Week 4 HW: Protein Design Part I
Part A: Conceptual Questions
Why do humans eat beef but do not become a cow, eat fish but do not become fish?
Consuming food does not alter our DNA. During digestion, the body breaks down proteins into individual amino acids, much like taking apart a Lego castle to get the generic bricks. Our cells then take these building blocks and, following the instructions in our own DNA, construct new human proteins. The amino acids don’t carry the “identity” of the animal they came from, they are simply raw materials.
Why are there only 20 natural amino acids?
This is a result of evolutionary optimization. These 20 amino acids provide enough chemical diversity to build almost any functional biological structure. Limiting the set to 20 makes the translation process in ribosomes more efficient and reduces the likelihood of tRNA matching errors, balancing complexity with genetic fidelity.
Can you make other non-natural amino acids? Design some new amino acids.
Yes, it is possible through synthetic biology by incorporating atoms rare in biology, such as fluorine, or functional groups like azides and alkynes for click chemistry. For example: designing p-fluorophenylalanine can increase a protein’s metabolic stability or alter its hydrophobic folding patterns
Where did amino acids come from before enzymes that make them, and before life started?
Amino acids formed abiotically. A famous example is the Miller-Urey experiment, which demonstrated that mixing primitive gases (methane, ammonia, hydrogen, and water vapor) with electrical discharges mimicking lightning causes amino acids to synthesize spontaneously. We also know they arrived from outer space via meteorites, suggesting the building blocks of life are widespread in the universe.
If you make an alpha-helix using D-amino acids, what handedness (right or left) would you expect?
It would be left-handed. Since natural L-amino acids form right-handed helices, using their mirror version (D-amino acids) causes the protein’s secondary structure to reflect that symmetry, resulting in a left-handed turn.
Can you discover additional helices in proteins?
Yes. While the classic alpha is the most common, researchers have identified others like 310 and the pi helix (wider and shorter). These often appear as short segments or transitions at the ends of regular alpha helices.
Why are most molecular helices right-handed?
This is due to the exclusive use of L-amino acids in life. When these are linked together, a right handed twist minimizes steric hindrance, allowing the side chains to point outward without bumping into each other, which creates the most stable configuration.
Why do $\beta$-sheets tend to aggregate?
The edges of beta have exposed hydrogen bond donors and acceptors (C=O and N-H groups) from the backbone. Because these bonds are not satisfied within the sheet itself, they seek to pair with other nearby polypeptide chains, making the sheets sticky.
What is the driving force for $\beta$-sheet aggregation?
The driving force is those hydrogen bonds and the fact that hydrophobic faces seek to hide from water by sticking together.
Why do many amyloid diseases form $\beta$-sheets?
This happens because when a protein misfolds, forming stacked beta-sheet structures is the most thermodynamically stable state possible. It acts like a super-strong glue that the body has no way to destroy, causing them to accumulate and lead to diseases such as Parkinson.
Can you use amyloid $\beta$-sheets as materials?
Yes, they can be used as materials due to their high resistance. In biotechnology and synthetic biology, they are being used to manufacture harder and more durable bioproducts, such as nanofibers.
Design a beta-sheet motif that forms a well-ordered structure.
The most typical method is to alternate amino acids. You can use a polar one, then a hydrophobic one, and repeat (e.g., Valine - Lysine - Valine - Lysine). This ensures the sheet has a “greasy” face to stick to another sheet and a hydrophilic face to interact with water, forming highly ordered structures.
Part B: Protein Analysis and Visualization
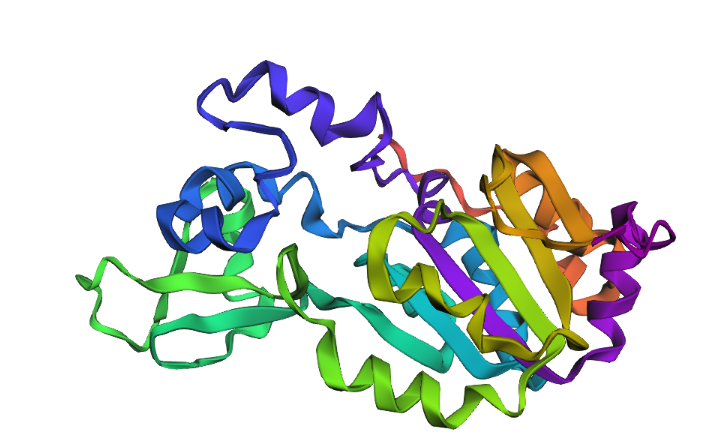
In this part of the homework, you will be using online resources and 3D visualization software to answer questions about proteins. Pick any protein (from any organism) of your interest that has a 3D structure and answer the following questions:
- Briefly describe the protein you selected and why you selected it.
