Biotechnologist with hands-on experience in environmental and agro-biotech projects.
I have worked on initiatives connecting biotechnology with sustainability, including bioeconomy applications and community-based environmental research in Latin America. My background combines wet lab techniques, project coordination, and policy-oriented research, with a strong focus on translating scientific knowledge into practical, real-world solutions.
I am also actively involved in building the Synthetic Biology Network for Latin America, promoting collaboration, policy development, and opportunities in SynBio across the region.
This week explores the read–write–edit toolkit: sequencing and synthesis workflows, restriction digests and gel electrophoresis, and early genome-editing frameworks.
Molecular weight calculation of eGFP via adjacent charge state analysis of LC-MS data and assessment of experimental measurement accuracy. The work includes the identification and quantification of Lysine and Arginine residues through bioinformatics-based sequence analysis. This approach integrates analytical chemistry and computational tools to achieve precise protein characterization.
Analysis of protein structure and sequence through 3D visualization and machine learning models such as ESMFold and ProteinMPNN. Implementation of mutational scanning and latent space analysis to explore protein design space and optimization. Development of computational strategies for bacteriophage engineering aimed at enhancing stability and functional outcomes.
In silico design of therapeutic peptides targeting mutant SOD1 using advanced generative models such as PepMLM and moPPIt. Structural validation and property prediction were conducted through AlphaFold3 and PeptiVerse to assess binding affinity and solubility. The work focused on multi-objective optimization to enhance the stability and folding of the MS2 phage lysis protein.
Exploration of molecular biology techniques for DNA assembly, focusing on PCR and Gibson Assembly protocols for genetic construct preparation. Implementation of the Asimov Kernel platform to design, simulate, and validate complex genetic circuits such as the repressilator. The work involves comparative analysis of assembly methods and the characterization of bacterial parts to engineer functional biological systems.
Investigation of neuromorphic genetic circuits and the implementation of Intracellular Artificial Neural Networks (IANNs) to perform "perceptron"-like computations. The work includes the design of multi-layer genetic perceptrons using endoribonucleases and an exploration of fungal materials as a chassis for synthetic biology. Additionally, it covers the initial DNA design and synthesis orders for the individual final project.
Study of cell-free protein synthesis (CFPS) systems for protein expression outside of cellular boundaries, emphasizing flexibility and control over experimental variables. Implementation of synthetic minimal cells for biosensing applications and the development of integrated biological sensors for architecture, textiles, and space exploration. The work includes optimizing energy regeneration and designing DNA constructs for specialized protein production in cell-free environments.
Characterization of recombinant eGFP protein using liquid chromatography and mass spectrometry to determine molecular weight, charge state distribution, and primary sequence via peptide mapping. Comparison of native versus denatured protein conformations and identification of oligomeric states in high-molecular-weight complexes through charge detection mass spectrometry.
Exploration of cloud laboratories for decentralized bioproduction and collaborative bioart experiments. The work focuses on optimizing cell-free protein synthesis (CFPS) through master mix design and biophysical characterization of fluorescent proteins using automated remote platforms.
Concept: I want to develop a small open-source, in-silico tool that explores synthetic biology “parts” (such as enzymes or simple pathways) with potential applications in environmental sustainability and circular economy.
Goal: The tool will collect and organize parts from open biological databases, classify them by environmental use case, and provide a simple interface to explore them. The goal is not to experimentally build organisms, but to support early-stage, responsible ideation and design in environmental synthetic biology.
2. Governance Goals
Main Goal: Ensure that this open, AI-assisted synthetic biology design tool contributes to a more equitable and inclusive future of biotechnology, particularly by lowering barriers to access for researchers and students in low- and middle-income countries.
Secondary Objectives:
Equitable Access: Reduce the gap in access to synthetic biology resources by making the tool fully open-source and low-cost.
Capacity Building: Support learning and innovation in contexts where access to wet-lab facilities is limited.
Responsible Use: Encourage users to consider local environmental and social contexts.
3. Governance Actions & Analysis
Action 1: Open-Source & Low-Resource Optimization
Purpose: Currently, many tools require paid licenses or high resources. I propose designing this tool to be runnable on basic laptops with minimal dependencies.
Risks: It could fail to reach users due to lack of visibility, or create frustration if users can design but not build (wet-lab gap).
Action 2: Targeted Incentives for Local Problems
Purpose: Funding often prioritizes global trends over local needs. I propose incentives/grants for projects using the tool to address local problems (e.g., agricultural waste).
Risks: Funding might favor projects that look good on paper but lack feasibility.
Action 3: Community-Led Learning Nodes
Purpose: SynBio training is concentrated in wealthy nations. I propose creating community-led “nodes” for peer mentorship and contextualized use of the tool.
Risks: Nodes may struggle to sustain activity without stable funding.
4. Evaluation Matrix
Does the option:
Action 1 (Open Source)
Action 2 (Incentives)
Action 3 (Nodes)
Enhance Biosecurity
• By preventing incidents
2
2
3
• By helping respond
1
2
n/a
Foster Lab Safety
• By preventing incidents
2
2
3
• By helping respond
n/a
2
n/a
Protect the Environment
• By preventing incidents
1
2
1
• By helping respond
3
2
3
Other Considerations
• Minimizing costs/burdens
1
2
2
• Feasibility
1
2
2
• Not impede research
1
1
2
• Promote constructive apps
1
1
1
(Scoring: 1 = Best, 3 = Least effective)
5. Conclusion & Recommendations
Priority: It is recommended to prioritize a combination of Action 1 and Action 3.
Why: Action 1 removes the technical barrier, while Action 3 ensures local human capital is trained to use the tools responsibly. Without mentorship, software access alone does not guarantee sovereign innovation.
Sustainability: Cloud-first software reduces infrastructure costs, and regional nodes act as knowledge multipliers.
6. Ethical Concerns & Proposed Governance
Emerging Issues:
Biopiracy Risk: Digitizing sequences from diverse biomes might allow high-resource institutions to commercialize products without benefiting source communities.
Info-Hazards: Democratizing design tools increases the risk of accidentally generating hazardous designs before physical synthesis.
Proposed Solutions:
Ethical Co-design: Funders should require local participation in agenda-setting, not just implementation.
Capacity as Governance: International initiatives must fund local training and safety culture, not just technical outputs.
Benefit-Sharing: Incorporate licenses that link tool access to the Nagoya Protocol, ensuring commercial derivatives contribute to a local sustainability fund.
Simulate Restriction Enzyme Digestion with the following Enzymes: EcoRI, HindIII, BamHI, KpnI, EcoRV, SacI, SalI.
Create a pattern/image in the style of Paul Vanouse’s Latent Figure Protocol artworks.
Part 3: DNA Design Challenge
In recitation, we discussed that you will pick a protein for your homework that you find interesting. Which protein have you chosen and why? Using one of the tools described in recitation (NCBI, UniProt, google), obtain the protein sequence for the protein you chose.
yo use la proteina SIRT1 por su potencial en …. y su relevancia en estudio antievenjecimiento y su relacion con el desarollo de cancer???. Obtuve su secuencia por medio de UniProt
1.2. Reverse Translate: Protein (amino acid) sequence to DNA (nucleotide) sequence.
The Central Dogma discussed in class and recitation describes the process in which DNA sequence becomes transcribed and translated into protein. The Central Dogma gives us the framework to work backwards from a given protein sequence and infer the DNA sequence that the protein is derived from. Using one of the tools discussed in class, NCBI or online tools (google “reverse translation tools”), determine the nucleotide sequence that corresponds to the protein sequence you chose above.
Using Benchling’s reverse translation tool, I converted the SIRT1 protein sequence into its corresponding coding DNA sequence (CDS) using the standard genetic code. The resulting DNA sequence is:
Using the Benchling's reverse translation tool, I converted the SIRT1 protein sequence into its corresponding coding DNA sequence (CDS) using the standard genetic code. The resulting DNA sequence is:
```text
ATGGCGGACGAGGCGGCGCTGGCGCTGCAGCCGGGCGGGAGCCCGAGCGCGGCGGGCGACCGCGAGGCGGCGAGCAGCCCGGCGGGCGAGCCGCTGCGCAAGCGCCCGCGCCGCGACGGCGCCGGCCTGGAGCGCAGCCCCGGCGAGCCGGGCGGCGCCGCCCCGGAGCGCGAGGTGCCGGCGGCGGCGCGCGGCTGCCCGGCGGCGGCGGCGGCGCTGTGGCGCGAGGCGGAGGCGGAGGCGGCGGCGGCGGGCGGCGAGCAGGAAGCGCAGGCGACGGCGGCGGCGGAGGGGGAGGACAACGGGCCGGGGCTGCAGGGCCCGAGCCGCGAGCCGCCGCTGGCGGACAACCTGTACGACGAGGACGACGACGACGAGGGCGAGGAGGAGGAGGCCGCGGCGGCGGCGATCGGCTACCGGCACAACCTGCTGTTCGGCGACGAGATCATCACCAACGGCTTCCACTCCTGCGAGAGCGACGAGGAGGACCGAGCCTCCCACGCCAGCAGCAGCGACTGGACCCCGAGGCCGAGGATCGGCCCGTACACCTTCGTGCAGCAGCATCTGATGATCGGGACCGACCCCAGGACGATACTGAAGGACCTGCTGCCGGAGACTATCCCGCCGCCGGAGCTGGACGACATGACTCTGTGGCAGATCGTGATCAATATCCTGAGCGAGCCGCCGAAACGGAAGAAGAGGAAAGACATCAACACTATTGAGGATGCGGTGAAACTGCTGCAGGAATGTAAAAAAATCATCGTCCTGACAGGTGCTGGAGTTTCTGTAAGTTGCGGCATTCCTGACTTTAGGTCACGAGACGGGATTTACGCGAGACTTGCTGTGGACTTTCCAGACCTTCCGGATCCGCAAGCAATGTTTGATATTGAATACTTCAGAAAGGACCCGCGCCCCTTCTTCAAGTTTGCAAAGGAAATATACCCAGGCCAGTTCCAACCAAGTTTGTGCCACAAGTTTATAGCCCTCTCAGACAAGGAAGGAAAACTGCTGAGAAATTATACCCAGAATATCGACACACTGGAGCAGGTTGCTGGGATCCAAAGAATCATTCAGTGTCATGGAAGTTTTGCCACTGCATCTTGCCTCATTTGCAAATACAAGGTGGATTGTGAAGCCGTCAGAGGGGACATTTTTAACCAAGTGGTCCCCCGGTGCCCCCGCTGCCCTGCCGATGAGCCGCTGGCAATAATGAAGCCAGAGATTGTGTTCTTTGGGGAAAACCTCCCTGAACAATTTCACAGAGCAATGAAGTATGATAAAGATGAAGTGGATCTACTGATAGTTATTGGCAGCAGCCTGAAAGTTCGTCCAGTGGCCTTAATACCAAGCAGCATTCCCCATGAAGTGCCTCAGATCCTGATCAATAGAGAGCCATTGCCACATCTCCACTTTGATGTGGAACTGCTGGGGGACTGTGACGTTATAATCAATGAACTGTGCCATAGACTGGGAGGGGAGTATGCAAAGCTGTGTTGCAATCCCGTAAAGTTATCCGAGATCACTGAAAAGCCCCCCCGGACTCAGAAGGAGCTGGCATATCTGTCAGAGCTGCCGCCTCCCACCCTCCACGTGTCCGAGGATAGTAGCAGCCCAGAGCGCACCTCCCCTCCGGACAGCAGTGTTATCGTCACACTGCTGGACCAGGCAGCTAAGAGCAATGATGATCTGGATGTGTCAGAGAGCAAAGGATGCATGGAGGAGAAGCCCCAGGAGGTCCAAACGTCGAGGAACGTGGAGAGCATCGCTGAGCAGATGGAGAATCCGGATTTGAAAAATGTTGGGAGTAGCACGGGGGAGAAGAATGAAAGAACTTCAGTCGCTGGGACGGTGAGAAAGTGCTGGCCAAACCGGGTTGCGAAAGAACAAATATCTCGCCGATTGGACGGGAATCAGTACCTGTTCCTGCCACCAAACCGATACATTTTCCATGGTGCTGAAGTTTACTCGGATGATTCAGAGGACGACGTGCTCTCTTCCTCCTCTTGTGGCTCCAATTCAGACTCAGGCACTTGCCAGTCACCCAGTCTAGAGGAACCTATGGAGGATGAGTCCGAAATAGAAGAGTTTTACAATGGGCTGGAGGATGAGCCTGATGTCCCTGAGCGTGCTGGGGGTGCAGGATTTGGAACAGATGGAGATGACCAAGAAGCCATAAATGAGGCAATCTCAGTAAAACAGGAAGTGACAGATATGAACTATCCGAGCAACAAATCA
1.3. Codon optimization.
Once a nucleotide sequence of your protein is determined, you need to codon optimize your sequence. You may, once again, utilize google for a “codon optimization tool”. In your own words, describe why you need to optimize codon usage. Which organism have you chosen to optimize the codon sequence for and why?
