Week 6 HW: Genetic Circuits Part I
Part A: Assignment: DNA Assembly
1. What are some components in the Phusion High-Fidelity PCR Master Mix and what is their purpose? Based on standard molecular biology principles, a High-Fidelity PCR Master Mix typically contains
- Phusion DNA Polymerase: A highly processive, proofreading polymerase responsible for synthesizing the new DNA strands accurately.
- Deoxynucleotide Triphosphates (dNTPs): The A, T, C, and G building blocks that the polymerase uses to build the new DNA strand.
- Reaction Buffer: Maintains the optimal pH and salt conditions for the enzyme.
- Magnesium Chloride: A crucial cofactor required for DNA polymerase activity.
- Specific template DNA, primers, and nuclease-free water.
2. What are some factors that determine primer annealing temperature during PCR?
- The core binding region should be 18-22 base pairs.GC
- The annealing temperature is typically set about 2-5°C below the lower melting temperature
- The primer binding region should ideally consist of 40-60% Guanine/Cytosine bases.
- The presence of 1-2 G or C bases at the 3’ end promotes specific binding, though having more than 3 G/C bases in the final 5 nucleotides should be avoided.
- The absence of strong hairpins or dimers (aiming for a Gibbs free energy above -10 kcal) ensures the primer binds to the template rather than itself.
3. There are two methods from this class that create linear fragments of DNA: PCR, and restriction enzyme digests. Compare and contrast these two methods, both in terms of protocol as well as when one may be preferable to use over the other.
PCR (Polymerase Chain Reaction), uses synthetic oligonucleotides (primers) and polymerase to amplify specific target sequences from a template, it is recommended to use it when you need to introduce intentional mutations (like the amilCP color changes), or when you need a massive quantity of a very specific sub-region of a larger template.The protocol requires thermal cycling to denature DNA, anneal primers, and extend new strands
Restriction Enzyme (RE) Digest, uses proteins to recognize and cleave specific DNA sequences. The document mentions this in the context of creating a “Linear vector (REase digested)” and using DpnI to digest methylated parental DNA. When to use it? preferable for quickly linearizing an entire circular plasmid , diagnosing assembly success, or destroying background template (like the DpnI step). It’s protocol involves incubating the DNA with the specific enzyme at its optimal temperature for a set time (30-60 minutes).
4. How can you ensure that the DNA sequences that you have digested and PCR-ed will be appropriate for Gibson cloning? To ensure your DNA sequences are ready for Gibson assembly, we must design fragments with 15-20 bp complementary overlapping ends and confirm they have the correct 5’ to 3’ orientation to cover the entire target region. Additionally we also need to perform a DpnI digest to eliminate the unmutated parental template and prevent background colonies. Finally, verify the size of your fragments by running a diagnostic agarose gel , and measure the purified DNA concentration to ensure it is sufficient (above ~30 ug/mL) to achieve the recommended 2:1 insert-to-vector molar ratio.
5. How does the plasmid DNA enter the E. coli cells during transformation? Plasmid DNA enters the bacterial cells through a process of diffusion. To make this possible, the cells must be shocked using either a sudden temperature change (heat shock) or a electroporation. This shock causes the bacterial cell membrane to open up and generate temporary pores. Once the plasmid diffuses inside, the cells are incubated in a nutrient-rich SOC medium to recover and close their membranes before plating.