I selected SIRT1 because of its fascinating potential in anti-aging research and its critical regenerative functions. This protein acts as a metabolic sensor that links a cell’s energy status with genome stability and epigenetic regulation.
The clinical importance of SIRT1 lies in its double-edged nature: while it promotes longevity and health in normal cells, this same survival mechanism is often hijacked by cancer cells. For instance, in pathologies like colorectal cancer (CRC), the overexpression of SIRT1 helps tumors resist chemotherapy and promotes metastasis. I chose this protein because understanding how it balances cell survival and disease progression is key to the future of precision medicine.
- Identify the amino acid sequence of your protein.
MADEAALALQPGGSPSAAGADREAASSPAGEPLRKRPRRDGPGLERSPGEPGGAAPEREVPAAARGCPGAAAAALWREAEAEAAAAGGEQEAQATAAAGEGDNGPGLQGPSREPPLADNLYDEDDDDEGEEEEEAAAAAIGYRDNLLFGDEIITNGFHSCESDEEDRASHASSSDWTPRP RIGPYTFVQHLMIGTDPRTILKDLLPETIPPPELDDMTLWQIVINILSEPPKRKKRKDINTIDAVKLLQECKKIIVLTGAGVSVSCGIPDFRSRDGIYARLAVDFPDLPDPQAMFDIEYFRKDPRPFFKFAKEIYPGQFQPSLCHKFIALSDKEGKLLRNYTQNIDTLEQVAGIQRII QCHGSFATASCLICKYKVDCEAVRGDIFNQVVPRCPRCPADEPLAIMKPEIVFFGENLPEQFHRAMKYDKDEVDLLIVIGSSLKVRPVALIPSNQYLFLPPNRYIFHGAEVYSDSEDDVLSSSSCGSNSDSGTCQSPSLEEPMEDESEIEEFYNGLEDEPDVPERAGGAGFGTDGDDQEAINEAISVKQEVTDMNYPSNKS
a. How long is it? What is the most frequent amino acid? You can use this Colab notebook to count the frequency of amino acids.
Protein Sequence Length: 559 amino acids
Amino Acid Frequencies: A: 59 (10.55%), E: 54 (9.66%), P: 48 (8.59%), D: 47 (8.41%), G: 44 (7.87%), L: 39 (6.98%), S: 38 (6.80%), I: 34 (6.08%), R: 31 (5.55%), K: 23 (4.11%), V: 23 (4.11%), F: 21 (3.76%), Q: 20 (3.58%), N: 17 (3.04%), T: 16 (2.86%), Y: 14 (2.50%), C: 13 (2.33%), M: 8 (1.43%), H: 7 (1.25%), W: 3 (0.54%)
b. How many protein sequence homologs are there for your protein? Hint: Use Uniprot’s BLAST tool to search for homologs.
Based on the UniProt BLAST search, there are at least 1,000 sequence homologs for this protein. The search reached the maximum display limit of 1,000 hits, which indicates that SIRT1 is a highly conserved protein across many species.