Codon optimization is required because the genetic code is degenerate-multiple codons encode the same amino acid, and different organisms have distinct codon usage biases. When expressing a human gene like SIRT1 in a foreign host such as E. coli, the presence of human-rare codons (which correspond to low-abundance tRNAs in E. coli) can lead to translational pausing, premature termination, mRNA instability, or protein misfolding. By replacing those rare codons with synonymous codons preferred by the expression host, I can dramatically increase protein yield, solubility, and functional expression without altering the amino acid sequence.
Organism chosen: Escherichia coli (E. coli)
I chose to optimize SIRT1 for E. coli for several reasons:
Cell-free system compatibility: The Week 11 cell-free protein synthesis system uses E. coli BL21(DE3) Star lysate, which is naturally optimized for E. coli codon preferences.
Speed and cost: E. coli is the fastest and most cost-effective microbial host for protein production.
Well-characterized codon bias: E. coli K-12 codon usage tables are extensively documented.
Twist synthesis standard: Twist Bioscience’s clonal gene service is optimized for E. coli expression vectors.
Downstream applications: Optimized genes work efficiently in both in vivo (E. coli transformation) and in vitro (cell-free lysate) platforms.
What technologies could be used to produce this protein from your DNA? Describe in your words the DNA sequence can be transcribed and translated into your protein. You may describe either cell-dependent or cell-free methods, or both.
Cell-dependent method (in vivo): The optimized SIRT1 gene would be cloned into an expression vector (e.g., pET-28a) containing a T7 promoter, ribosome binding site (RBS), and terminator. This plasmid is transformed into E. coli BL21(DE3), where IPTG induction drives T7 RNA polymerase to transcribe the gene into mRNA, which is then translated by bacterial ribosomes into SIRT1 protein. The protein can be purified using affinity tags (e.g., 6xHis tag).
Cell-free method (in vitro):
The DNA template (linear or plasmid) is added directly to an E. coli lysate-based cell-free system (as in Week 11). The lysate provides all necessary machinery — RNA polymerase, ribosomes, tRNAs, energy regeneration systems, and amino acids — to perform coupled transcription and translation in a test tube within hours, without living cells. This method is faster, allows precise control over reaction conditions, and avoids toxicity issues.
1.5. [Optional] How does it work in nature/biological systems?
A single gene can produce multiple protein variants through a process called alternative splicing. In higher eukaryotes like humans, the initial pre-mRNA transcript contains both exons (coding regions) and introns (non-coding regions). During splicing, different combinations of exons can be joined together, producing multiple mature mRNA isoforms from the same gene. Each isoform translates into a different protein variant with potentially distinct functions, localization, or regulatory properties.
For SIRT1, alternative splicing generates at least five known isoforms:
Isoform
Exon combination
Protein length
Functional difference
Isoform 1 (canonical)
Exons 1-9
747 aa
Full-length, nuclear localization
Isoform 2
Missing exon 3
~650 aa
Truncated, altered deacetylase activity
Isoform 3
Alternative exon 5
~500 aa
Cytoplasmic, different substrate specificity
Isoform 4
Missing exons 3 & 5
~400 aa
Catalytically inactive, dominant negative
Isoform 5
Alternative 3’ end
685 aa
Different C-terminus, altered stability
Central Dogma: DNA → RNA → Protein alignment
Below is the alignment for the first 30 amino acids of SIRT1, showing the flow of information from DNA to RNA to protein.
Color legend:
🔵 Blue: DNA template strand (coding sequence)
🟢 Green: RNA transcript (T → U)
🟡 Yellow: Protein (amino acids from codons)
Complete alignment for the first 30 amino acids in table format:
DNA (5’ → 3')
RNA (5’ → 3')
Codon
Amino Acid (3-letter)
Amino Acid (1-letter)
ATG
AUG
AUG
Methionine
M
GCG
GCG
GCG
Alanine
A
GAC
GAC
GAC
Aspartic acid
D
GAG
GAG
GAG
Glutamic acid
E
GCA
GCA
GCA
Alanine
A
GCA
GCA
GCA
Alanine
A
CTG
CUG
CUG
Leucine
L
GCA
GCA
GCA
Alanine
A
CTG
CUG
CUG
Leucine
L
CAG
CAG
CAG
Glutamine
Q
CCG
CCG
CCG
Proline
P
GGC
GGC
GGC
Glycine
G
GGC
GGC
GGC
Glycine
G
AGC
AGC
AGC
Serine
S
CCG
CCG
CCG
Proline
P
AGC
AGC
AGC
Serine
S
GCA
GCA
GCA
Alanine
A
GCC
GCC
GCC
Alanine
A
GGT
GGU
GGU
Glycine
G
GAT
GAU
GAU
Aspartic acid
D
CGT
CGU
CGU
Arginine
R
GAA
GAA
GAA
Glutamic acid
E
GCA
GCA
GCA
Alanine
A
GCA
GCA
GCA
Alanine
A
TCT
UCU
UCU
Serine
S
TCT
UCU
UCU
Serine
S
CCG
CCG
CCG
Proline
P
GCA
GCA
GCA
Alanine
A
GGT
GGU
GGU
Glycine
G
GAA
GAA
GAA
Glutamic acid
E
Key observations from the alignment:
Feature
Description
Transcription
All “T” (thymine) in DNA are replaced by “U” (uracil) in RNA
Translation
Each set of 3 nucleotides (codon) specifies one amino acid
Reading frame
The sequence is read continuously from the start codon (ATG/AUG/Met)
Genetic code
Follows the standard genetic code (same for humans and E. coli for most codons)
Start codon
ATG (DNA) / AUG (RNA) codes for Methionine (M)
Additional notes on SIRT1 biology:
In nature, SIRT1 expression is regulated at multiple levels:
Transcriptional regulation: The SIRT1 promoter contains binding sites for transcription factors like p53, FOXO3a, and HIC1
Alternative splicing: As shown above, multiple isoforms arise from the same gene
Post-transcriptional regulation: miRNAs (e.g., miR-34a, miR-9) bind to the 3’ UTR and repress translation
Post-translational modification: The protein itself can be phosphorylated, SUMOylated, or acetylated
This multi-level regulation allows a single gene to respond to diverse cellular signals (NAD+ levels, oxidative stress, caloric restriction) and produce context-specific functional outcomes.
Week 3 HW: Lab Automation
Python Script for Opentrons Artwork
Exploring the Spiral of Ouro.
Artistic Concept and Motivation
For this week’s assignment, I developed a “Spiral of Ouro.” This design is based on phyllotaxis principles, using the Golden Angle to distribute points across a 96-well plate. This choice reflects the core philosophy of my project: using mathematical and computational frameworks to simplify biological design. By translating natural patterns into a functional robotic protocols.
Technical Implementation
The implementation uses a Python script for the Opentrons liquid handling robot. Instead of manual plotting, I developed an algorithmic approach to calculate Cartesian coordinates for the ninety-six wells. The script utilizes the simulate module to ensure precise movement. This methodology aligns with my goal of creating high-fidelity digital blueprints that can be shared globally, reducing the need for expensive proprietary software and making advanced laboratory automation more accessible to researchers in low-resource settings.
Python Protocol Code
Below is the complete protocol used to generate the spiral. It includes the necessary metadata and the mathematical loop that defines the dispensing pattern.
importmathimportmatplotlib.pyplotaspltimportnumpyasnpfromopentronsimportsimulatemetadata={'protocolName':'Spiral of Ouro - Environmental SynBio Design','author':'Zaira Fabiola Guzman','description':'An algorithmic design for environmental ideation and accessible biotechnology.','apiLevel':'2.13'}# Initial setup of the simulator and canvasprotocol=simulate.get_protocol_api('2.13')plate=protocol.load_labware('corning_96_wellplate_360ul_flat','1')# Parameters for the Spiral of Ouron_points=96golden_angle=137.508*(math.pi/180)scale_factor=3.5x_coords=[]y_coords=[]# Coordinate generation and movement simulationforiinrange(n_points):r=scale_factor*math.sqrt(i)theta=i*golden_anglex=r*math.cos(theta)y=r*math.sin(theta)x_coords.append(x)y_coords.append(y)well=plate.wells()[i]protocol.comment(f"Moving to spiral coordinate {i}: ({x:.2f}, {y:.2f})")# Visualization block for Colabplt.figure(figsize=(7,7))forwellinplate.wells():plt.plot(well.geometry.position.x,well.geometry.position.y,'ko',fillstyle='none',alpha=0.1)plt.scatter(x_coords,y_coords,c=range(n_points),cmap='magma',s=100,edgecolors='white')plt.title("Artistic Visualization: Spiral of Ouro")plt.axis('equal')plt.show()
Post-Lab Questions
Lab Automation and Biological Design
Part 1: Published Paper on Automation Tool
The paper titled “A low-cost, open-source Turbidostat design for in-vivo control experiments in Synthetic Biology” by Guarino et al.(2019) describes the design and implementation of an open-source, highly flexible turbidostat built with 3D printed parts and controlled by an Arduino board. This automation tool continuously monitors the optical density of a bacterial culture using an LED and photodiode circuit, and it autonomously triggers custom 3D-printed syringe or peristaltic pumps to inject fresh media when required.
The authors propose using this automated, modular machine for multicellular control experiments. In this novel application, the automation hardware is extended to connect two different culture chambers separated by a nanoporous membrane. This setup allows two distinct bacterial populations to be physically separated while exchanging sensing molecules, automating the precise maintenance of specific population ratios for synthetic microbial consortia.
Part 2: Final Project Automation Plan
I will automate the translation of the conceptual biological design into an executable liquid-handling script. Currently, a researcher must manually translate a metabolic pathway design into a step-by-step pipetting protocol. I will program my Python tool so that, once the AI and database search finalize the parts for a cell-free system (e.g., specific enzymes for plastic degradation), the software automatically generates and outputs a ready-to-run Opentrons Python script or a JSON payload formatted for Ginkgo Nebula’s cloud laboratory.
Part 2: Final Project Automation Plan
I will automate the translation of the conceptual biological design into an executable liquid-handling script. Currently, a researcher must manually translate a metabolic pathway design into a step-by-step pipetting protocol. I will program my Python tool so that, once the AI and database search finalize the parts for a cell-free system (e.g., specific enzymes for plastic degradation), the software automatically generates and outputs a ready-to-run Opentrons Python script or a JSON payload formatted for Ginkgo Nebula’s cloud laboratory.
importjsonfromopentronsimportprotocol_api# AUTOMATICALLY GENERATED BY BIOCIRCULAR DESIGN TOOL# Target: Ginkgo Nebula / Opentrons OT-2 Cloud Lab# Design: Cell-free PET Degradation Pathwaymetadata={'protocolName':'Automated Cell-Free Assembly','author':'BioCircular Tool Compiler','description':'Automated liquid handling protocol for testing a computationally designed cell-free system.','apiLevel':'2.13'}defrun(protocol:protocol_api.ProtocolContext):# The tool automatically calculated these volumes based on the DB sequence sizereaction_setup={'cell_free_extract':15.0,'enzyme_A_DNA':3.5,'enzyme_B_DNA':4.2,'buffer_solution':7.3}# Labware setupplate=protocol.load_labware('corning_96_wellplate_360ul_flat','1')tuberack=protocol.load_labware('opentrons_24_tuberack','2')pipette=protocol.load_instrument('p300_single_gen2','left')# Automated dispensing steps compiled by the in-silico toolprotocol.comment("Initiating automated cell-free assembly based on digital design.")pipette.transfer(reaction_setup['cell_free_extract'],tuberack['A1'],plate['A1'])pipette.transfer(reaction_setup['enzyme_A_DNA'],tuberack['B1'],plate['A1'])pipette.transfer(reaction_setup['enzyme_B_DNA'],tuberack['C1'],plate['A1'])pipette.transfer(reaction_setup['buffer_solution'],tuberack['D1'],plate['A1'])pipette.mix(3,20,plate['A1'])protocol.comment("Assembly complete. Ready for incubation and reading.")
Week 4 HW: Protein Design Part I
Part A: Conceptual Questions
Why do humans eat beef but do not become a cow, eat fish but do not become fish?
Consuming food does not alter our DNA. During digestion, the body breaks down proteins into individual amino acids, much like taking apart a Lego castle to get the generic bricks. Our cells then take these building blocks and, following the instructions in our own DNA, construct new human proteins. The amino acids don’t carry the “identity” of the animal they came from, they are simply raw materials.