6. Describe another assembly method in detail (such as Golden Gate Assembly) Golden Gate Assembly is a highly efficient cloning method that relies on Type IIS restriction enzymes (such as BsaI or BsmBI) and T4 DNA Ligase. Unlike traditional restriction enzymes, Type IIS enzymes bind to a specific recognition sequence but cut the DNA several base pairs completely outside of that site.
a. Explain the other method in 5 - 7 sentences plus diagrams (either handmade or online)
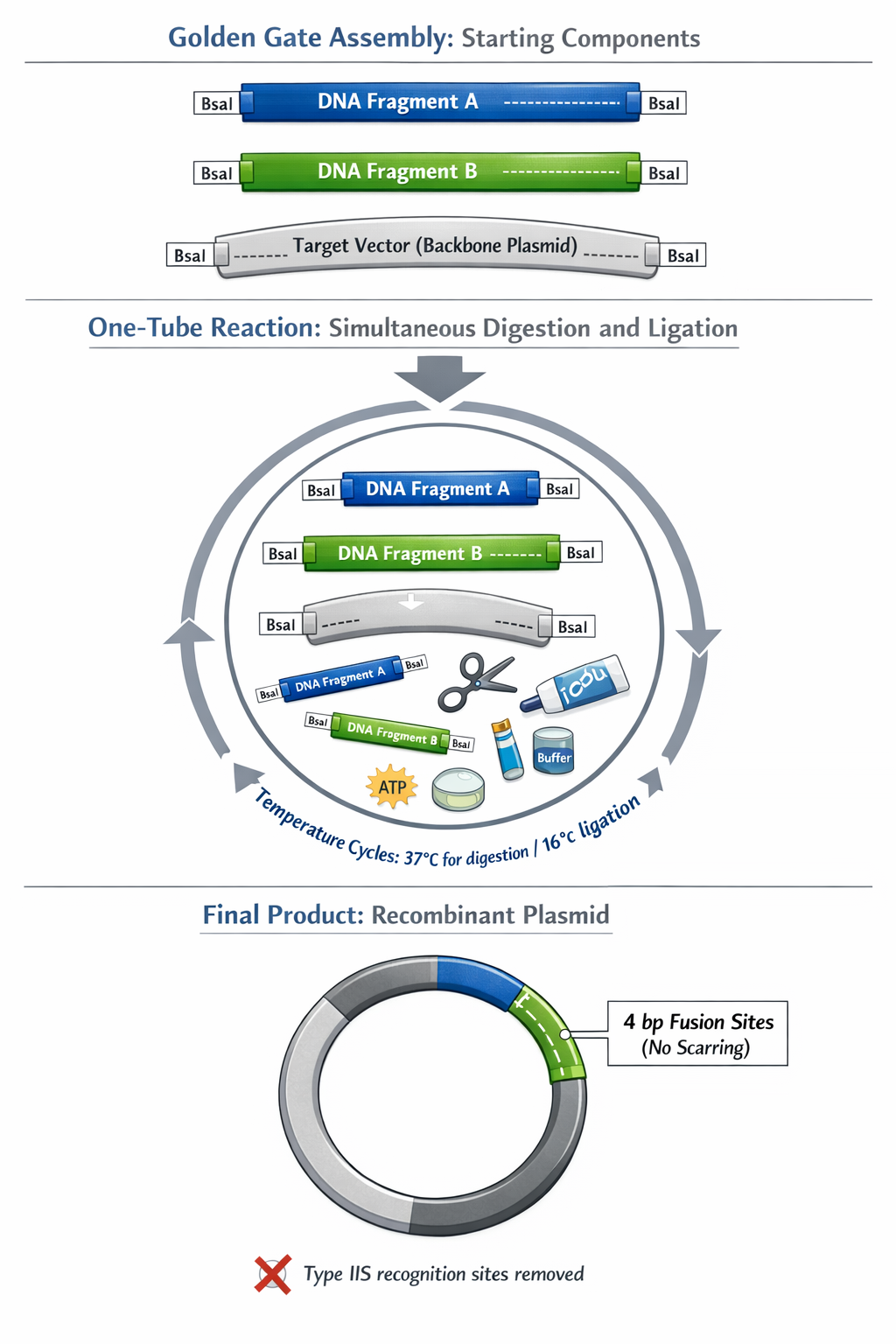
Import Sequences: Upload the DNA sequences of your backbone and all inserts into your Benchling workspace.
Identify Enzymes: Ensure your backbone has the appropriate Type IIS recognition sites (e.g., BsaI) flanking the insertion region.