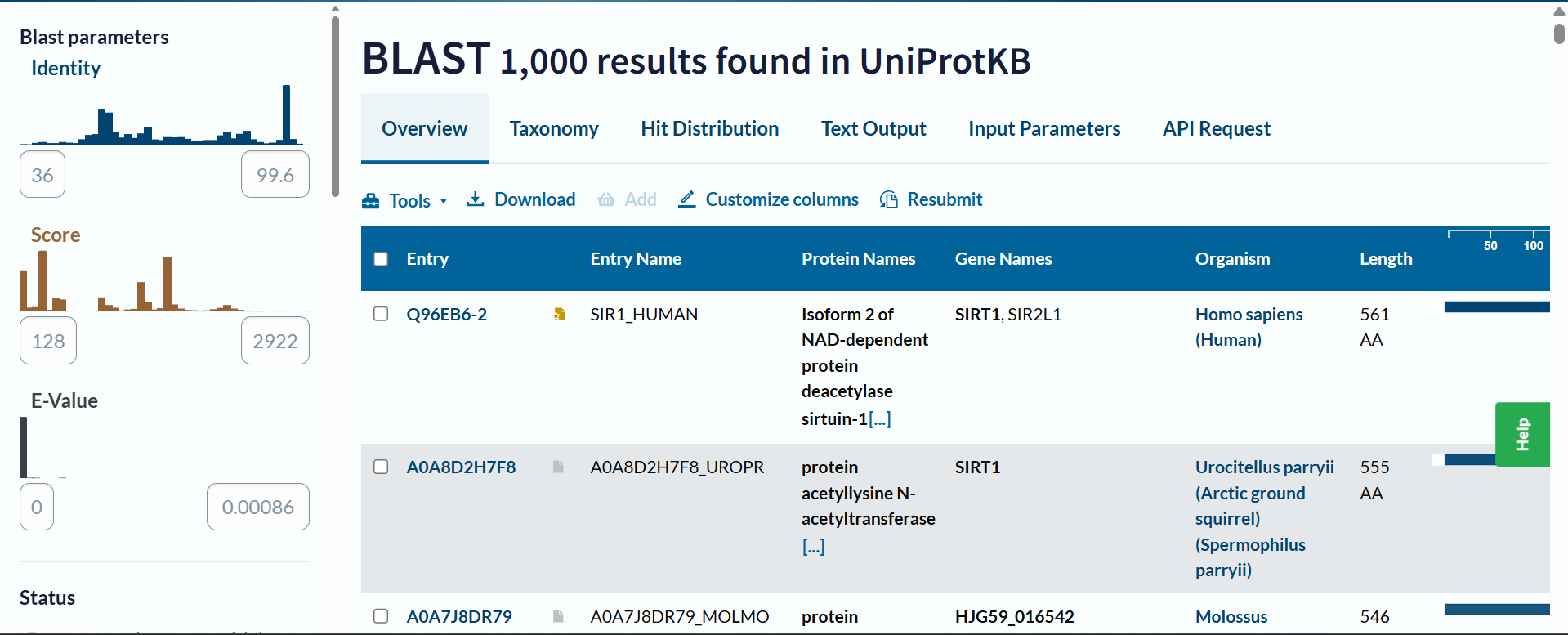
c. Does your protein belong to any protein family?
Yes, it belongs to the Sirtuin (Sir2) mammal’s family
Identify the structure page of your protein in RCSB. When was the structure solved? Is it a good quality structure? Good quality structure is the one with good resolution. Smaller the better (Resolution: 2.70 Å)
 Deposited: 2012-12-14 Released: 2013-10-23
Deposition Author(s): Davenport, A.M., Huber, F.M., Hoelz, A.
Resolution: 2.25 Å => It is a good quality structure
https://www.rcsb.org/structure/4IF6
Deposited: 2012-12-14 Released: 2013-10-23
Deposition Author(s): Davenport, A.M., Huber, F.M., Hoelz, A.
Resolution: 2.25 Å => It is a good quality structure
https://www.rcsb.org/structure/4IF6a. Are there any other molecules in the solved structure apart from protein?
Yes, apart from the protein, the solved structure contains two other molecules (ligands): an Adenosine-5-diphosphoribose (APR) and a Zinc ion (ZN).
b. Does your protein belong to any structure classification family?
Yes. According to structural classification databases like CATH and SCOP, its core catalytic domain belongs to the Rossmann fold family (typical for NAD-binding). Additionally, sequence and protein family databases (like InterPro) classify it within the Sir2 (sirtuin) family.
- Open the structure of your protein in any 3D molecule visualization software:PyMol Tutorial Here (hint: ChatGPT is good at PyMol commands)
Visualize the protein as “cartoon”, “ribbon” and “ball and stick”.
Color the protein by secondary structure. Does it have more helices or sheets?
Color the protein by residue type. What can you tell about the distribution of hydrophobic vs hydrophilic residues?
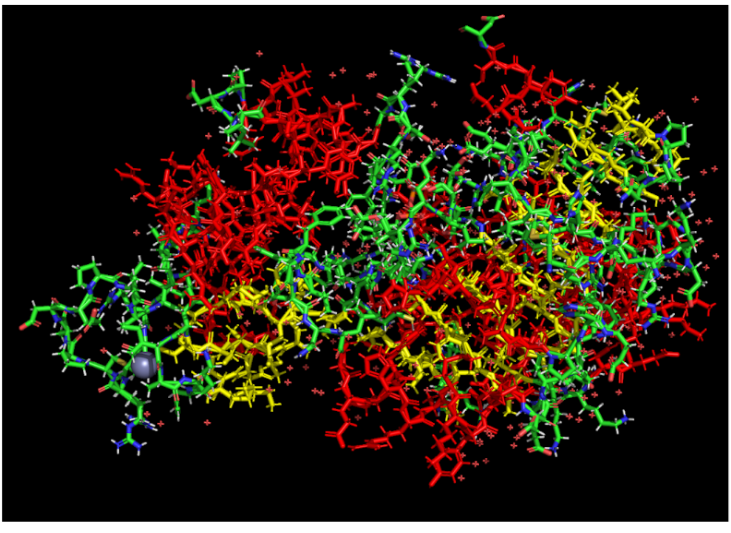
The hydrophobic residues (non-polar) are clustered in the inner core of the protein to hide from water. The hydrophilic residues (polar and charged) are distributed on the outer surface, exposed to the aqueous environment.