Why are there only 20 natural amino acids?
This is a result of evolutionary optimization. These 20 amino acids provide enough chemical diversity to build almost any functional biological structure. Limiting the set to 20 makes the translation process in ribosomes more efficient and reduces the likelihood of tRNA matching errors, balancing complexity with genetic fidelity.
Can you make other non-natural amino acids? Design some new amino acids.
Yes, it is possible through synthetic biology by incorporating atoms rare in biology, such as fluorine, or functional groups like azides and alkynes for click chemistry. For example: designing p-fluorophenylalanine can increase a protein’s metabolic stability or alter its hydrophobic folding patterns
Where did amino acids come from before enzymes that make them, and before life started?
Amino acids formed abiotically. A famous example is the Miller-Urey experiment, which demonstrated that mixing primitive gases (methane, ammonia, hydrogen, and water vapor) with electrical discharges mimicking lightning causes amino acids to synthesize spontaneously. We also know they arrived from outer space via meteorites, suggesting the building blocks of life are widespread in the universe.
If you make an alpha-helix using D-amino acids, what handedness (right or left) would you expect?
It would be left-handed. Since natural L-amino acids form right-handed helices, using their mirror version (D-amino acids) causes the protein’s secondary structure to reflect that symmetry, resulting in a left-handed turn.
Can you discover additional helices in proteins?
Yes. While the classic alpha is the most common, researchers have identified others like 310 and the pi helix (wider and shorter). These often appear as short segments or transitions at the ends of regular alpha helices.
Why are most molecular helices right-handed?
This is due to the exclusive use of L-amino acids in life. When these are linked together, a right handed twist minimizes steric hindrance, allowing the side chains to point outward without bumping into each other, which creates the most stable configuration.
Why do $\beta$-sheets tend to aggregate?
The edges of beta have exposed hydrogen bond donors and acceptors (C=O and N-H groups) from the backbone. Because these bonds are not satisfied within the sheet itself, they seek to pair with other nearby polypeptide chains, making the sheets sticky.
What is the driving force for $\beta$-sheet aggregation?
The driving force is those hydrogen bonds and the fact that hydrophobic faces seek to hide from water by sticking together.
Why do many amyloid diseases form $\beta$-sheets?
This happens because when a protein misfolds, forming stacked beta-sheet structures is the most thermodynamically stable state possible. It acts like a super-strong glue that the body has no way to destroy, causing them to accumulate and lead to diseases such as Parkinson.
Can you use amyloid $\beta$-sheets as materials?
Yes, they can be used as materials due to their high resistance. In biotechnology and synthetic biology, they are being used to manufacture harder and more durable bioproducts, such as nanofibers.
Design a beta-sheet motif that forms a well-ordered structure.
The most typical method is to alternate amino acids. You can use a polar one, then a hydrophobic one, and repeat (e.g., Valine - Lysine - Valine - Lysine). This ensures the sheet has a “greasy” face to stick to another sheet and a hydrophilic face to interact with water, forming highly ordered structures.
Part B: Protein Analysis and Visualization
In this part of the homework, you will be using online resources and 3D visualization software to answer questions about proteins. Pick any protein (from any organism) of your interest that has a 3D structure and answer the following questions:
Briefly describe the protein you selected and why you selected it.
I selected SIRT1 because of its fascinating potential in anti-aging research and its critical regenerative functions. This protein acts as a metabolic sensor that links a cell’s energy status with genome stability and epigenetic regulation.
The clinical importance of SIRT1 lies in its double-edged nature: while it promotes longevity and health in normal cells, this same survival mechanism is often hijacked by cancer cells. For instance, in pathologies like colorectal cancer (CRC), the overexpression of SIRT1 helps tumors resist chemotherapy and promotes metastasis. I chose this protein because understanding how it balances cell survival and disease progression is key to the future of precision medicine.
b. How many protein sequence homologs are there for your protein? Hint: Use Uniprot’s BLAST tool to search for homologs.
Based on the UniProt BLAST search, there are at least 1,000 sequence homologs for this protein. The search reached the maximum display limit of 1,000 hits, which indicates that SIRT1 is a highly conserved protein across many species.
c. Does your protein belong to any protein family?
Yes, it belongs to the Sirtuin (Sir2) mammal’s family
Identify the structure page of your protein in RCSB. When was the structure solved? Is it a good quality structure? Good quality structure is the one with good resolution. Smaller the better (Resolution: 2.70 Å)
Deposited: 2012-12-14 Released: 2013-10-23
Deposition Author(s): Davenport, A.M., Huber, F.M., Hoelz, A.
Resolution: 2.25 Å => It is a good quality structure
https://www.rcsb.org/structure/4IF6
a. Are there any other molecules in the solved structure apart from protein?
Yes, apart from the protein, the solved structure contains two other molecules (ligands): an Adenosine-5-diphosphoribose (APR) and a Zinc ion (ZN).
b. Does your protein belong to any structure classification family?
Yes. According to structural classification databases like CATH and SCOP, its core catalytic domain belongs to the Rossmann fold family (typical for NAD-binding). Additionally, sequence and protein family databases (like InterPro) classify it within the Sir2 (sirtuin) family.
Open the structure of your protein in any 3D molecule visualization software:PyMol Tutorial Here (hint: ChatGPT is good at PyMol commands)
Visualize the protein as “cartoon”, “ribbon” and “ball and stick”.
Color the protein by secondary structure. Does it have more helices or sheets?
Color the protein by residue type. What can you tell about the distribution of hydrophobic vs hydrophilic residues?
The hydrophobic residues (non-polar) are clustered in the inner core of the protein to hide from water. The hydrophilic residues (polar and charged) are distributed on the outer surface, exposed to the aqueous environment.
Visualize the surface of the protein. Does it have any “holes” (aka binding pockets)?
The protein has distinct ‘holes’ or binding pockets. As shown by the cavity visualization, there is a very prominent, large pocket right in the center of the structure (which is the main active site cleft), along with a few smaller pockets distributed across the protein.
Part C. Using ML-Based Protein Design Tools
Part C1: Using ML-Based Protein Design Tools
Deep Mutational Scans (ESM2)
a. Use ESM2 to generate an unsupervised deep mutational scan of your protein based on language model likelihoods.
A clear horizontal pattern stands out for the mutation to Proline (P), which shows predominantly dark purple (negative) scores across the entire sequence. This aligns perfectly with protein biophysics: Proline has a unique, rigid cyclic structure that makes it a notorious ‘helix breaker’.The model correctly predicts that forcing a Proline into random positions will disrupt the secondary structure (especially alpha helices) and destabilize the overall protein.Additionally, the vertical dark purple columns interspersed between lighter regions clearly distinguish the highly conserved, intolerant catalytic core residues from the more flexible, mutation-tolerant surface loops.
Additionally, the vertical dark purple columns interspersed between lighter regions clearly distinguish the highly conserved, intolerant catalytic core residues from the more flexible, mutation-tolerant surface loops
Latent Space Analysis
a. Use the provided sequence dataset to embed proteins in reduced dimensionality.
b. Analyze the different formed neighborhoods: do they approximate similar proteins?
Yes, the formed neighborhoods strongly approximate similar proteins. Dimensionality reduction algorithms like t-SNE take the high-dimensional embeddings generated by the ESM2 language model and group them based on learned sequence patterns. Because the model captures evolutionary and structural ‘grammar’, proteins that share similar domains, 3D folds, or biological functions naturally cluster together into distinct neighborhoods, even if their raw sequences are not identical.
c. Place your protein in the resulting map and explain its position and similarity to its neighbors.
My protein, SIRT1, was successfully projected into the latent space and placed within a specific cluster (the yellow group). Upon inspecting its immediate neighborhood, it clustered closely with proteins such as the NTF2-like domain of Tip associating protein (TAP), the Nonprocessive cellulase Cel9M, and the Nisin biosynthesis protein NisC. This placement makes biological sense because, although their specific functions vary, these neighbors share core structural features (complex mixed folds and extensive alpha-helical regions) with SIRT1, demonstrating that the language model correctly grouped them based on their underlying structural ‘grammar’ and similarities.
Part C2: Protein Folding
Fold your protein with ESMFold. Do the predicted coordinates match your original structure?
No, the predicted coordinates do not match the experimental structure perfectly; there are significant and noticeable differences. While the central catalytic core shares some similarities, the ESMFold prediction lacks several beta-sheets (arrows) that are clearly visible in the PyMOL experimental structure, replacing them with unstructured loops.
Additionally, the predicted model has extremely long, uncoiled chains at the ends (purple and blue regions). This is because ESMFold predicts the full-length sequence, which includes highly disordered terminal tails that were likely truncated in the experimental crystal structure. Finally, ESMFold only predicts the protein backbone, so it completely misses the Zinc ion (the sphere) and any other bound ligands present in the PyMOL visualization.
Try changing the sequence, first try some mutations, then large segments. Is your protein structure resilient to mutations?
To test the structural resilience, I drastically modified the sequence by replacing a large unstructured segment with a massive Poly-Alanine sequence. The results show that the protein structure is highly resilient in its core, but globally altered. As expected, the Poly-Alanine mutation forced the formation of a massive, unnaturally long and coiled alpha-helix (visible as the extended purple/blue structure). However, despite this drastic appendage, the central catalytic domain demonstrated remarkable resilience. It folded into its characteristic globular shape, maintaining the core beta-sheets and the surrounding native helices. This proves that the SIRT1 core fold is highly stable even under extreme mutational stress at its terminals.
Part C3: Protein Generation
Analyze the predicted sequence probabilities and compare the predicted sequence vs the original one.
ProteinMPNN created a sequence recovery of approximately 54% (0.5396) for the structured regions. The generated sequence prominently features a massive block of ‘X’s at the N-terminus. This reflects the limitations of the input experimental structure (4IF6), which lacks 3D coordinates for the highly flexible, intrinsically disordered tails.
For the resolved catalytic core, the AI designed a novel sequence that is only 54% identical to the wild-type, successfully finding alternative amino acids that can hypothetically stabilize the same Rossmann-fold topology
Input this sequence into ESMFold and compare the predicted structure to your original.
I input the clean, AI-generated sequence from ProteinMPNN (excluding the unresolved ‘X’ terminal regions) back into ESMFold. The resulting predicted structure successfully adopts the exact same core topology as the original experimental SIRT1 structure.
As observed, the overall chains appear shorter and lack the massive uncoiled loops seen in the wild-type prediction, which is expected since we only folded the core domain. The structure also lacks forced unnatural coils, displaying a natural distribution of secondary structures.
Part D. Group Brainstorm on Bacteriophage Engineering
HTGAA Final Project Proposal: Engineering the L Protein for Viral Titer Optimization
The objective of this project is to computationally redesign the bacteriophage lysis (L) protein to slightly delay the lysis event. In phage therapy, obtaining high viral concentrations is critical for clinical efficacy. By “tuning” the L protein’s kinetics, the goal is to maximize the burst size (the number of virions released per cell), allowing the host machinery to complete more viral assembly cycles before cell death occurs.
Based on the provided reading materials, this proposal addresses several key concepts:
Host-Phage Interaction: The literature highlights that lysis is a finely regulated process where the L protein must interact with specific bacterial host chaperones, such as DnaJ.
Clinical Need: As noted in reviews regarding the past, present, and future of phage therapy, one of the most significant bottlenecks for therapeutic translation is the industrial-scale production of sufficiently high titers required to combat systemic infections.
Developability: Protein engineering to improve stability and controlled function is a prerequisite for these therapies to be viable and scalable.
I will utilize an “anti-black-box” computational design pipeline based on protein language models (pLMs) and generative diffusion design:
ESM-2 & ESMFold: First, I will predict the structure of the wild-type L protein and perform an in silico Deep Mutational Scanning (DMS) to identify key residues that govern its stability and lytic activity.
moPPIt (Multi-Objective-Guided Discrete Flow Matching): I will use moPPIt to design specific motifs that optimize the L protein’s interaction with the E. coli membrane or its natural host inhibitors. This will guide the generation toward sequences that maintain target engagement but feature controlled, delayed kinetics.
ProteinMPNN: I will generate a library of sequence variants with tuned thermodynamic stability to find the exact “sweet spot” where the protein remains functional but is not prematurely hyperactive.
PeptiVerse & AlphaFold3: I will validate the top candidates by predicting their binding affinity and therapeutic properties (like solubility) in PeptiVerse, followed by evaluating structural confidence (ipTM scores) of the host-target complexes using AlphaFold3.
These approaches enable precision structural design that far surpasses traditional random mutagenesis. By utilizing moPPIt, I can explicitly constrain my design to target specific functional motifs on the host machinery. Furthermore, using foundational models like ESM-2 allows me to leverage rich, learned representations that capture the evolutionary “grammar” of lysis proteins, exploring massive sequence spaces strictly in silico before any wet-lab synthesis.
