Week 5 HW: Protein Design Part II
Part A: SOD1 Binder Peptide Design
🔹 Part 1 — Generate Binders with PepMLM
!!! info “Description” Using PepMLM, four 12‑amino‑acid peptides were generated conditioned on the SOD1 A4V mutant sequence. SOD1‑binding peptide FLYRWLPSRRGG was added as a control, and the perplexity scores were recorded to estimate model confidence.
📊 Perplexity Scores
| Peptide | Perplexity |
|---|---|
| WTRGDEEEEPWL | 23.743201 |
| RRTGDTEEEEPE | 10.299139 |
| KTTGDETEEEGR | 11.090010 |
| WTEGDEELGEWR | 14.360410 |
| FLYRWLPSRRGG | — |
🔹 Part 2 — Evaluate Binders with AlphaFold3
📈 AlphaFold3 Results
| Peptide | Perplexity | ipTM | pTM |
|---|---|---|---|
| WTRGDEEEEPWL | 23.743201 | 0.37 | 0.86 |
| RRTGDTEEEEPE | 10.299139 | 0.51 | 0.84 |
| KTTGDETEEEGR | 11.090010 | 0.46 | 0.89 |
| WTEGDEELGEWR | 14.360410 | 0.42 | 0.88 |
| FLYRWLPSRRGG | — | 0.33 | 0.83 |
!!! info “AlphaFold3 Model — Figure 51”
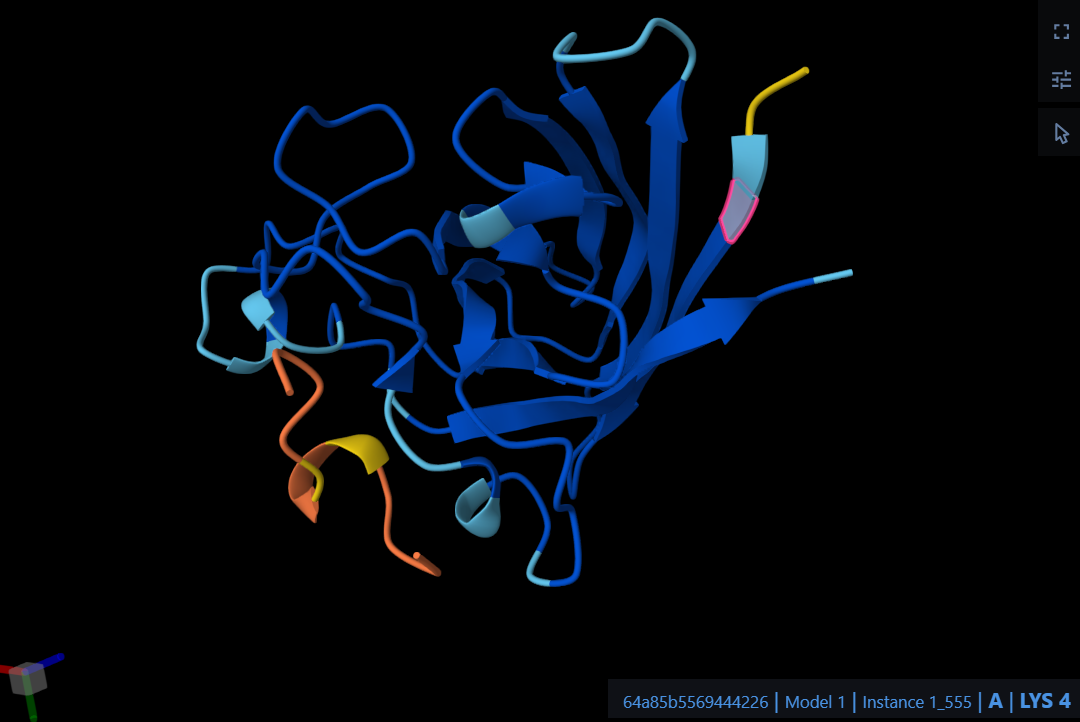
The peptide WTRGDEEEEPWL localizes near the N-terminus where the A4V mutation sits, specifically interacting with the initial residues of the protein chain. It predominantly engages the beta-barrel region of the SOD1 mutant rather than approaching the dimer interface. Structurally, the peptide appears to be entirely surface-bound and does not appear to be partially buried within the protein’s core.