Visualize the surface of the protein. Does it have any “holes” (aka binding pockets)?
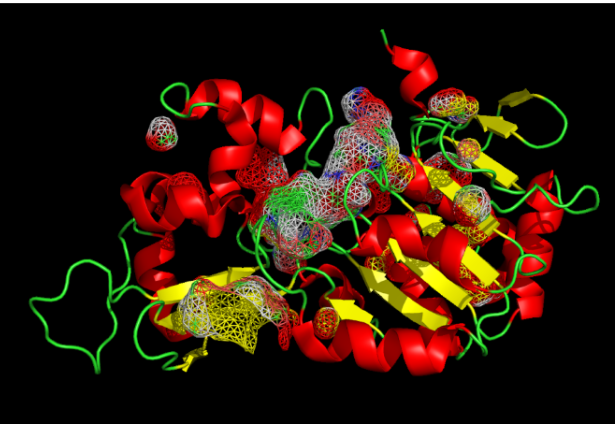
The protein has distinct ‘holes’ or binding pockets. As shown by the cavity visualization, there is a very prominent, large pocket right in the center of the structure (which is the main active site cleft), along with a few smaller pockets distributed across the protein.
Part C. Using ML-Based Protein Design Tools
Part C1: Using ML-Based Protein Design Tools
- Deep Mutational Scans (ESM2)
a. Use ESM2 to generate an unsupervised deep mutational scan of your protein based on language model likelihoods.
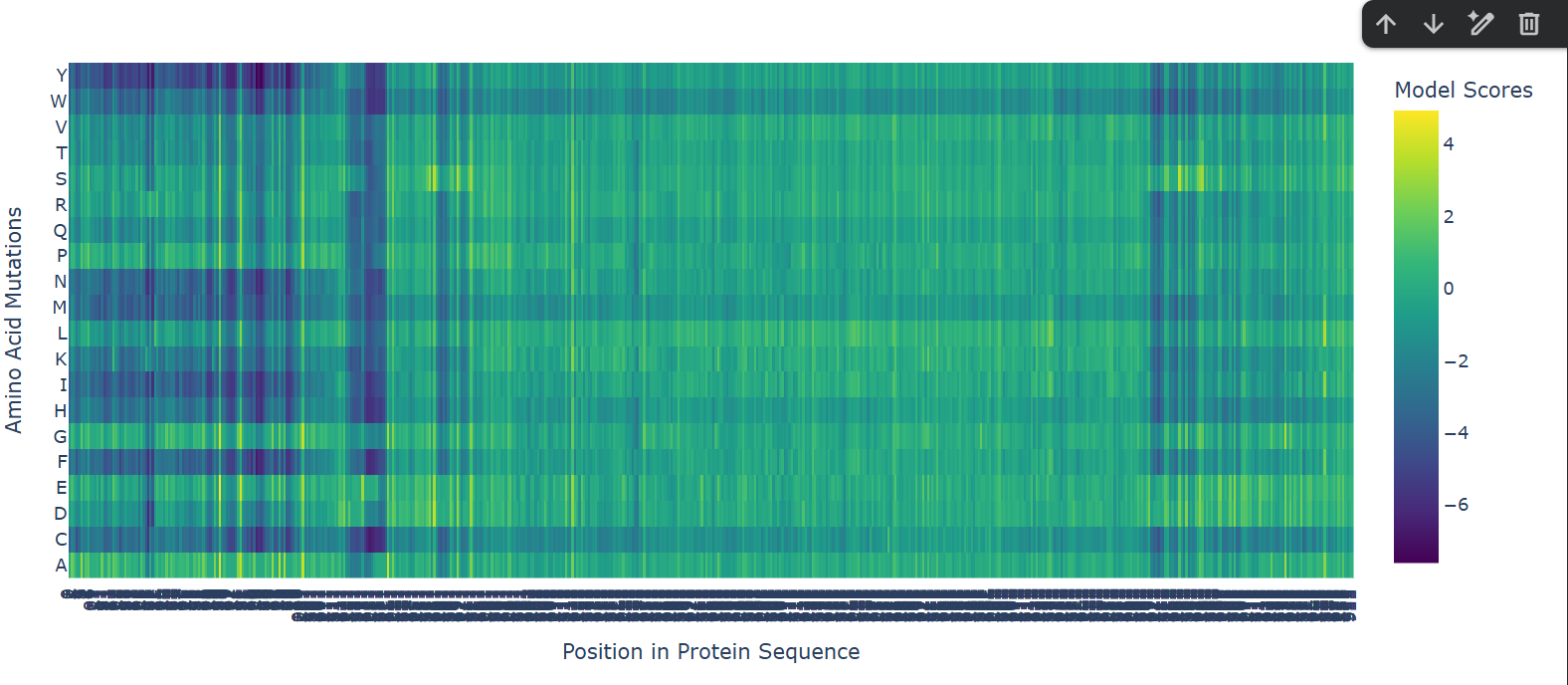
A clear horizontal pattern stands out for the mutation to Proline (P), which shows predominantly dark purple (negative) scores across the entire sequence. This aligns perfectly with protein biophysics: Proline has a unique, rigid cyclic structure that makes it a notorious ‘helix breaker’.The model correctly predicts that forcing a Proline into random positions will disrupt the secondary structure (especially alpha helices) and destabilize the overall protein.Additionally, the vertical dark purple columns interspersed between lighter regions clearly distinguish the highly conserved, intolerant catalytic core residues from the more flexible, mutation-tolerant surface loops.