Potential Pitfalls
Lysis Complexity: The relationship between the L protein’s static structure and the highly dynamic, exact timing of cell lysis is difficult to model precisely using only static structural predictors.
Production Toxicity Paradox: There is a risk that a kinetically altered L protein could still prove highly toxic to the bacterial production strains. If the expression timing is not carefully regulated experimentally, this could paradoxically lead to premature lysis and lower overall titers.
Pipeline Schematic
[Phase 1: Structural & Mutational Analysis]
Wild-Type L Sequence -> ESMFold (Structure) -> ESM-2 (in silico DMS)
|
v
[Phase 2: Affinity & Motif Redesign]
moPPIt (Optimize binding to targeted E. coli host motifs)
|
v
[Phase 3: Developability & Validation]
PeptiVerse (Solubility & Affinity Prediction) -> AlphaFold3 (ipTM scoring)
|
v
[Phase 4: Output]
Final Candidate: Engineered L Protein with optimized lysis kinetics
Week 5 HW: Protein Design Part II
Part A: SOD1 Binder Peptide Design
🔹 Part 1 — Generate Binders with PepMLM
!!! info “Description”
Using PepMLM, four 12‑amino‑acid peptides were generated conditioned on the SOD1 A4V mutant sequence. SOD1‑binding peptide FLYRWLPSRRGG was added as a control, and the perplexity scores were recorded to estimate model confidence.
📊 Perplexity Scores
Peptide
Perplexity
WTRGDEEEEPWL
23.743201
RRTGDTEEEEPE
10.299139
KTTGDETEEEGR
11.090010
WTEGDEELGEWR
14.360410
FLYRWLPSRRGG
—
🔹 Part 2 — Evaluate Binders with AlphaFold3
📈 AlphaFold3 Results
Peptide
Perplexity
ipTM
pTM
WTRGDEEEEPWL
23.743201
0.37
0.86
RRTGDTEEEEPE
10.299139
0.51
0.84
KTTGDETEEEGR
11.090010
0.46
0.89
WTEGDEELGEWR
14.360410
0.42
0.88
FLYRWLPSRRGG
—
0.33
0.83
!!! info “AlphaFold3 Model — Figure 51”
The peptide WTRGDEEEEPWL localizes near the N-terminus where the A4V mutation sits, specifically interacting with the initial residues of the protein chain. It predominantly engages the beta-barrel region of the SOD1 mutant rather than approaching the dimer interface. Structurally, the peptide appears to be entirely surface-bound and does not appear to be partially buried within the protein’s core.
!!! info “AlphaFold3 Model — Figure 51”
The peptide RRTGDTEEEEPE localizes near the N-terminus at the A4V mutation site, specifically engaging with the initial residues and the adjacent beta-barrel region. It appears to be primarily surface-bound rather than buried within the protein core, and it does not show a significant approach to the dimer interface.
!!! info “AlphaFold3 Model — Figure 51”
The peptide KTTGDETEEEGR localizes near the N-terminus at the A4V mutation site, engaging the initial loop and the upper beta-barrel region. Similar to the previous candidates, it remains primarily surface-bound and does not appear to penetrate the protein core or approach the dimer interface.

The peptide WTEGDEELGEWR localizes near the N-terminus at the A4V mutation site, engaging the initial loop and the upper beta-barrel region. It appears strictly surface-bound and does not show significant penetration into the protein core or approach toward the dimer interface.

The control peptide FLYRWLPSRRGG localizes near the N-terminus of the SOD1 mutant, specifically interacting with the initial loop region where the A4V mutation is situated. It remains surface-bound, engaging the outer edge of the $\beta$-barrel structure without approaching the dimer interface.
The observed ipTM values for the PepMLM-generated peptides range from 0.37 to 0.51, while the known binder FLYRWLPSRRGG yielded a surprisingly low score of 0.33. Notably, all four generated peptides exceeded the structural confidence of the known binder, with Peptide 2 (RRTGDTEEEEPE) achieving the highest score of 0.51. This suggests that the AI-generated candidates may offer improved structural complementarity to the A4V mutant compared to the original control sequence.
🧩 Summary of ipTM
All PepMLM‑generated peptides outperform the known binder in ipTM. P2 (0.51) shows the strongest structural confidence.
🔹 Part 3 — Evaluate Properties with PeptiVerse
🧪 PeptiVerse Predictions
Property
P1
P2
P3
P4
Control
Sequence
WTRGDEEEEPWL
RRTGDTEEEEPE
KTTGDETEEEGR
WTEGDEELGEWR
FLYRWLPSRRGG
Solubility
1
1
1
1
1
Hemolysis
0.057
0.076
0.055
0.104
0.047
Net Charge (pH 7)
-4.23
-4.22
-3.23
-4.23
2.76
Molecular Weight
1546.6
1447.4
1351.3
1506.5
1507.7
ipTM
0.37
0.51
0.46
0.42
0.33
Binding Affinity
6.092
4.824
4.823
5.708
5.968
🔍 Comparison with AlphaFold3
Key Insight: Higher ipTM does not correlate with stronger predicted affinity.
Comparing the AlphaFold3 structural observations with PeptiVerse predictions reveals a clear divergence between structural confidence and actual binding strength. Peptides with higher ipTM scores do not show stronger predicted affinity; for instance, Peptide 2 had the highest ipTM (0.51) but the lowest binding affinity (4.824). Conversely, Peptide 1 achieved the highest affinity (6.092) despite a lower ipTM (0.37). Fortunately, none of the strong binders are predicted to be hemolytic or poorly soluble; all generated candidates achieved perfect solubility scores (1.000) and safely low hemolysis probabilities.
⭐ Selected Peptide
**P1 — WTRGDEEEEPWL** -> Best balance of predicted affinity, solubility, and safety.
WTRGDEEEEPWL: It best balances the required properties by achieving the highest predicted binding affinity even outperforming the known control peptide (5.968) while maintaining a remarkably safe therapeutic profile that is highly soluble and non-hemolytic.
🔹 Part 4 — Optimized Peptides with moPPIt
🧬 Differences from PepMLM
The peptide generated by moPPIt differs fundamentally from those generated by PepMLM in the level of design control. While PepMLM unconditionally samples plausible binders conditioned solely on the target sequence, moPPIt utilizes a Multi-Objective-Guided Discrete Flow Matching (MOG-DFM) framework. This allowed me to explicitly constrain the generation process, aiming directly at specific residues (like the A4V mutation site) while simultaneously optimizing for high binding affinity, perfect solubility, and low hemolysis, rather than relying on random sampling.
🧪 Evaluation Before Clinical Advancement
Before advancing this engineered peptide to clinical studies, computational confidence must be validated through rigorous experimental assays. First, developability metrics such as protease resistance, half-life, and immunogenicity must be thoroughly evaluated, potentially exploring non-canonical or cyclic modifications to improve pharmacokinetic stability. Finally, in vitro functional assays (such as cellular binding assays and toxicity screens) must be performed to confirm that the peptide safely and effectively engages the targeted A4V mutant SOD1 in a biological environment
🧪 Part C — Final Project: L‑Protein Mutants
High level summary: The objective of this assignment is to improve the stability and auto-folding of the lysis protein of a MS2-phage. This mechanism is key to the understanding of how phages can potentially solve antibiotic-resistance.
After running the in silico Deep Mutational Scanning (ESM-2) and cross-referencing the predicted LLR Scores with the provided experimental dataset (L-Protein Mutants - Sheet1.csv). The results shows a that evolutionary stability (high LLR scores) does not perfectly correlate with lytic toxicity. Therefore, I prioritized mutants that showed empirical success (Lysis = 1 in the experimental data) and analyzed the biophysical rationale behind their efficacy to disrupt DnaJ dependency and enhance membrane pore formation.
Proposed Sequence Variants and Justifications
L44P (Leucine to Proline at position 44): Proline is a known “helix breaker” that introduces a rigid kink into the peptide backbone. While the AI models might penalize this for structural instability, in the context of a transmembrane lytic pore, this structural kink likely alters the oligomerization geometry, favoring an irreversible disruption of the bacterial lipid bilayer, leading to the successful cell lysis observed in the experimental data.
I46F (Isoleucine to Phenylalanine at position 46): Justification: Phenylalanine is a bulky, aromatic amino acid. Substituting a smaller aliphatic side chain (Isoleucine) with a bulky aromatic one increases the overall hydrophobicity and bulkiness within the membrane core. This enhances the anchoring strength of the L-protein in the E. coli inner membrane, stabilizing the lytic pore complex.
R18I (Arginine to Isoleucine at position 18): Justification: The soluble domain relies on positively charged residues (like Arginine) to interact with the host chaperone DnaJ. Mutating Arginine to Isoleucine replaces a highly charged, hydrophilic residue with a purely hydrophobic one. This effectively breaks the electrostatic interaction with DnaJ, allowing the lysis protein to evade the host’s resistance mechanism and auto-fold.
E25V (Glutamate to Valine at position 25): Justification: Similar to the previous rationale, Glutamate is a negatively charged amino acid that participates in polar contacts. Swapping it for Valine (hydrophobic and uncharged) disrupts the native binding interface with bacterial chaperones. The experimental data confirms that neutralizing these charges in the soluble domain leads to successful lysis (Lysis = 1), proving chaperone independence.
R30L (Arginine to Leucine at position 30): Justification: This mutation reinforces the pattern discovered during the data analysis: eliminating positively charged Arginines (R18, R30) in the soluble domain consistently yields functional, lytic phages. Replacing Arginine 30 with Leucine reduces the solubility of the N-terminal tail, potentially accelerating its collapse and auto-folding without needing E. coli’s DnaJ assistance.
Week 6 HW: Genetic Circuits Part I
Part A: Assignment: DNA Assembly
1. What are some components in the Phusion High-Fidelity PCR Master Mix and what is their purpose?
Based on standard molecular biology principles, a High-Fidelity PCR Master Mix typically contains
Phusion DNA Polymerase: A highly processive, proofreading polymerase responsible for synthesizing the new DNA strands accurately.
Deoxynucleotide Triphosphates (dNTPs): The A, T, C, and G building blocks that the polymerase uses to build the new DNA strand.
Reaction Buffer: Maintains the optimal pH and salt conditions for the enzyme.
Magnesium Chloride: A crucial cofactor required for DNA polymerase activity.
Specific template DNA, primers, and nuclease-free water.
2. What are some factors that determine primer annealing temperature during PCR?
The core binding region should be 18-22 base pairs.GC
The annealing temperature is typically set about 2-5°C below the lower melting temperature
The primer binding region should ideally consist of 40-60% Guanine/Cytosine bases.
The presence of 1-2 G or C bases at the 3’ end promotes specific binding, though having more than 3 G/C bases in the final 5 nucleotides should be avoided.
The absence of strong hairpins or dimers (aiming for a Gibbs free energy above -10 kcal) ensures the primer binds to the template rather than itself.
3. There are two methods from this class that create linear fragments of DNA: PCR, and restriction enzyme digests. Compare and contrast these two methods, both in terms of protocol as well as when one may be preferable to use over the other.
PCR (Polymerase Chain Reaction), uses synthetic oligonucleotides (primers) and polymerase to amplify specific target sequences from a template, it is recommended to use it when you need to introduce intentional mutations (like the amilCP color changes), or when you need a massive quantity of a very specific sub-region of a larger template.The protocol requires thermal cycling to denature DNA, anneal primers, and extend new strands
Restriction Enzyme (RE) Digest, uses proteins to recognize and cleave specific DNA sequences. The document mentions this in the context of creating a “Linear vector (REase digested)” and using DpnI to digest methylated parental DNA. When to use it? preferable for quickly linearizing an entire circular plasmid , diagnosing assembly success, or destroying background template (like the DpnI step). It’s protocol involves incubating the DNA with the specific enzyme at its optimal temperature for a set time (30-60 minutes).
4. How can you ensure that the DNA sequences that you have digested and PCR-ed will be appropriate for Gibson cloning?
To ensure your DNA sequences are ready for Gibson assembly, we must design fragments with 15-20 bp complementary overlapping ends and confirm they have the correct 5’ to 3’ orientation to cover the entire target region. Additionally we also need to perform a DpnI digest to eliminate the unmutated parental template and prevent background colonies. Finally, verify the size of your fragments by running a diagnostic agarose gel , and measure the purified DNA concentration to ensure it is sufficient (above ~30 ug/mL) to achieve the recommended 2:1 insert-to-vector molar ratio.