Design Overhangs: Modify the ends of your insert sequences (usually via PCR primer design within Benchling) to include the Type IIS recognition site followed by the specific 4-bp fusion sites that match the backbone and adjacent fragments.
Simulate Assembly: Click the “Assembly” tool in Benchling, select “Golden Gate,” and define your specific Type IIS enzyme.
Set Fragments: Select your backbone and inserts in the correct order. Benchling will automatically detect the matching 4-bp overhangs, simulate the digestion/ligation, and generate the final scarless plasmid map for you to save.
b. Model this assembly method with Benchling or Asimov Kernel!
Part A: Assignment: DNA Assembly
- Create a Repository for your work
- Create a blank Notebook entry to document the homework and save it to that Repository
Interactive Notebook: View my complete design on Asimov Kernel
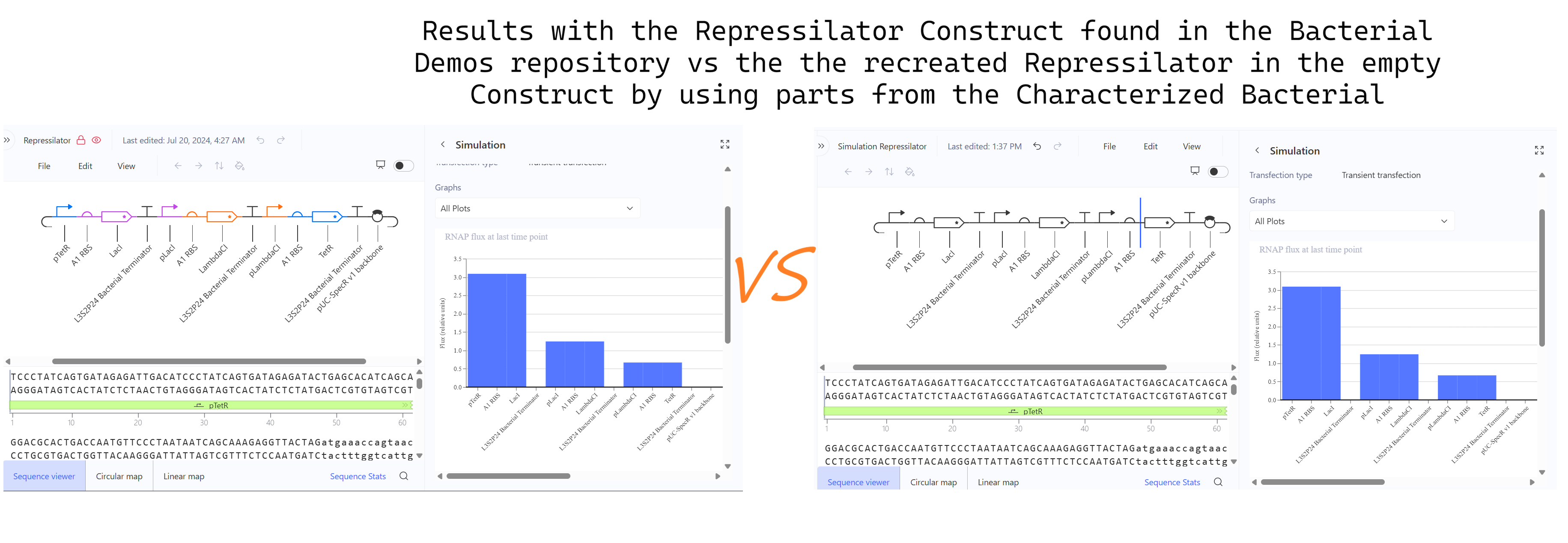
3. Create a blank Construct and save it to your Repository a. Recreate the Repressilator in that empty Construct by using parts from the Characterized Bacterial Parts repository b. Search the parts using the Search function in the right menu c. Drag and drop the parts into the Construct d. Confirm it works as expected by running the Simulator (“play” button) and compare your results with the Repressilator Construct found in the Bacterial Demos repository
4. Build three of your own Constructs using the parts in the Characterized Bacterials Parts Repo
**First Attempt:** In my first design, I used generic RBS_strong parts and standard repressor genes without degradation tags, I followed the Promoter → RBS → CDS → Terminator structure.