!!! info “AlphaFold3 Model — Figure 51”
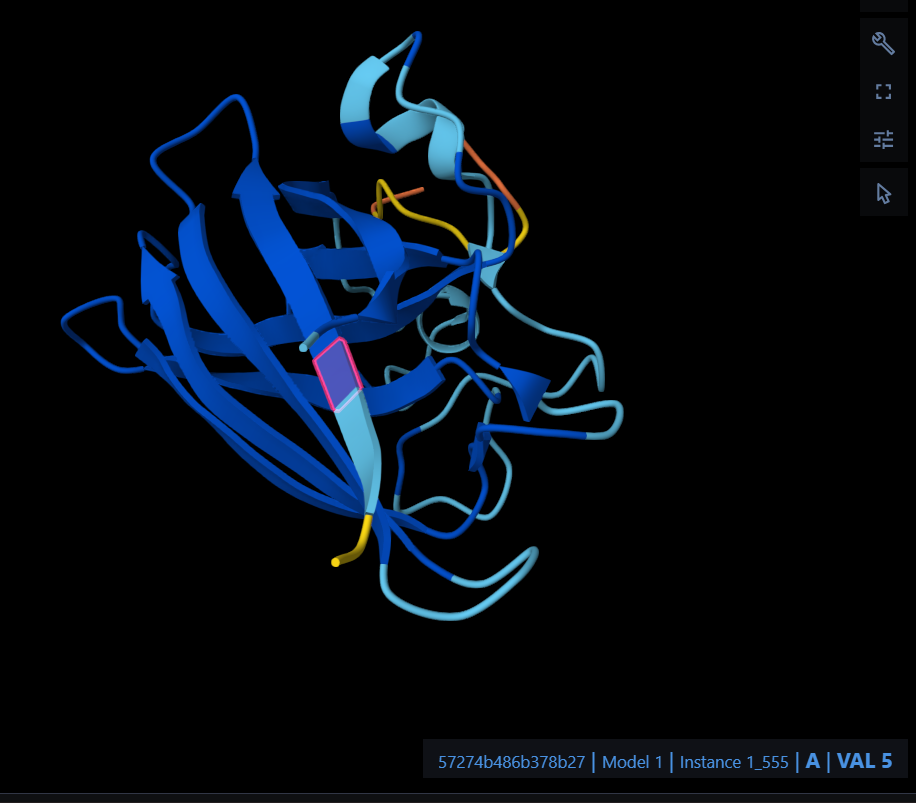
The peptide RRTGDTEEEEPE localizes near the N-terminus at the A4V mutation site, specifically engaging with the initial residues and the adjacent beta-barrel region. It appears to be primarily surface-bound rather than buried within the protein core, and it does not show a significant approach to the dimer interface.
!!! info “AlphaFold3 Model — Figure 51”
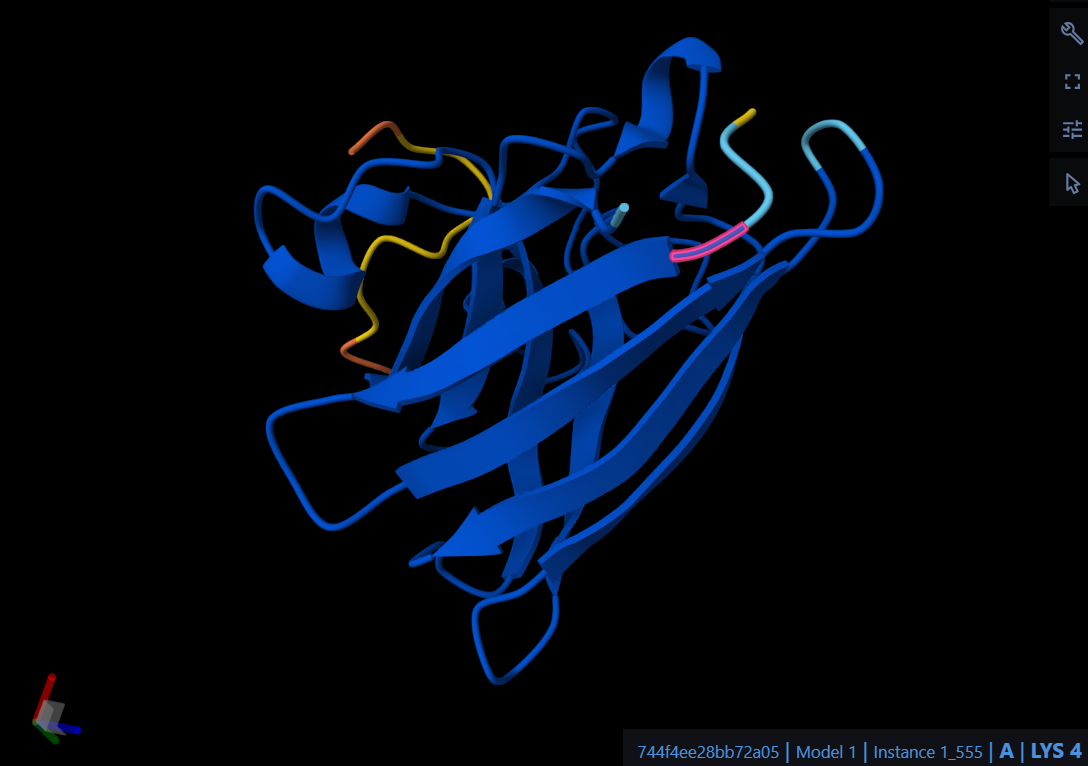
The peptide KTTGDETEEEGR localizes near the N-terminus at the A4V mutation site, engaging the initial loop and the upper beta-barrel region. Similar to the previous candidates, it remains primarily surface-bound and does not appear to penetrate the protein core or approach the dimer interface.