5. How does the plasmid DNA enter the E. coli cells during transformation?
Plasmid DNA enters the bacterial cells through a process of diffusion. To make this possible, the cells must be shocked using either a sudden temperature change (heat shock) or a electroporation. This shock causes the bacterial cell membrane to open up and generate temporary pores. Once the plasmid diffuses inside, the cells are incubated in a nutrient-rich SOC medium to recover and close their membranes before plating.
6. Describe another assembly method in detail (such as Golden Gate Assembly)
Golden Gate Assembly is a highly efficient cloning method that relies on Type IIS restriction enzymes (such as BsaI or BsmBI) and T4 DNA Ligase. Unlike traditional restriction enzymes, Type IIS enzymes bind to a specific recognition sequence but cut the DNA several base pairs completely outside of that site.
a. Explain the other method in 5 - 7 sentences plus diagrams (either handmade or online)
Import Sequences: Upload the DNA sequences of your backbone and all inserts into your Benchling workspace.
Identify Enzymes: Ensure your backbone has the appropriate Type IIS recognition sites (e.g., BsaI) flanking the insertion region.
Design Overhangs: Modify the ends of your insert sequences (usually via PCR primer design within Benchling) to include the Type IIS recognition site followed by the specific 4-bp fusion sites that match the backbone and adjacent fragments.
Simulate Assembly: Click the “Assembly” tool in Benchling, select “Golden Gate,” and define your specific Type IIS enzyme.
Set Fragments: Select your backbone and inserts in the correct order. Benchling will automatically detect the matching 4-bp overhangs, simulate the digestion/ligation, and generate the final scarless plasmid map for you to save.
b. Model this assembly method with Benchling or Asimov Kernel!
Part A: Assignment: DNA Assembly
Create a Repository for your work
Create a blank Notebook entry to document the homework and save it to that Repository
3. Create a blank Construct and save it to your Repository
a. Recreate the Repressilator in that empty Construct by using parts from the Characterized Bacterial Parts repository
b. Search the parts using the Search function in the right menu
c. Drag and drop the parts into the Construct
d. Confirm it works as expected by running the Simulator (“play” button) and compare your results with the Repressilator Construct found in the Bacterial Demos repository
4. Build three of your own Constructs using the parts in the Characterized Bacterials Parts Repo
**First Attempt:** In my first design, I used generic RBS_strong parts and standard repressor genes without degradation tags, I followed the Promoter → RBS → CDS → Terminator structure.
Second Attempt: Trying to fix the gridlock, I swapped the parts for generic RBS components.
Third Attempt (The Final Success): Finally, I debugged the construct by looking at the reference demo and updating my parts to match the specific kinetics required for oscillation.
Use of Chatgpt to create the diagram
Week 7 HW: Genetic Circuits Part II
Assignment Part 1: Intracellular Artificial Neural Networks
1. What advantages do IANNs have over traditional genetic circuits, whose input/output behaviors are Boolean functions?
IANNs provide a massive leap forward by allowing cells to process analog information instead of being stuck with binary logic. Traditional genetic circuits are brittle because they depend on strict thresholds, whereas these networks use weighted inputs to handle fuzzy or noisy data with much higher precision. This compact design enables sophisticated decision making using fewer genetic parts, which is essential for working within the tight constraints of a living organism.
2. Describe a useful application for an IANN; include a detailed description of input/output behavior, as well as any limitations an IANN might face to achieve your goal.
A powerful application is a smart therapeutic cell designed to identify and destroy tumors. The inputs consist of the concentrations of multiple biomarkers like specific micro RNAs found in malignant tissue. The network assigns weights to these signals to calculate a weighted sum, and if that sum reaches a certain level, the output triggers the production of a protein that kills the cell. The primary limitation is the heavy metabolic burden. Running such complex synthetic logic drains the energy reserves of the cell, which can lead to slower growth or the eventual loss of the circuit through natural mutations as the host tries to survive.
3. Below is a diagram depicting an intracellular single-layer perceptron where the X1 input is DNA encoding for the Csy4 endoribonuclease and the X2 input is DNA encoding for a fluorescent protein output whose mRNA is regulated by Csy4. Tx: transcription; Tl: translation.
*Note: This diagram was drawn by Chatgpt
This architecture creates a molecular hierarchy where Layer 1 integrates its DNA inputs to produce Endo 1. That enzyme then acts as the inhibitory weight for Layer 2, fighting against the $X_3$ activation signal. The final production of protein $Y$ depends on the combined logic of both stages, showing how information flows through a multi layer neural network.
Assignment Part 2: Fungal Materials
1. What are some examples of existing fungal materials and what are they used for? What are their advantages and disadvantages over traditional counterparts?2.What might you want to genetically engineer fungi to do and why? What are the advantages of doing synthetic biology in fungi as opposed to bacteria?
Mycelium leather and bio-composites used in construction are prime examples of how fungi are replacing leather and synthetic foams. The main draw is their ability to turn agricultural waste into high-value products with a tiny carbon footprint. While they beat traditional materials on environmental impact, these fungal alternatives can be less consistent and more sensitive to environmental changes like humidity, which currently limits their use in high-stress industrial applications.
Engineering fungi to behave like a living computer using the intracellular perceptron logic from the research you shared, could revolutionize environmental sensing. You might program a mycelium network to monitor soil health, where it calculates a weighted sum of moisture, pH, and nitrogen levels before deciding whether to stimulate plant growth or remain dormant. This smart behavior is far more efficient than a simple on/off switch because it protects the fungi from the metabolic exhaustion that usually comes with heavy synthetic biology modifications.
The reason to pick fungi over bacteria for this work comes down to architecture and chemistry. Fungi are eukaryotes, meaning they can fold and modify proteins in ways bacteria simply cannot, making them better factories for complex molecules. Beyond the chemistry, their hyphal networks provide a physical structure that is inherently stronger and more adaptable for creating 3D bio-materials. Bacteria are great for liquid-phase chemistry, but if you want to build a smart, solid object that can sense and react, fungi are the clear winners.
Assignment Part 3: First DNA Twist Order
Review Part 3: DNA Design Challenge of the week 2 homework. Design at least 1 insert sequence and place it into the Benchling/Kernel/Other folder you shared in the Google Form above. Document the backbone vector it will be synthesized in on your website.
Week 9 HW: Cell-Free Systems
Homework Part A: General and Lecturer-Specific Questions
General homework questions
Explain the main advantages of cell-free protein synthesis over traditional in vivo methods, specifically in terms of flexibility and control over experimental variables. Name at least two cases where cell-free expression is more beneficial than cell production.
Cell-free systems provide an open-access biological engine where you have direct control over every molecular dial. The main advantage over traditional in vivo methods is the lack of a cellular wall, which allows for the direct addition of non-canonical amino acids or specific inhibitors. This transparency makes it perfect for producing proteins that are normally toxic to a living host or for rapid prototyping where results are needed in hours rather than days. Two key cases where this is superior include the production of antimicrobial peptides that would lyses a host cell and the synthesis of proteins containing site-specific labels for structural NMR analysis
Describe the main components of a cell-free expression system and explain the role of each component.
Setting up a cell-free reaction requires a few fundamental pillars to function. You need the cellular hardware, which is the lysate containing ribosomes, tRNAs, and translation factors extracted from a host organism. Then you provide the software in the form of a DNA or mRNA template, along with the raw materials like amino acids and a specialized buffer containing the salts and ions required to keep the machinery stable. Each component serves as a critical gear in the molecular assembly line that turns genetic code into a physical product.
Why is energy provision regeneration critical in cell-free systems? Describe a method you could use to ensure continuous ATP supply in your cell-free experiment.
Energy is the primary bottleneck in these reactions because the synthesis process consumes ATP at an incredible rate. Without a way to recycle that energy, the reaction would grind to a halt within minutes as byproduct phosphates build up. To solve this, you can implement a regeneration system such as the creatine phosphate and creatine kinase pathway. This setup acts like a secondary battery that constantly recharges spent ADP back into functional ATP, ensuring the ribosomes have a steady power supply to finish building long protein chains.
Compare prokaryotic versus eukaryotic cell-free expression systems. Choose a protein to produce in each system and explain why.
Choosing between a prokaryotic or eukaryotic system is a balance of speed versus sophistication. If you are producing a simple reporter like GFP, a bacterial system (E. coli) is the best bet because it is fast and cost-effective. However, if your goal is a complex human protein like a glycosylated antibody, a eukaryotic system such as Rabbit Reticulocyte Lysate is necessary. Bacteria lack the sophisticated molecular chaperones and post-translational machinery required to fold and modify these high-order proteins correctly.
How would you design a cell-free experiment to optimize the expression of a membrane protein? Discuss the challenges and how you would address them in your setup.
Designing a setup for membrane proteins requires addressing their intense dislike for water. In a standard reaction, these proteins would aggregate into a useless clump upon synthesis. To fix this, you must provide a synthetic lipid environment like nanodiscs or liposomes directly in the reaction tube so the protein has a stable anchor to fold into. The main challenge is managing the detergent concentrations; too little and the protein aggregates, while too much poisons the translation machinery itself.
Imagine you observe a low yield of your target protein in a cell-free system. Describe three possible reasons for this and suggest a troubleshooting strategy for each.
If your yield is low, you must treat the process as a diagnostic puzzle. One likely reason is poor DNA quality, where residual salts from a purification kit are poisoning the reaction; this is fixed by performing an additional ethanol precipitation. Another culprit is the presence of RNases that shred the message before it can be read, requiring the addition of RNase inhibitors. Finally, magnesium levels might be slightly off since ribosomes are extremely sensitive to their ionic environment, so running a titration to find the optimal salt concentration is the standard recovery strategy.
Homework question from Kate Adamala
Design an example of a useful synthetic minimal cell as follows:
a. Pick a function and describe it.
This synthetic minimal cell (SMC) acts as a specialized “Heavy Metal Scout” designed to detect and neutralize lead contamination in industrial wastewater. The core idea is to create a biological containment unit that performs a specific cleanup task without the ecological risks associated with releasing self-replicating, genetically modified organisms into the wild.
The primary function of this SMC is to sense lead ions (Pb^{2+}) in the surrounding environment and respond by synthesizing and releasing metallothioneins, which are small proteins that act like molecular magnets to bind and sequester heavy metals. While this metabolic logic could technically happen in a cell-free Tx/Tl reaction without a membrane, the encapsulation is what makes it a tool rather than just a solution. The lipid bilayer protects the fragile transcription machinery from the harsh, varying pH levels of wastewater and prevents the enzymes from being washed away or diluted. A natural cell could certainly be engineered to do this, but the SMC provides a unique “fail-safe” because it lacks the genome required to reproduce or evolve, making it a much more acceptable choice for environmental bioremediation. The desired outcome is a localized reduction in bioavailable lead, turning a toxic site into a safer environment through programmed protein synthesis.
b. Design all components that would need to be part of your synthetic cell.
The physical boundary of this artificial cell consists of a mixture of POPC (1-palmitoyl-2-oleoyl-glycero-3-phosphocholine) and cholesterol to provide the necessary structural integrity and fluidity for membrane-bound processes. Inside this lipid shell, we encapsulate a complete bacterial transcription and translation system, such as the PURE system, which contains all the purified ribosomes and factors needed to turn DNA into functional proteins.
Communication with the external environment is handled by a selective gate. We incorporate the gene for $\alpha$-hemolysin ($\alpha$HL), a protein that forms non-selective nanopores in the membrane. This allows small lead ions to diffuse into the cell and ensures that the produced metallothioneins can be secreted back out into the water to do their work. A bacterial Tx/Tl system is perfectly suited for this application because the lead-sensing logic we are using is derived from the pbr operon found in lead-resistant bacteria, which is already optimized for prokaryotic machinery.
Design Choice: Utilizing a bacterial-derived PURE system ensures high compatibility with the pbrR regulatory circuit, minimizing the need for complex eukaryotic promoter engineering.
c. Experimental details
To build this system, we need a specific set of genetic and lipid components. The lipid phase requires POPC and cholesterol in a specific molar ratio to ensure the vesicles are stable enough for field use. The genetic payload consists of two primary constructs: the pbrR gene, which encodes the lead-sensitive transcription factor, and the MT1 gene, which encodes the metallothionein protein. The pbrR protein stays bound to the DNA until it encounters Pb^{2+}; once the metal binds to the protein, it triggers the high-level expression of the MT1 “cleanup” proteins.
Measuring the success of the system happens through a dual-track approach. First, we include a fluorescent reporter like sfGFP (superfolder Green Fluorescent Protein) fused to the metallothionein sequence. This allows us to use flow cytometry or simple fluorometry to see if the cells “light up” when lead is present, confirming that the internal computer is working. Second, we can perform a functional assay by measuring the decrease in lead concentration in the surrounding medium using atomic absorption spectroscopy. If the cells are working as intended, we should see a clear correlation between the increase in green fluorescence and the decrease in dissolved heavy metals in the water sample.