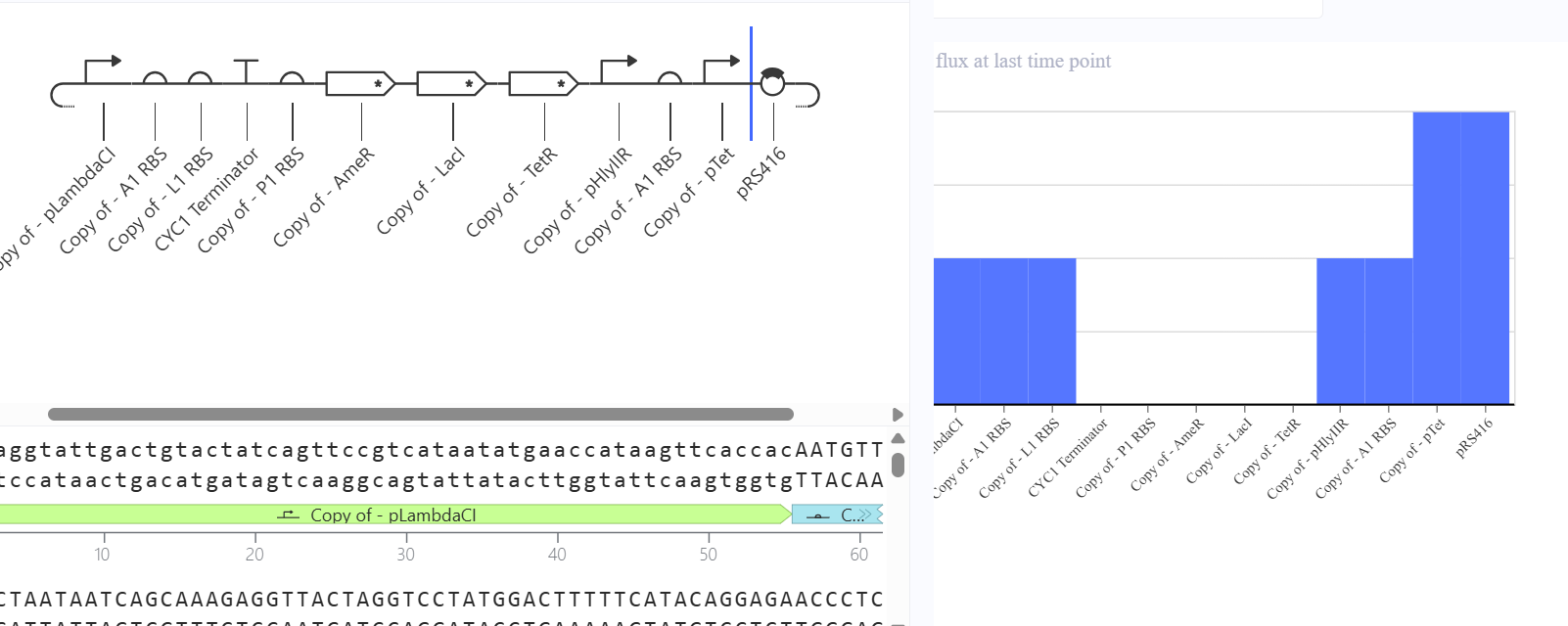
Second Attempt: Trying to fix the gridlock, I swapped the parts for generic RBS components.