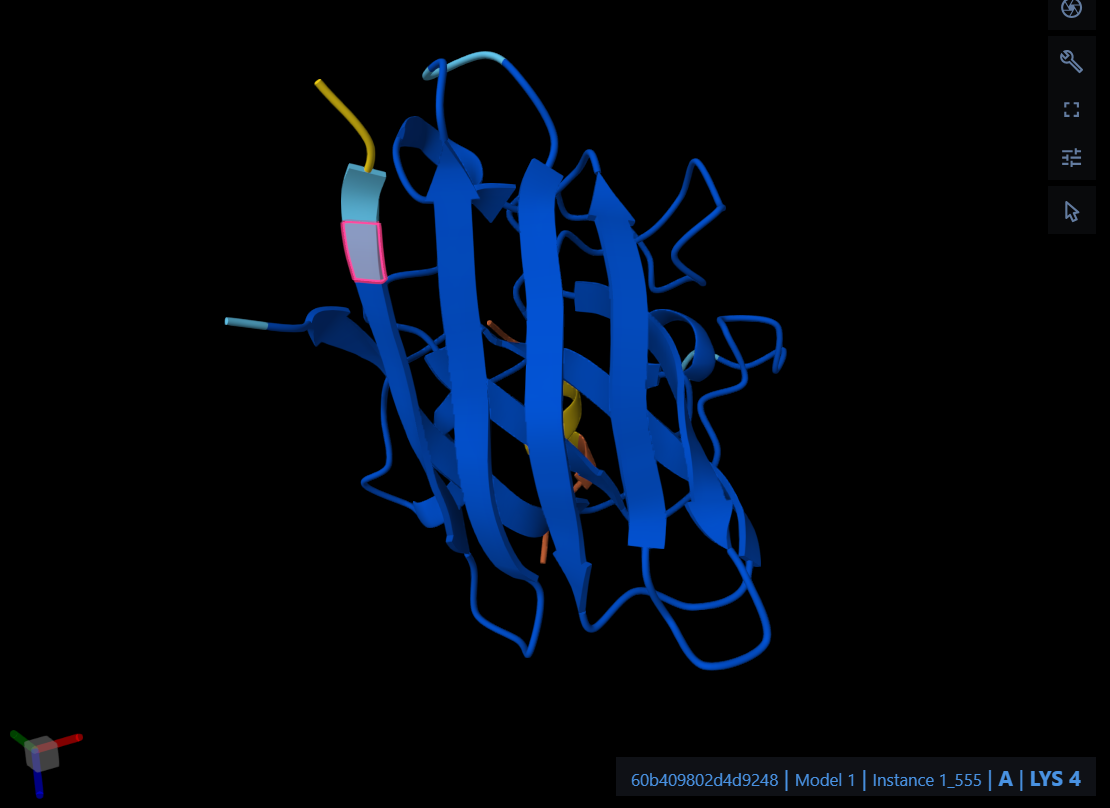
The peptide WTEGDEELGEWR localizes near the N-terminus at the A4V mutation site, engaging the initial loop and the upper beta-barrel region. It appears strictly surface-bound and does not show significant penetration into the protein core or approach toward the dimer interface.