Homework question from Peter Nguyen
Pitch Sentence
This concept introduces a bio-responsive architectural textile that detects and visualizes structural stress or moisture-induced pathogens by producing localized bioluminescence without any external power source.
Detailed Mechanism
The material consists of a porous architectural mesh embedded with micro-capsules containing freeze-dried cell-free TX-TL components and a specific DNA logic circuit. Upon detecting moisture or specific fungal enzymes, these capsules act as the activation site by absorbing local humidity to kickstart the translation of reporter proteins. This allows the building facade to perform localized computation and provide a visual output, like a color change, exactly where the structural risk is highest.
Societal Challenge
Silent building failures and toxic mold growth lead to billions in property damage and severe respiratory health issues, yet these problems remain invisible to the naked eye until the damage is irreversible and costly to fix.
Addressing Limitations
Stability is maintained through a specialized lyoprotectant matrix that keeps the enzymes functional in a dry state for over a year during storage and installation. To overcome the one-time use bottleneck, the material is designed to be recharged by a localized spray containing a fresh energy-rich buffer, while the biological output is engineered to be transient, allowing the system to reset once the environment stabilizes.
Homework question from Ally Huang
Extended space missions face a critical pharmacy problem because medicines degrade rapidly under cosmic radiation and have limited shelf lives. Carrying a massive stockpile for a multiyear mission to Mars is inefficient and risky if supplies spoil or new pathogens emerge. Developing on-demand biological manufacturing is essential for astronaut autonomy. Understanding how cell-free systems like BioBits function in microgravity is scientifically fascinating because it tests the fundamental limits of molecular machinery outside the protective environment of Earth’s gravity, offering a decentralized way to maintain human health during deep space exploration.
The genetic target is a synthetic DNA sequence encoding the antimicrobial peptide Magainin 2, specifically designed for expression within the BioBits cell-free protein synthesis system. In the confined, high-stress environment of a spacecraft, bacteria can undergo rapid mutations, potentially increasing virulence or antibiotic resistance. Since antimicrobial peptides like Magainin 2 kill bacteria by physically disrupting their membranes rather than targeting specific metabolic pathways, they are less prone to resistance. By using BioBits to produce these peptides on-demand, astronauts could synthesize fresh, targeted treatments for skin or surface infections without relying on a pre-packaged pharmacy that might have lost its potency during long transit through high-radiation environments.
The primary goal is to demonstrate that the BioBits cell-free system can consistently produce functional, therapeutic-grade antimicrobial peptides in a microgravity environment using minimal resources. I hypothesize that the lack of buoyancy-driven convection in space will not significantly inhibit the localized molecular interactions required for transcription and translation in the BioBits freeze-dried matrix. Furthermore, the use of the P51 fluorescence viewer will show that the peptide production remains high enough to be clinically relevant. If successful, this research proves that we can replace heavy, degrading medical cargo with lightweight, stable DNA templates and freeze-dried molecular machinery, transforming how we provide healthcare on the lunar surface or during the journey to Mars.
The experiment will compare three samples: a positive control with BioBits expressing GFP, a test sample expressing the Magainin 2 peptide, and a negative control with no DNA. After activation with water, the miniPCR thermal cycler will maintain a constant 37 degrees Celsius incubation. We will measure protein production using the P51 Molecular Fluorescence Viewer to track a fluorescently-tagged version of the peptide. Data collection involves periodic visual checks and photographs to quantify fluorescence intensity, providing a real-time read on the efficiency of protein synthesis in the unique environment of the International Space Station.
Based on the predicted amino acid sequence and known modifications (chromophore formation + His-tag + LE linker):
eGFP (239 aa, including initial Met): ~27,021 Da (monoisotopic)
LE linker: +242.13 Da
HHHHHH His-tag: +822.35 Da
Chromophore formation (loss of 2H₂O): -36 Da
Theoretical monoisotopic MW ≈ 28,049 Da
2. Molecular weight using adjacent charge state method
3. Accuracy of the measurement
4. Can you observe the charge state for the zoomed-in peak?
No, it is not possible to determine the charge state from a single zoomed-in peak alone.
Why? The mass spectrometer measures only the mass-to-charge ratio (m/z). For a single peak, infinite combinations of mass (M) and charge (z) yield the same m/z. To determine z, at least two adjacent peaks from the same charge envelope are required, using the formula shown in section 2 above.
Waters Part III — Peptide Mapping - primary structure
How many Lysines (K) and Arginines (R) are in eGFP? Please circle or highlight them in the eGFP sequence given in Waters Part I question 1 above. (Note: adding the sequence to Benchling as an amino acid file and clicking biochemical properties tab will show you a count for each amino acid).
-> eGFP contains a total of: 20 lysines (K) and 7 arginines (R), for a total of 27 trypsin cleavage residues.
How many peptides will be generated from tryptic digestion of eGFP?
Note: Use Figure 4 as a guide for the relevant parameters to predict peptides from eGFP.
Based on the LC-MS data for the Peptide Map data generated in lab please use Figure 5a as a reference how many chromatographic peaks do you see in the eGFP peptide map between 0.5 and 6 minutes? You may count all peaks that are >10% relative abundance.
Using the criteria of counting only peaks with >10% relative abundance (comparing their height to the tallest peak in the chromatogram within the specified time window), I observe a total of 21 chromatographic peaks between 0.5 and 6 minutes. 19 peaks (if counting only those clearly separated) or 21 (if including small but visible shoulders like at 0.43 and 1.25). The most conservative and precise count based on a strict “>10% of max” interpretation is 21.
Assuming all the peaks are peptides, does the number of peaks match the number of peptides predicted from question 2 above? Are there more peaks in the chromatogram or fewer?
The chromatogram shows FEWER peaks than the theoretical number of tryptic peptides from eGFP. This is expected due to very small peptides eluting before 0.5 min, co-elution of multiple peptides into single peaks, and some peptides being below the 10% abundance threshold or eluting after 6 minutes.
Identify the mass-to-charge (m/z) of the peptide shown in Figure 5b. What is the charge (z) of the most abundant charge state of the peptide (use the separation of the isotopes to determine the charge state). Calculate the mass of the singly charged form of the peptide ([M+H]+) based on its m/z and z.
a) Mass-to-charge ratio (m/z):** 465.74 (most abundant isotope peak)
b) Charge state (z):** 2+ Rationale: The spacing between adjacent isotopic peaks is approximately 0.5 Da. Since Δ(m/z) = 1/z, therefore z = 1/0.5 = 2.
c) Mass of [M+H]⁺:** [[M+H]^+ = (m/z \times z) - 1.0078 = (465.74 \times 2) - 1.0078 = 930.5 \text{ Da}]
Identify the peptide based on comparison to expected masses in the PeptideMass tool. What is mass accuracy of measurement? Please calculate the error in ppm. ($\text{Accuracy} = \frac{TP + TN}{TP + TN + FP + FN}$) ($$\text{Accuracy} = \frac{TP + TN}{TP + TN + FP + FN}$$)
`DGNYK` (from eGFP tryptic digest)
930.45 Da (calculated using PeptideMass)
930.50 Da (from Figure 5b)
Mass accuracy (ppm error):
\[\text{ppm error} = \frac{|930.45 - 930.50|}{930.45} \times 10^6 = \frac{0.05}{930.45} \times 10^6 \approx 54 \text{ ppm}\]
The measurement has an error of approximately **54 parts per million**, which is excellent for a standard LC-TOF instrument.
What is the percentage of the sequence that is confirmed by peptide mapping? see Figure 6
Based on Figure 6 (coverage map), 88% of the eGFP sequence (including the His-tag) was confirmed by peptide mapping. his means that peptides covering 216 out of 246 amino acids were detected and matched to the predicted sequence. The remaining 12% were not observed, likely due to:
Very small peptides eluting in the void volume (<0.5 min)
Hydrophobic peptides not eluting under the LC conditions
Peptides with poor ionization efficiency
Low abundance peptides below the detection threshold
Waters Part IV — Oligomers
We will determine Keyhole Limpet Hemocyanin (KLH)’s oligomeric states using charge detection mass spectrometry (CDMS). CDMS single-particle measurements of KLH allow us to make direct mass measurements to determine what oligomeric states (that is, how many protein subunits combine) are present in solution. Using the known masses of the polypeptide subunits (Table 1) for KLH, identify where the following oligomeric species are on the spectrum shown below from the CDMS (Figure 7):
The following oligomeric species were identified on the CDMS spectrum:
Species
Subunit
Subunits
Calculated Mass
Peak (observed)
7FU Decamer
7FU
10
3.4 MDa
~3.4 MDa
8FU Didecamer
8FU
20
8.0 MDa
~8.0 MDa
8FU 3-Decamer
8FU
30
12.0 MDa
~12.0 MDa
8FU 4-Decamer
8FU
40
16.0 MDa
~16.0 MDa
The spectrum shows a clear series of peaks corresponding to multimers of the 8FU subunit at approximately 4, 8, 12, and 16 MDa, with an additional peak at 3.4 MDa corresponding to the 7FU decamer.
Explanation: The theoretical MW (28.006 kDa) was calculated using the eGFP sequence including the chromophore modification, LE linker, and His-tag. The observed MW (27.982 kDa) was deconvoluted from the intact LC-MS data. The resulting mass error of 857 ppm is larger than the typical <50 ppm required for confident protein identification. Possible reasons include:
Incomplete calibration of the mass spectrometer
Partial loss of the His-tag or linker during sample preparation
Alternative post-translational modifications not accounted for
Waters Part V — Did I make GFP?
Fill out this table with the data acquired from the lab work (or using the screenshots in this document):
Theoretical
Observed/measured on the Intact LC-MS
PPM Mass Error
Molecular weight (kDa)
28.049
27.367
~24,300
Note: The theoretical MW (28.049 kDa) includes the chromophore modification, LE linker, and His-tag. The observed MW (27.367 kDa) was calculated from the adjacent charge state method using peaks from Figure 1 (m/z 1520.4 and 1440.9). The PPM error is calculated as:
[
\frac{|27,367 - 28,049|}{28,049} \times 10^6 \approx 24,300 \text{ ppm}
]
Week 11 HW: Bioproduction & Cloud Labs
Part A: The 1,536 Pixel Artwork Canvas | Collective Artwork
What you contributed to the community bioart project (e.g., “I made part of the DNA on the bottom right plate”)
Even though my contribution was just a few pixels, I liked that I could at least do my part in such an interesting global project.
What you liked about the project, and what about this collaborative art experiment could be improved for next year?
I liked seeing how everyone had different ideas and how they all came together into a single global artwork, how each person gave a very special and personal touch to their work. I think what could be improved for the next event is a guide or more details about the collaborative process and how the process itself works, because I personally didn’t quite understand what it was about or where I was in the drawing process.
Part B: Cell-Free Protein Synthesis | Cell-Free Reagents
Referencing the cell-free protein synthesis reaction composition the middle box outlined in yellow on the image above, also listed below), provide a 1-2 sentence description of what each component’s role is in the cell-free reaction.
E. coli Lysate-BL21 (DE3) Star Lysate (includes T7 RNA Polymerase)
Provides the cellular machinery (ribosomes, tRNAs, initiation/elongation factors) required for transcription and translation. The BL21(DE3) strain includes T7 RNA polymerase, which specifically transcribes genes cloned under a T7 promoter.
Maintain optimal pH (HEPES), ionic strength, and provide essential ions like Mg²⁺, a critical cofactor for RNA polymerase and ribosome stability. Potassium glutamate mimics the cytoplasmic environment and acts as an osmoprotectant.
Supplies energy (via glucose and ribose metabolism) and nucleotide monophosphate building blocks that are metabolically converted into NTPs (ATP, GTP, CTP, UTP). These NTPs serve as substrates for RNA polymerase during transcription.
Provides all 20 essential amino acids required by ribosomes to synthesize the target protein during translation. Tyrosine and cysteine are added separately because they can be limiting or chemically sensitive.
Additives: Nicotinamide
Acts as a mild nuclease inhibitor, protecting mRNA and DNA template from degradation. It may also support redox balance and serve as a precursor for NAD+.
Backfill: Nuclease Free Wate
Adjusts the final reaction volume to achieve the correct concentration of all components without introducing contaminating nucleases.
Describe the main differences between the 1-hour optimized PEP-NTP master mix and the 20-hour NMP-Ribose-Glucose master mix shown in the Google Slide above. (2-3 sentences)
The 1-hour optimized PEP-NTP master mix uses pre-formed phosphoenolpyruvate (PEP) and nucleotide triphosphates (NTPs) for rapid energy regeneration and immediate transcription, enabling fast protein production but exhausting energy quickly. The 20-hour NMP-Ribose-Glucose master mix uses nucleotide monophosphates (NMPs) plus ribose and glucose, which are metabolized through glycolysis and salvage pathways to slowly regenerate NTPs, providing sustained energy for prolonged protein synthesis over many hours.
