DNA) offer greater design flexibility but typically require an assembly or cloning step prior to transformation. An advantage is If designed with the appropriate exonuclease protection, gene fragments can be used directly in cell-free expression.
4.4. Import your sequence
- You just took an amino acid sequence of interest and converted it into DNA, codon optimized it, and built an expression cassette around it! Choose the Nucleotide Sequence option and Upload Sequence File to upload your FASTA file.
4.5. Choose Your Vector
Since we’re ordering a clonal gene, you will need to refer to Twist’s Vector Catalog to choose your circular backbone. You can think of this as taking your linear expression cassette for your protein of interest, and completing the rest of the circle!
The backbone confers many special properties like antibiotic resistance, an origin of replication, and more. Discuss with your node to decide on appropriate antibiotic options. At MIT/Harvard, you can use Ampicillin, Chloramphenicol, or Kanamycin resistance.
Twist vectors do not contain restriction sites near the insert fragment, so make sure to flank your design with cut sites if you are intending to extract this DNA insert fragment later.
For this demonstration, choose a Twist cloning vectors like pTwist Amp High Copy.
Part 5: DNA Read/Write/Edit
5.1 DNA Read
What DNA would you want to sequence (e.g., read) and why? This could be DNA related to human health (e.g. genes related to disease research), environmental monitoring (e.g., sewage waste water, biodiversity analysis), and beyond (e.g. DNA data storage, biobank).
In lecture, a variety of sequencing technologies were mentioned. What technology or technologies would you use to perform sequencing on your DNA and why?
Also answer the following questions:
Is your method first-, second- or third-generation or other? How so?
What is your input? How do you prepare your input (e.g. fragmentation, adapter ligation, PCR)? List the essential steps.
What are the essential steps of your chosen sequencing technology, how does it decode the bases of your DNA sample (base calling)?
What is the output of your chosen sequencing technology?
5.2 DNA Write
What DNA would you want to synthesize (e.g., write) and why? These could be individual genes, clusters of genes or genetic circuits, whole genomes, and beyond. As described in class thus far, applications could range from therapeutics and drug discovery (e.g., mRNA vaccines and therapies) to novel biomaterials (e.g. structural proteins), to sensors (e.g., genetic circuits for sensing and responding to inflammation, environmental stimuli, etc.), to art (DNA origamis). If possible, include the specific genetic sequence(s) of what you would like to synthesize! You will have the opportunity to actually have Twist synthesize these DNA constructs! :)
What technology or technologies would you use to perform this DNA synthesis and why?
5.3 DNA Edit
What DNA would you want to edit and why? In class, George shared a variety of ways to edit the genes and genomes of humans and other organisms. Such DNA editing technologies have profound implications for human health, development, and even human longevity and human augmentation. DNA editing is also already commonly leveraged for flora and fauna, for example in nature conservation efforts, (animal/plant restoration, de-extinction), or in agriculture (e.g. plant breeding, nitrogen fixation). What kinds of edits might you want to make to DNA (e.g., human genomes and beyond) and why?
What technology or technologies would you use to perform these DNA edits and why?