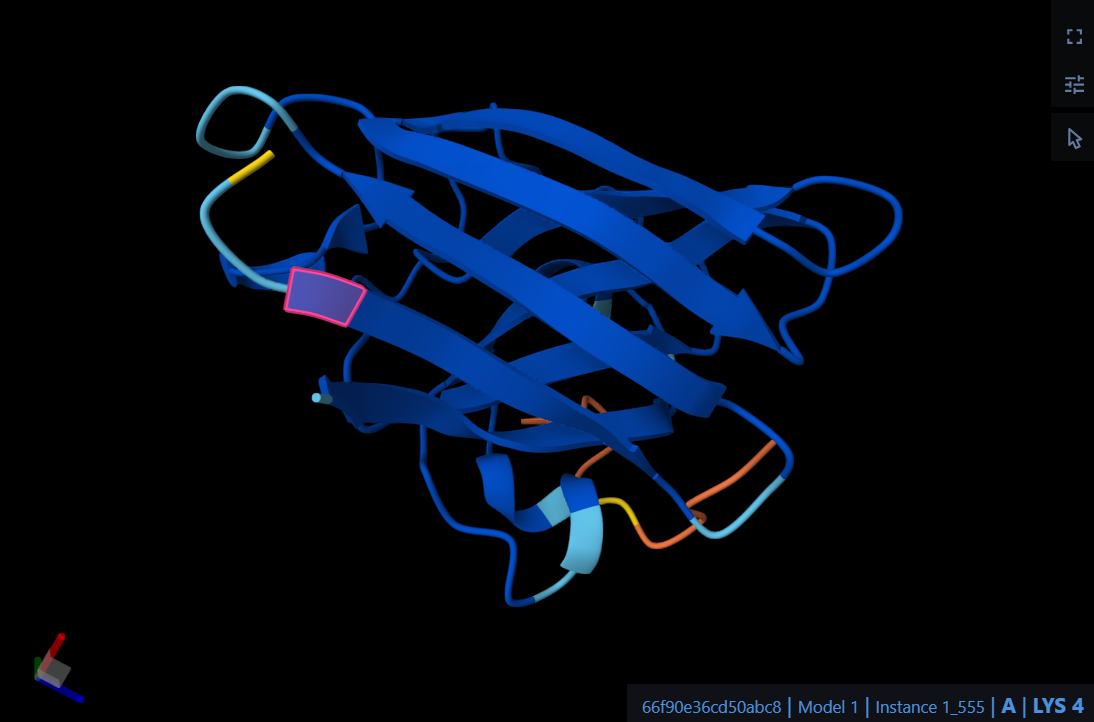
The control peptide FLYRWLPSRRGG localizes near the N-terminus of the SOD1 mutant, specifically interacting with the initial loop region where the A4V mutation is situated. It remains surface-bound, engaging the outer edge of the $\beta$-barrel structure without approaching the dimer interface.
The observed ipTM values for the PepMLM-generated peptides range from 0.37 to 0.51, while the known binder FLYRWLPSRRGG yielded a surprisingly low score of 0.33. Notably, all four generated peptides exceeded the structural confidence of the known binder, with Peptide 2 (RRTGDTEEEEPE) achieving the highest score of 0.51. This suggests that the AI-generated candidates may offer improved structural complementarity to the A4V mutant compared to the original control sequence.
🧩 Summary of ipTM
All PepMLM‑generated peptides outperform the known binder in ipTM.
P2 (0.51) shows the strongest structural confidence.
🔹 Part 3 — Evaluate Properties with PeptiVerse
🧪 PeptiVerse Predictions
| Property | P1 | P2 | P3 | P4 | Control |
|---|---|---|---|---|---|
| Sequence | WTRGDEEEEPWL | RRTGDTEEEEPE | KTTGDETEEEGR | WTEGDEELGEWR | FLYRWLPSRRGG |
| Solubility | 1 | 1 | 1 | 1 | 1 |
| Hemolysis | 0.057 | 0.076 | 0.055 | 0.104 | 0.047 |
| Net Charge (pH 7) | -4.23 | -4.22 | -3.23 | -4.23 | 2.76 |
| Molecular Weight | 1546.6 | 1447.4 | 1351.3 | 1506.5 | 1507.7 |
| ipTM | 0.37 | 0.51 | 0.46 | 0.42 | 0.33 |
| Binding Affinity | 6.092 | 4.824 | 4.823 | 5.708 | 5.968 |
🔍 Comparison with AlphaFold3
Key Insight: Higher ipTM does not correlate with stronger predicted affinity.