Bonus question: How can transcription occur if GMP is not included but Guanine is?
Transcription can still occur because the E. coli lysate contains guanine phosphoribosyltransferase, an enzyme in the purine salvage pathway that converts free guanine into GMP (guanosine monophosphate). Cellular kinases then phosphorylate GMP to GDP and finally to GTP, which is the direct substrate required by RNA polymerase for incorporating guanine into RNA during transcription.
Part C: Planning the Global Experiment | Cell-Free Master Mix Design
Biophysical/Functional Property of Each Fluorescent Protein
sfGFP (Superfolder GFP)
Property: Extremely fast and robust folding with high solubility, even under non-optimal conditions.
Effect on expression/readout: Its rapid maturation (minutes) allows for early fluorescence readout, and its folding efficiency makes it ideal for cell-free systems where chaperone availability may be limited. It is primarily translation-limited rather than folding-limited.
mRFP1 (Monomeric Red Fluorescent Protein 1)
Property: Slower chromophore maturation compared to sfGFP, but faster than its tetrameric parent DsRed. It is monomeric.
Effect on expression/readout: The slower maturation delays fluorescence onset, requiring longer incubation for signal detection. Its monomeric nature prevents aggregation artifacts, enabling more accurate quantitative measurements.
mKO2 (Monomeric Kusabira Orange 2)
Property: High brightness and relatively fast maturation, but moderate sensitivity to acidic pH.
Effect on expression/readout: Provides strong orange signal suitable for multiplexing, but its fluorescence intensity can drop if the cell-free reaction becomes acidic over long (36-hour) incubations due to metabolic byproducts.
mTurquoise2
Property: Very high quantum yield (0.93) and mono-exponential fluorescence lifetime, but requires precise oxidative folding.
Effect on expression/readout: Its brightness gives excellent signal-to-noise ratio, but its sensitivity to redox conditions means that suboptimal buffer composition can severely reduce the fraction of properly folded, fluorescent protein.
mScarlet_I
Property: High molecular brightness and fast maturation for a red fluorescent protein, but still oxygen-dependent for chromophore formation.
Effect on expression/readout: Provides bright, red-shifted fluorescence with minimal spectral overlap, making it ideal for multiplexed assays. However, oxygen availability in the reaction vessel can become limiting over 36 hours, reducing final yield.
Electra2
Property: Engineered for brightness and stability in the blue spectrum, but can form aggregates (puncta) under certain ionic conditions.
Effect on expression/readout: Its blue fluorescence avoids common autofluorescence, but aggregation reduces soluble fluorescent protein. It is also sensitive to redox balance and ionic strength.
Hypothesis for Maximizing Fluorescence Over 36 Hours
Protein: mScarlet_I
Hypothesis: Supplementing the master mix with additional glucose (to 2.5 g/L)** and magnesium glutamate (to 9.0 mM) , while **increasing reaction surface area (e.g., using a low-retention 384-well plate) to improve oxygen diffusion, will maximize mScarlet_I fluorescence over 36 hours.
Expected effect: The increased glucose extends ATP regeneration beyond 20 hours, sustaining translation. Higher magnesium enhances ribosome activity and translation rate. Improved oxygen availability overcomes the oxygen-dependent chromophore maturation bottleneck specific to red FPs. Together, these changes will yield higher peak fluorescence and slower signal decay compared to the standard master mix.