Additionally, the vertical dark purple columns interspersed between lighter regions clearly distinguish the highly conserved, intolerant catalytic core residues from the more flexible, mutation-tolerant surface loops
- Latent Space Analysis
a. Use the provided sequence dataset to embed proteins in reduced dimensionality.
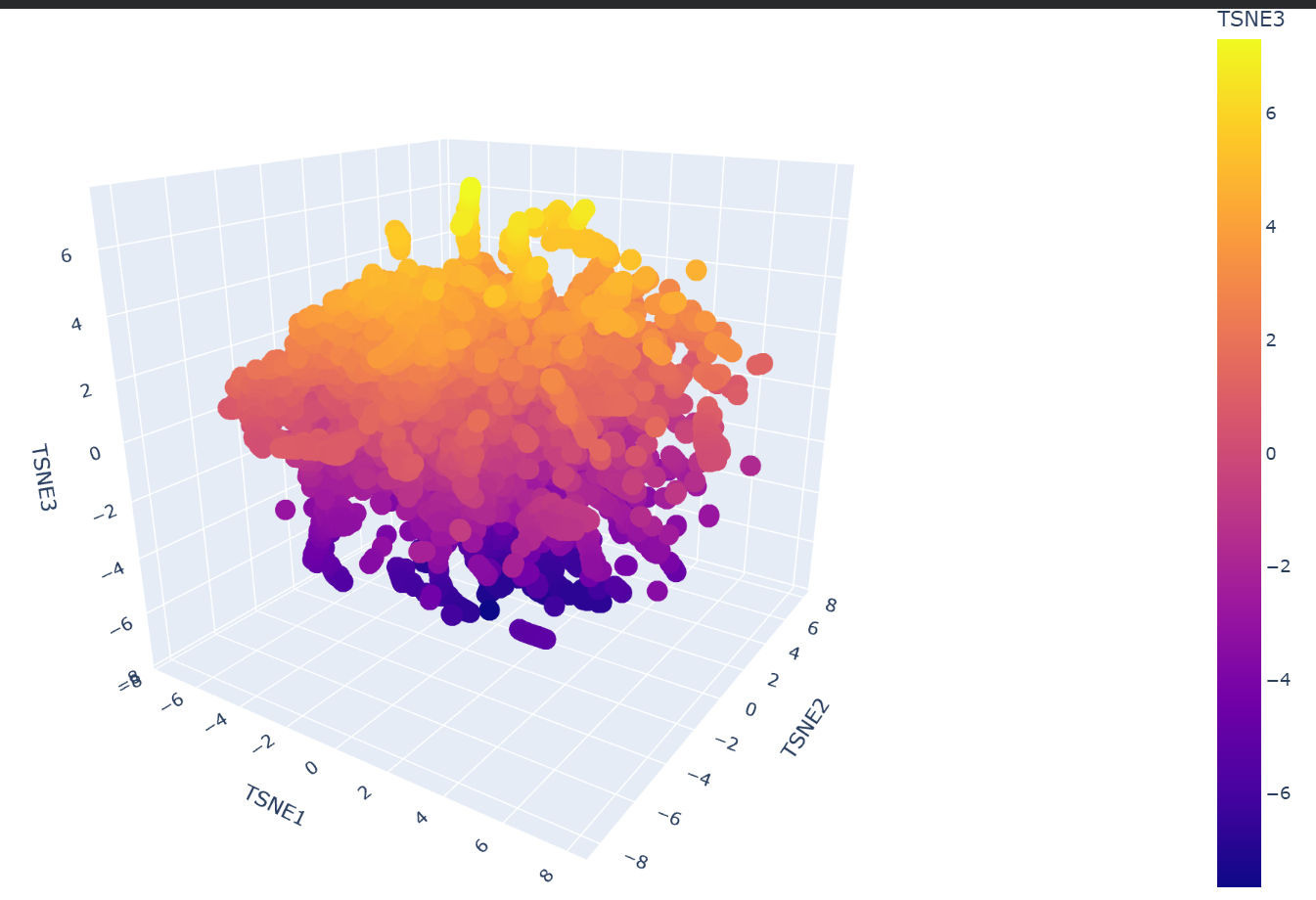
b. Analyze the different formed neighborhoods: do they approximate similar proteins?
Yes, the formed neighborhoods strongly approximate similar proteins. Dimensionality reduction algorithms like t-SNE take the high-dimensional embeddings generated by the ESM2 language model and group them based on learned sequence patterns. Because the model captures evolutionary and structural ‘grammar’, proteins that share similar domains, 3D folds, or biological functions naturally cluster together into distinct neighborhoods, even if their raw sequences are not identical.
c. Place your protein in the resulting map and explain its position and similarity to its neighbors.
My protein, SIRT1, was successfully projected into the latent space and placed within a specific cluster (the yellow group). Upon inspecting its immediate neighborhood, it clustered closely with proteins such as the NTF2-like domain of Tip associating protein (TAP), the Nonprocessive cellulase