Comparing the AlphaFold3 structural observations with PeptiVerse predictions reveals a clear divergence between structural confidence and actual binding strength. Peptides with higher ipTM scores do not show stronger predicted affinity; for instance, Peptide 2 had the highest ipTM (0.51) but the lowest binding affinity (4.824). Conversely, Peptide 1 achieved the highest affinity (6.092) despite a lower ipTM (0.37). Fortunately, none of the strong binders are predicted to be hemolytic or poorly soluble; all generated candidates achieved perfect solubility scores (1.000) and safely low hemolysis probabilities.
⭐ Selected Peptide
**P1 — WTRGDEEEEPWL** -> Best balance of predicted affinity, solubility, and safety.
WTRGDEEEEPWL: It best balances the required properties by achieving the highest predicted binding affinity even outperforming the known control peptide (5.968) while maintaining a remarkably safe therapeutic profile that is highly soluble and non-hemolytic.
🔹 Part 4 — Optimized Peptides with moPPIt
🧬 Differences from PepMLM
The peptide generated by moPPIt differs fundamentally from those generated by PepMLM in the level of design control. While PepMLM unconditionally samples plausible binders conditioned solely on the target sequence, moPPIt utilizes a Multi-Objective-Guided Discrete Flow Matching (MOG-DFM) framework. This allowed me to explicitly constrain the generation process, aiming directly at specific residues (like the A4V mutation site) while simultaneously optimizing for high binding affinity, perfect solubility, and low hemolysis, rather than relying on random sampling.
🧪 Evaluation Before Clinical Advancement
Before advancing this engineered peptide to clinical studies, computational confidence must be validated through rigorous experimental assays. First, developability metrics such as protease resistance, half-life, and immunogenicity must be thoroughly evaluated, potentially exploring non-canonical or cyclic modifications to improve pharmacokinetic stability. Finally, in vitro functional assays (such as cellular binding assays and toxicity screens) must be performed to confirm that the peptide safely and effectively engages the targeted A4V mutant SOD1 in a biological environment
🧪 Part C — Final Project: L‑Protein Mutants
High level summary: The objective of this assignment is to improve the stability and auto-folding of the lysis protein of a MS2-phage. This mechanism is key to the understanding of how phages can potentially solve antibiotic-resistance.
After running the in silico Deep Mutational Scanning (ESM-2) and cross-referencing the predicted LLR Scores with the provided experimental dataset (L-Protein Mutants - Sheet1.csv). The results shows a that evolutionary stability (high LLR scores) does not perfectly correlate with lytic toxicity. Therefore, I prioritized mutants that showed empirical success (Lysis = 1 in the experimental data) and analyzed the biophysical rationale behind their efficacy to disrupt DnaJ dependency and enhance membrane pore formation.
Proposed Sequence Variants and Justifications
L44P (Leucine to Proline at position 44): Proline is a known “helix breaker” that introduces a rigid kink into the peptide backbone. While the AI models might penalize this for structural instability, in the context of a transmembrane lytic pore, this structural kink likely alters the oligomerization geometry, favoring an irreversible disruption of the bacterial lipid bilayer, leading to the successful cell lysis observed in the experimental data.
I46F (Isoleucine to Phenylalanine at position 46): Justification: Phenylalanine is a bulky, aromatic amino acid. Substituting a smaller aliphatic side chain (Isoleucine) with a bulky aromatic one increases the overall hydrophobicity and bulkiness within the membrane core. This enhances the anchoring strength of the L-protein in the E. coli inner membrane, stabilizing the lytic pore complex.
R18I (Arginine to Isoleucine at position 18): Justification: The soluble domain relies on positively charged residues (like Arginine) to interact with the host chaperone DnaJ. Mutating Arginine to Isoleucine replaces a highly charged, hydrophilic residue with a purely hydrophobic one. This effectively breaks the electrostatic interaction with DnaJ, allowing the lysis protein to evade the host’s resistance mechanism and auto-fold.
E25V (Glutamate to Valine at position 25): Justification: Similar to the previous rationale, Glutamate is a negatively charged amino acid that participates in polar contacts. Swapping it for Valine (hydrophobic and uncharged) disrupts the native binding interface with bacterial chaperones. The experimental data confirms that neutralizing these charges in the soluble domain leads to successful lysis (Lysis = 1), proving chaperone independence.
R30L (Arginine to Leucine at position 30): Justification: This mutation reinforces the pattern discovered during the data analysis: eliminating positively charged Arginines (R18, R30) in the soluble domain consistently yields functional, lytic phages. Replacing Arginine 30 with Leucine reduces the solubility of the N-terminal tail, potentially accelerating its collapse and auto-folding without needing E. coli’s DnaJ assistance.