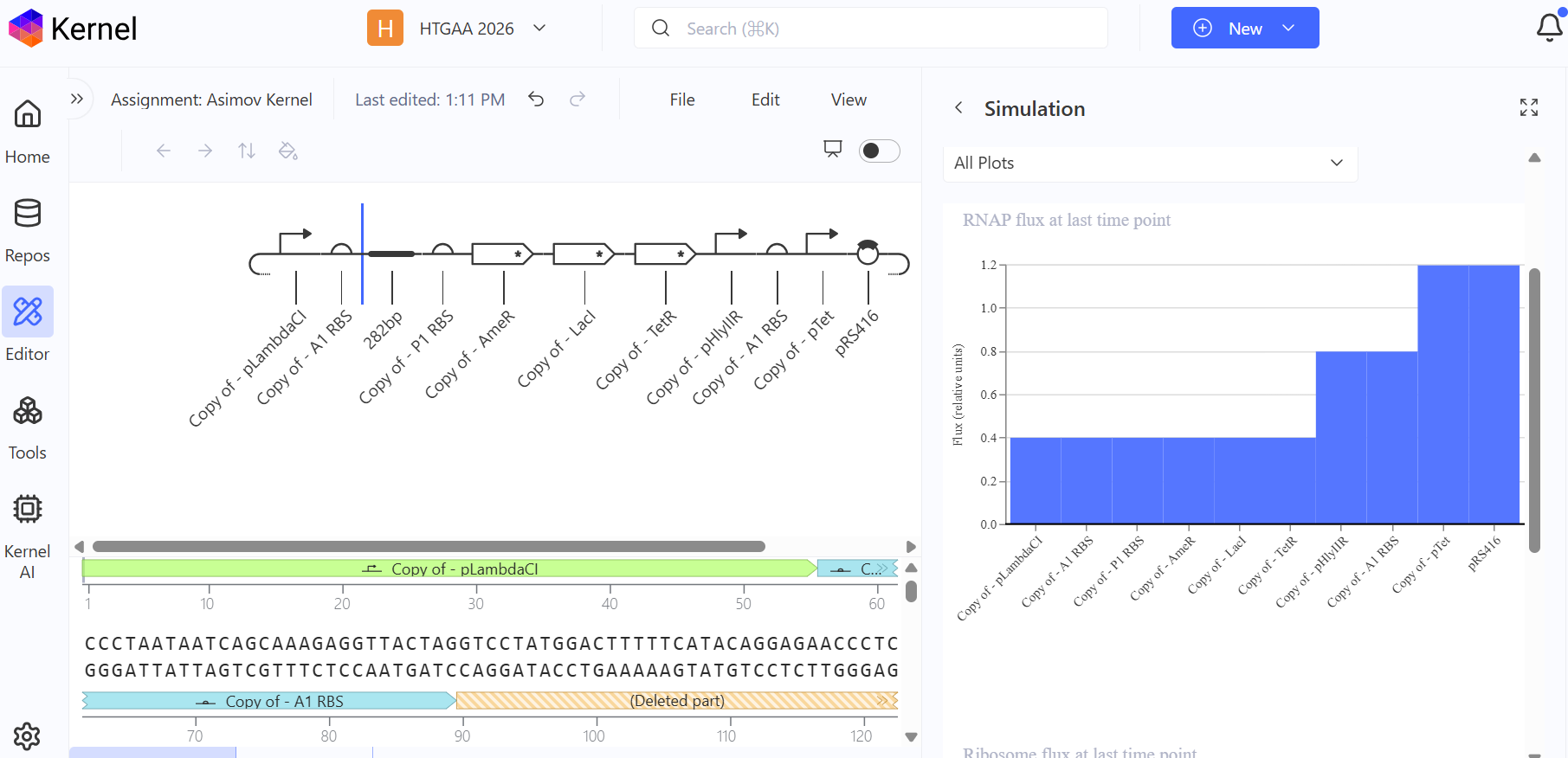
Third Attempt (The Final Success): Finally, I debugged the construct by looking at the reference demo and updating my parts to match the specific kinetics required for oscillation.