Cel9M, and the Nisin biosynthesis protein NisC. This placement makes biological sense because, although their specific functions vary, these neighbors share core structural features (complex mixed folds and extensive alpha-helical regions) with SIRT1, demonstrating that the language model correctly grouped them based on their underlying structural ‘grammar’ and similarities.

Part C2: Protein Folding
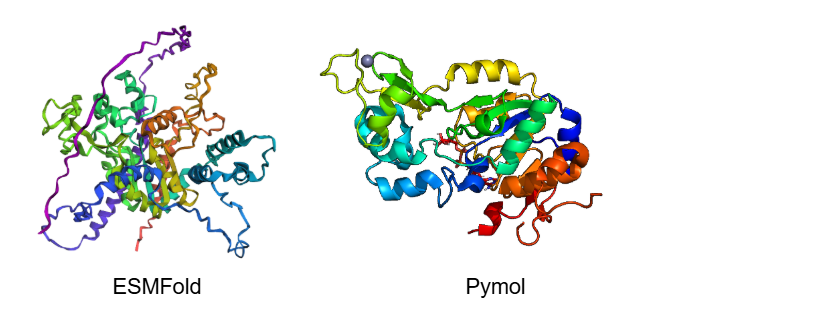
- Fold your protein with ESMFold. Do the predicted coordinates match your original structure?
No, the predicted coordinates do not match the experimental structure perfectly; there are significant and noticeable differences. While the central catalytic core shares some similarities, the ESMFold prediction lacks several beta-sheets (arrows) that are clearly visible in the PyMOL experimental structure, replacing them with unstructured loops.
Additionally, the predicted model has extremely long, uncoiled chains at the ends (purple and blue regions). This is because ESMFold predicts the full-length sequence, which includes highly disordered terminal tails that were likely truncated in the experimental crystal structure. Finally, ESMFold only predicts the protein backbone, so it completely misses the Zinc ion (the sphere) and any other bound ligands present in the PyMOL visualization.
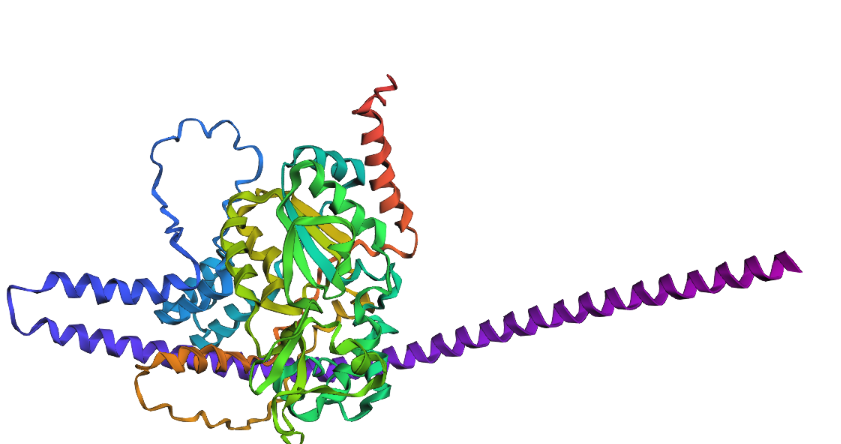
- Try changing the sequence, first try some mutations, then large segments. Is your protein structure resilient to mutations?
To test the structural resilience, I drastically modified the sequence by replacing a large unstructured segment with a massive Poly-Alanine sequence. The results show that the protein structure is highly resilient in its core, but globally altered. As expected, the Poly-Alanine mutation forced the formation of a massive, unnaturally long and coiled alpha-helix (visible as the extended purple/blue structure). However, despite this drastic appendage, the central catalytic domain demonstrated remarkable resilience. It folded into its characteristic globular shape, maintaining the core beta-sheets and the surrounding native helices. This proves that the SIRT1 core fold is highly stable even under extreme mutational stress at its terminals.
Part C3: Protein Generation
- Analyze the predicted sequence probabilities and compare the predicted sequence vs the original one.
ProteinMPNN created a sequence recovery of approximately 54% (0.5396) for the structured regions. The generated sequence prominently features a massive block of ‘X’s at the N-terminus. This reflects the limitations of the input experimental structure (4IF6), which lacks 3D coordinates for the highly flexible, intrinsically disordered tails.
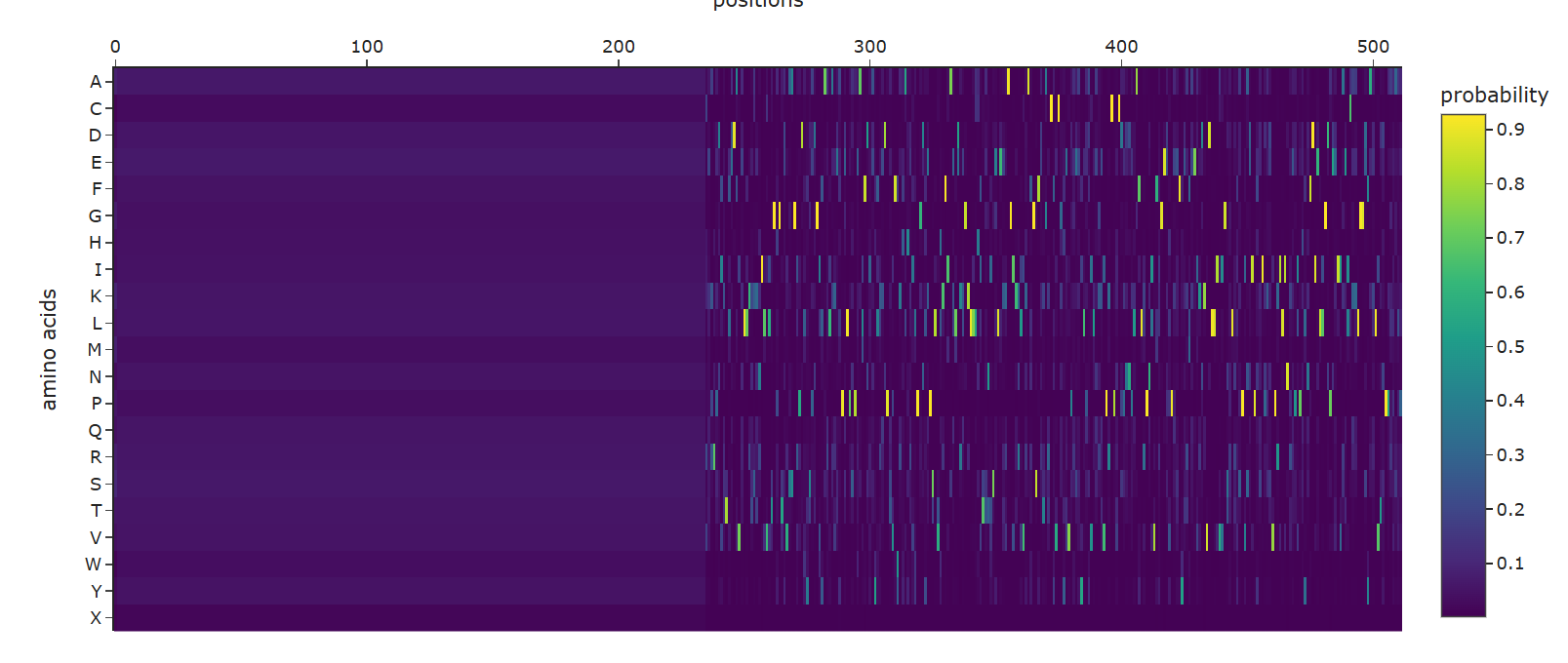
For the resolved catalytic core, the AI designed a novel sequence that is only 54% identical to the wild-type, successfully finding alternative amino acids that can hypothetically stabilize the same Rossmann-fold topology
- Input this sequence into ESMFold and compare the predicted structure to your original.