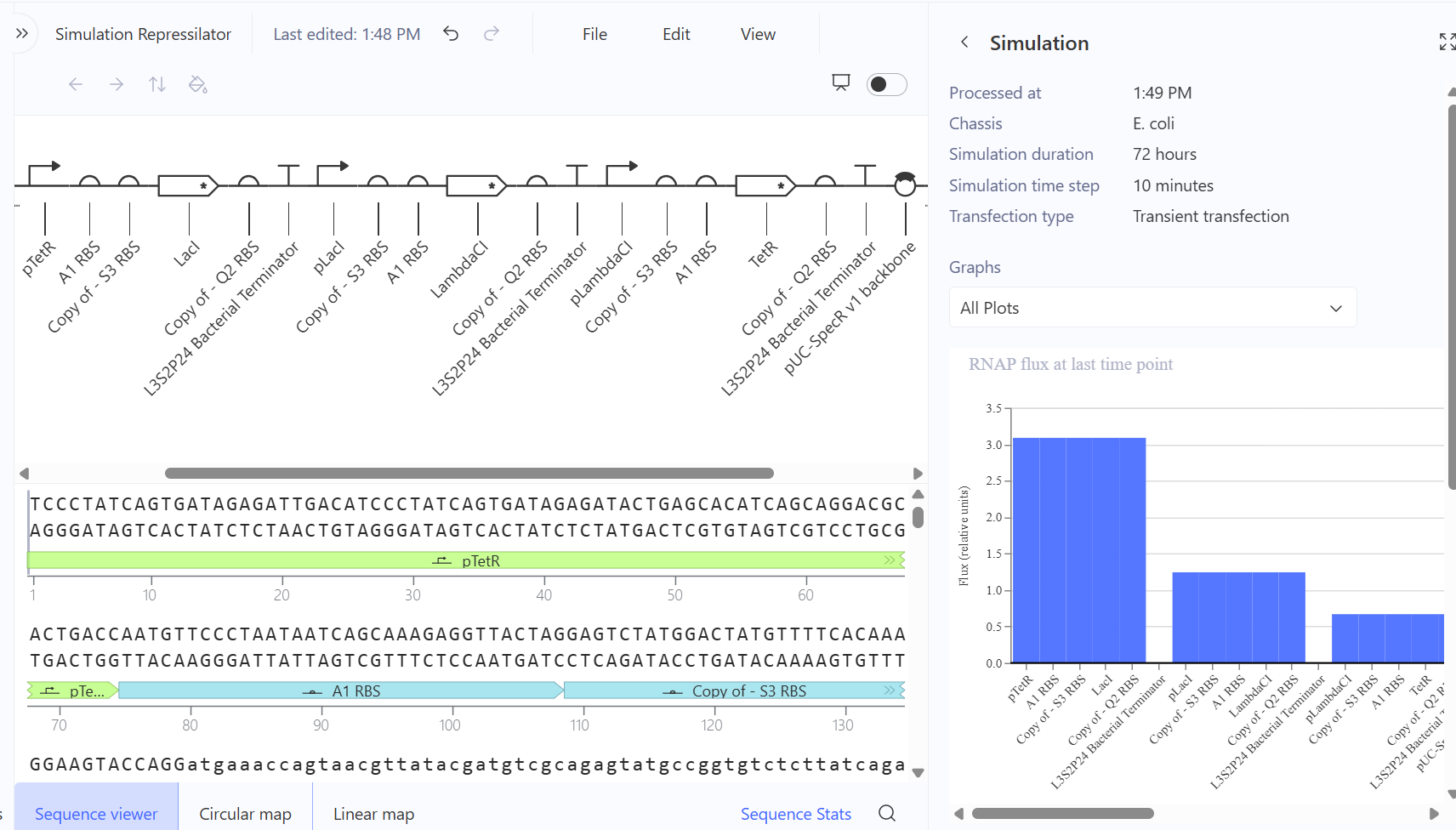
Use of Chatgpt to create the diagram