I input the clean, AI-generated sequence from ProteinMPNN (excluding the unresolved ‘X’ terminal regions) back into ESMFold. The resulting predicted structure successfully adopts the exact same core topology as the original experimental SIRT1 structure.
As observed, the overall chains appear shorter and lack the massive uncoiled loops seen in the wild-type prediction, which is expected since we only folded the core domain. The structure also lacks forced unnatural coils, displaying a natural distribution of secondary structures.
Part D. Group Brainstorm on Bacteriophage Engineering
HTGAA Final Project Proposal: Engineering the L Protein for Viral Titer Optimization
The objective of this project is to computationally redesign the bacteriophage lysis (L) protein to slightly delay the lysis event. In phage therapy, obtaining high viral concentrations is critical for clinical efficacy. By “tuning” the L protein’s kinetics, the goal is to maximize the burst size (the number of virions released per cell), allowing the host machinery to complete more viral assembly cycles before cell death occurs.
Based on the provided reading materials, this proposal addresses several key concepts:
- Host-Phage Interaction: The literature highlights that lysis is a finely regulated process where the L protein must interact with specific bacterial host chaperones, such as DnaJ.
- Clinical Need: As noted in reviews regarding the past, present, and future of phage therapy, one of the most significant bottlenecks for therapeutic translation is the industrial-scale production of sufficiently high titers required to combat systemic infections.
- Developability: Protein engineering to improve stability and controlled function is a prerequisite for these therapies to be viable and scalable.
I will utilize an “anti-black-box” computational design pipeline based on protein language models (pLMs) and generative diffusion design:
- ESM-2 & ESMFold: First, I will predict the structure of the wild-type L protein and perform an in silico Deep Mutational Scanning (DMS) to identify key residues that govern its stability and lytic activity.
- moPPIt (Multi-Objective-Guided Discrete Flow Matching): I will use moPPIt to design specific motifs that optimize the L protein’s interaction with the E. coli membrane or its natural host inhibitors. This will guide the generation toward sequences that maintain target engagement but feature controlled, delayed kinetics.
- ProteinMPNN: I will generate a library of sequence variants with tuned thermodynamic stability to find the exact “sweet spot” where the protein remains functional but is not prematurely hyperactive.
- PeptiVerse & AlphaFold3: I will validate the top candidates by predicting their binding affinity and therapeutic properties (like solubility) in PeptiVerse, followed by evaluating structural confidence (ipTM scores) of the host-target complexes using AlphaFold3.
These approaches enable precision structural design that far surpasses traditional random mutagenesis. By utilizing moPPIt, I can explicitly constrain my design to target specific functional motifs on the host machinery. Furthermore, using foundational models like ESM-2 allows me to leverage rich, learned representations that capture the evolutionary “grammar” of lysis proteins, exploring massive sequence spaces strictly in silico before any wet-lab synthesis.
Potential Pitfalls
- Lysis Complexity: The relationship between the L protein’s static structure and the highly dynamic, exact timing of cell lysis is difficult to model precisely using only static structural predictors.
- Production Toxicity Paradox: There is a risk that a kinetically altered L protein could still prove highly toxic to the bacterial production strains. If the expression timing is not carefully regulated experimentally, this could paradoxically lead to premature lysis and lower overall titers.
Pipeline Schematic