Alternative hypothesis (for mTurquoise2):
Adding *nicotinamide (to 5 mM) and *cysteine (to 5 mM) while slightly reducing potassium glutamate (to 290 mM) will improve redox balance and reduce oxidative stress. Because mTurquoise2 requires precise oxidative folding, these changes will increase the fraction of properly folded protein, resulting in brighter and more stable cyan fluorescence over 36 hours.
Master Mix Composition Example (for mScarlet_I)
Based on the reaction format (6 µL lysate + 10 µL 2X master mix + 2 µL DNA + 2 µL custom supplements = 20 µL total):
Supplement
Volume (nL) in 2 µL
Final concentration in 20 µL reaction
Rationale
Nuclease-free water
1350
(balance)
Backfill
Glucose
150
2.5 g/L
Extends energy regeneration
Magnesium glutamate
100
9.0 mM
Boosts translation rate
Ribose
75
~12 g/L (standard)
Maintains nucleotide regeneration
AMP
50
0.5 mM
NTP precursor
GMP
50
0.5 mM
Direct GTP precursor
Cysteine
50
5.0 mM
Supports redox and prevents oxidation
Nicotinamide
50
4.0 mM
Redox balance
Potassium glutamate
75
~315 mM (standard)
Ionic strength
HEPES-KOH
25
~45 mM (standard)
pH buffering
Total
2000 nL
JSON format for the custom supplement (to submit):
HTGAA 2026: Final Project Selection Hey there! Welcome to the BioLoop Engine 🌱 You know how we see tons of agricultural waste—like fruit peels or leftover crops—just rotting away in the streets or fields? It’s a huge problem, and honestly, it always felt like a massive waste of potential to me.
You know how we see tons of agricultural waste—like fruit peels or leftover crops—just rotting away in the streets or fields? It’s a huge problem, and honestly, it always felt like a massive waste of potential to me.
I really wanted to build something to actually fix it. That’s how the idea for BioLoop was born!
Think of it like a smart recipe generator, but for biology. If you have a bunch of, let’s say, mango peels, you just type that into the app. The AI then acts like a detective: it searches through biological databases to find the perfect microscopic “Pac-Man” (an enzyme) that loves to break down those specific peels. Once it finds it, the system automatically writes the exact DNA “instruction manual” so we can build that enzyme in the lab.
The best part? I didn’t want this to be some expensive, secret corporate tool. It’s completely open-source. The goal is to make it super easy for anyone, anywhere, to turn their local organic trash into valuable, eco-friendly products!
📄 Part 1: Project Proposal & Validation
This is where I put all the official, nerdy details of the project: the biological validation, the data, how much it costs, and the bioethics behind it.
---
🔬 Part 2: How the Magic Happens (Step-by-Step)
Curious about how we actually go from typing “mango peels” on a computer to getting a real DNA sequence? This section breaks down the entire process from the software code to the actual lab work.

.png)
.png)
.png)
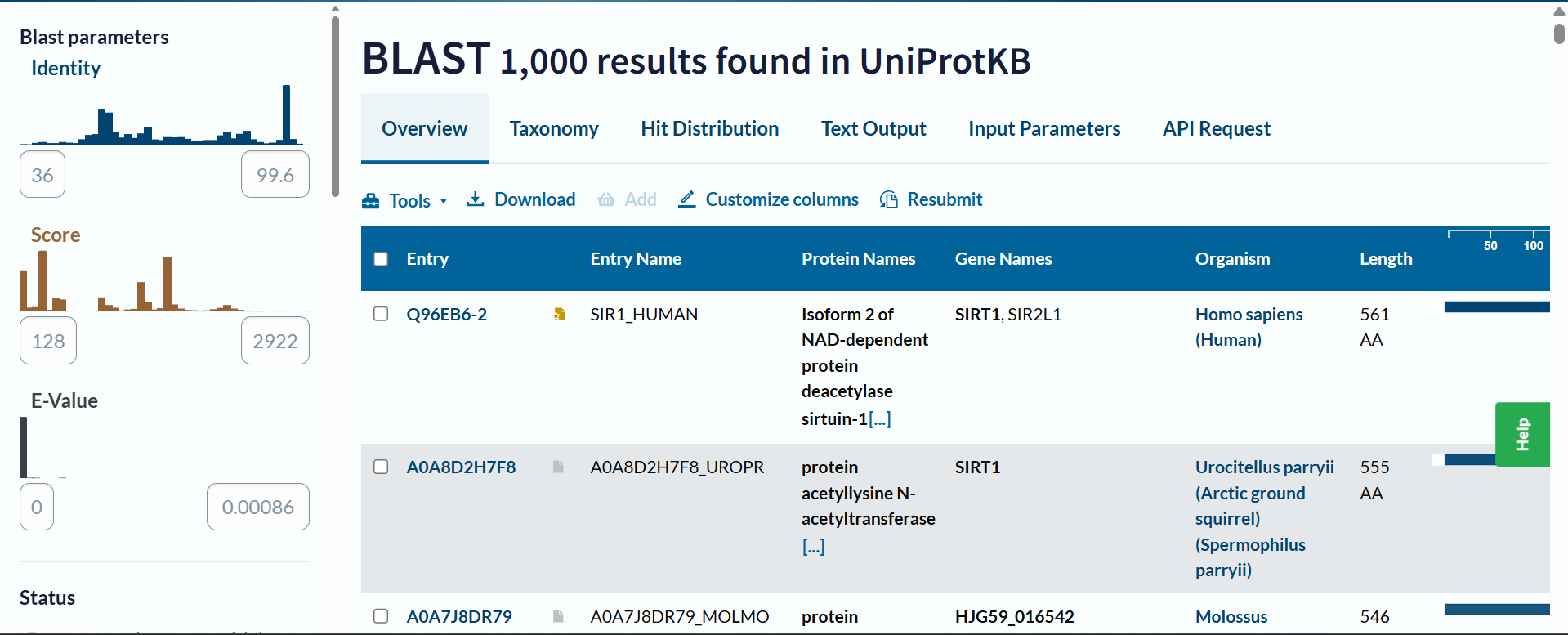

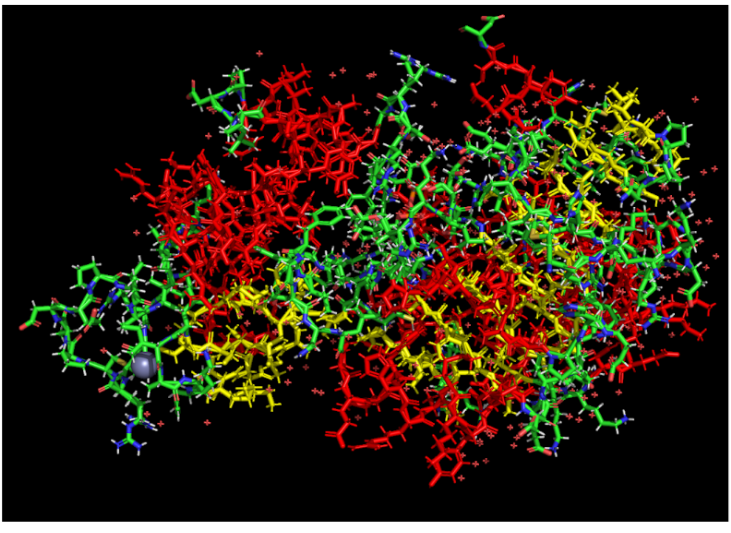
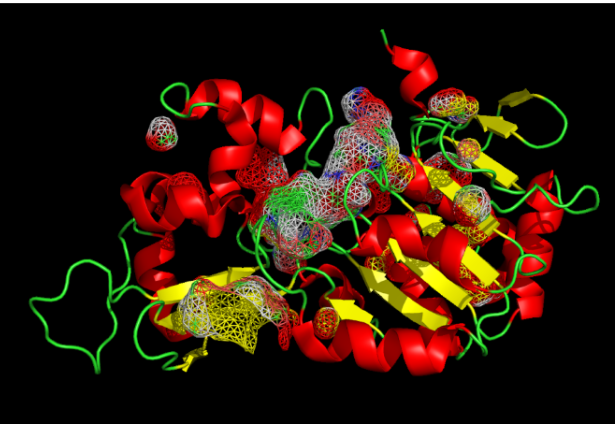
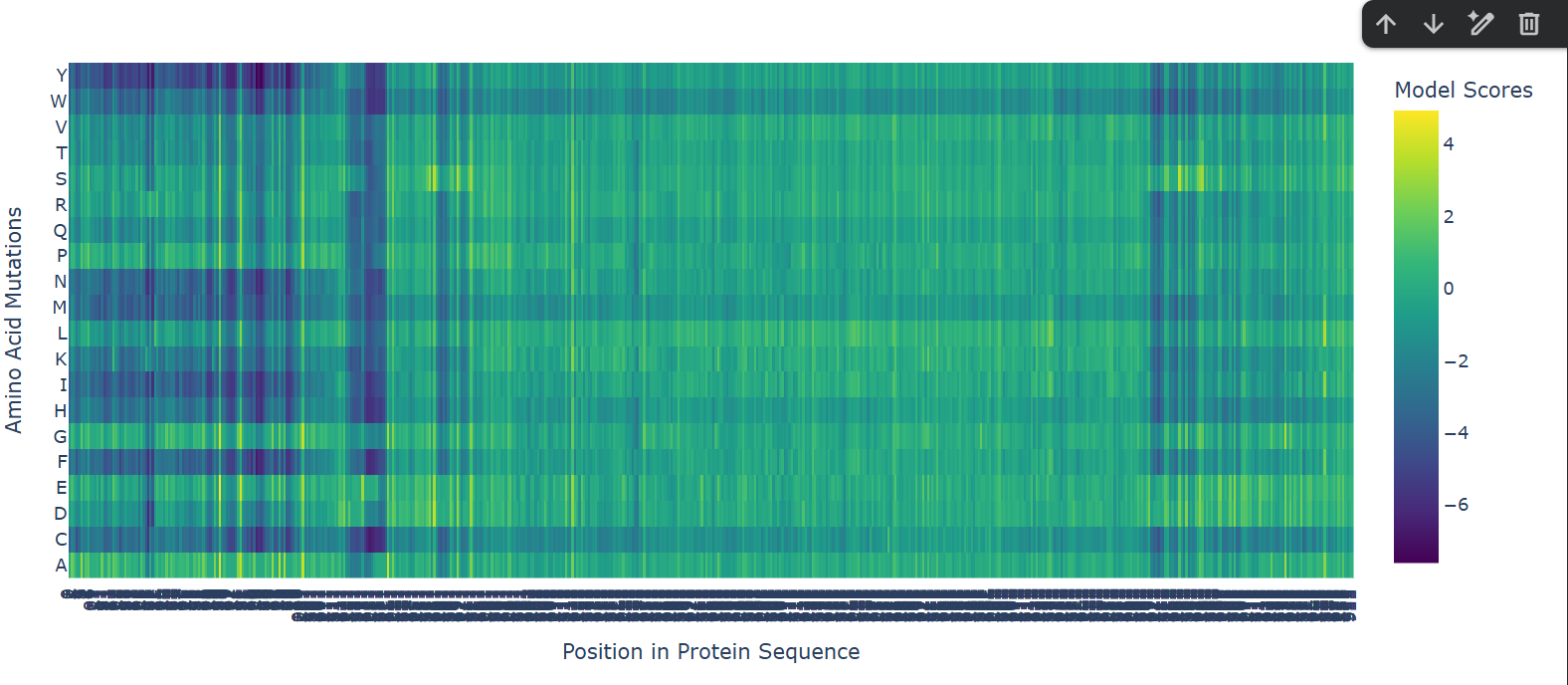
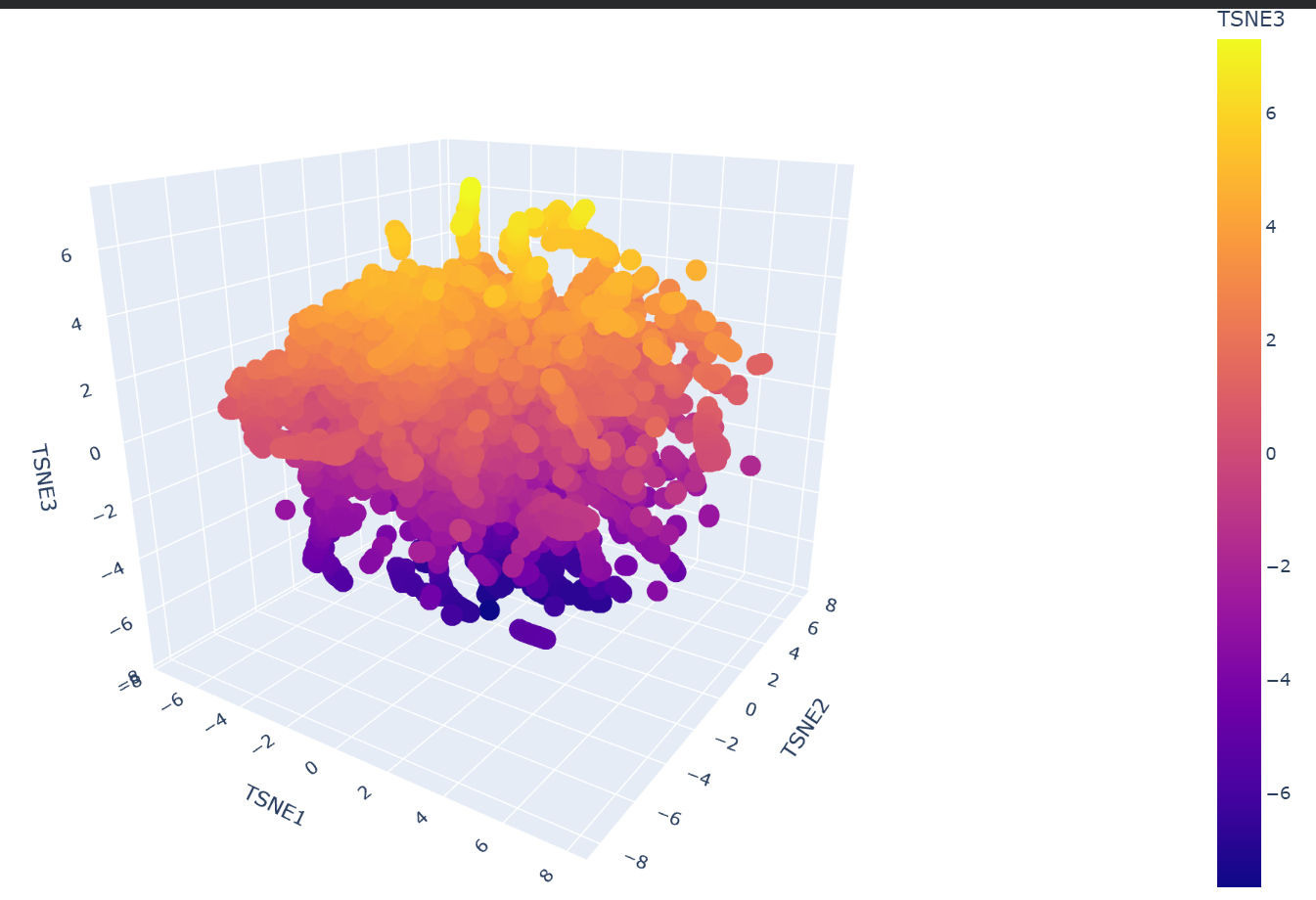

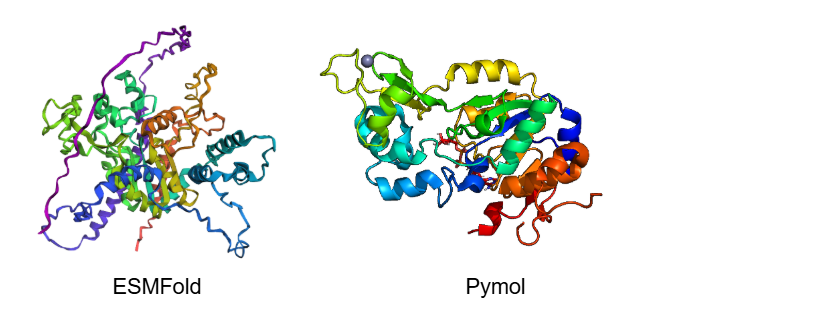
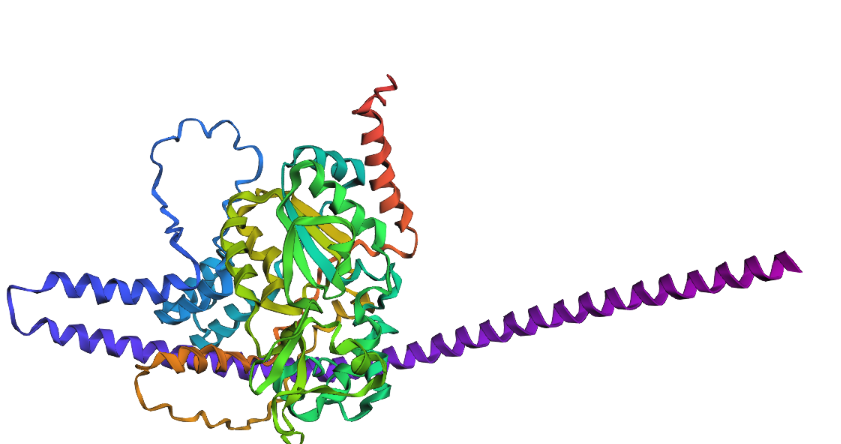
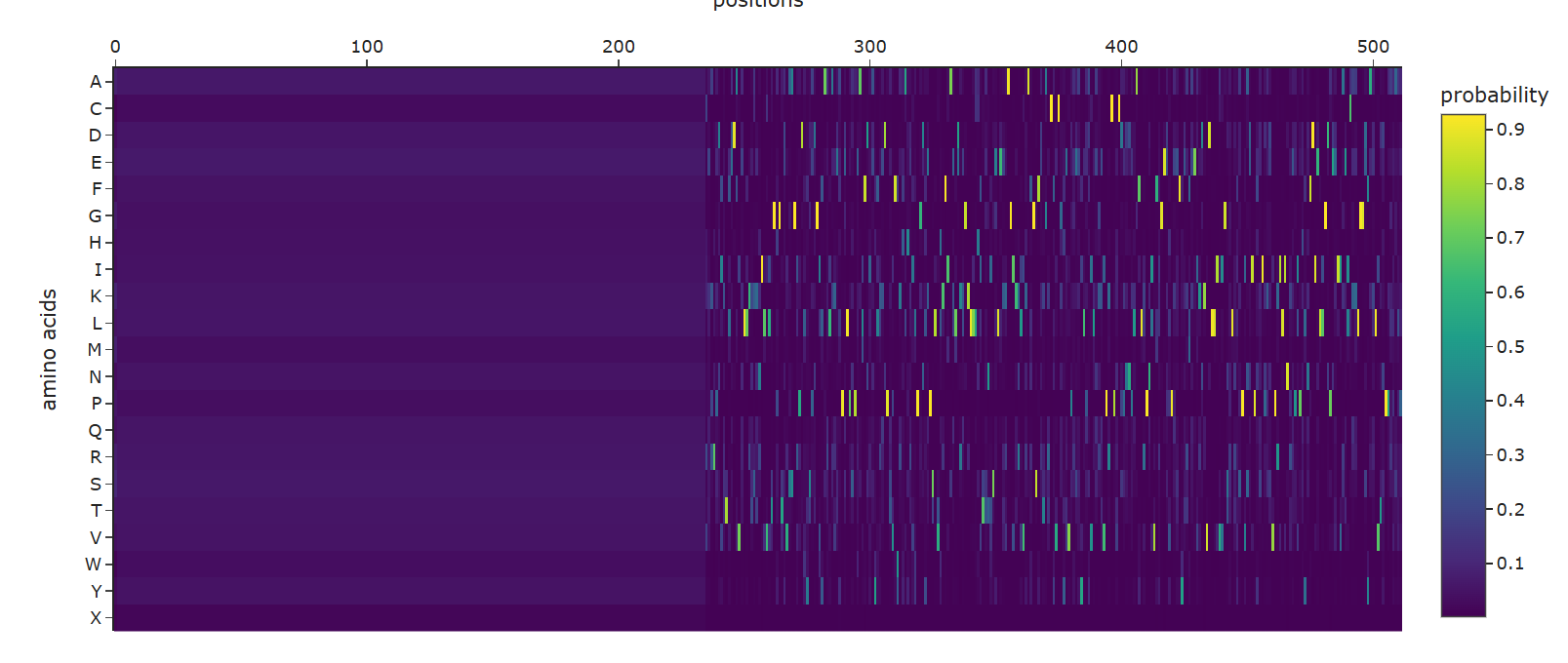
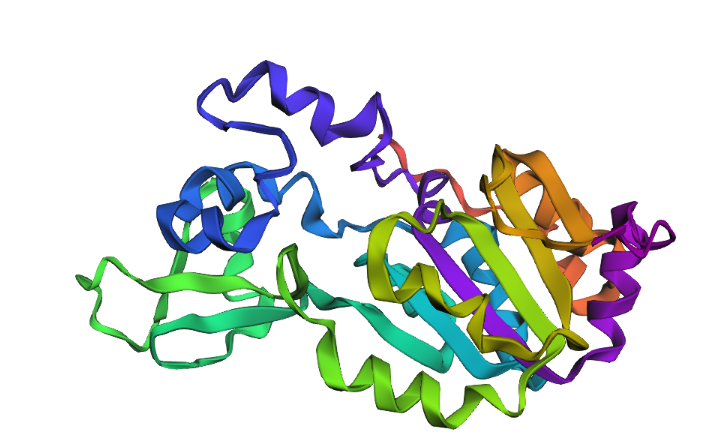
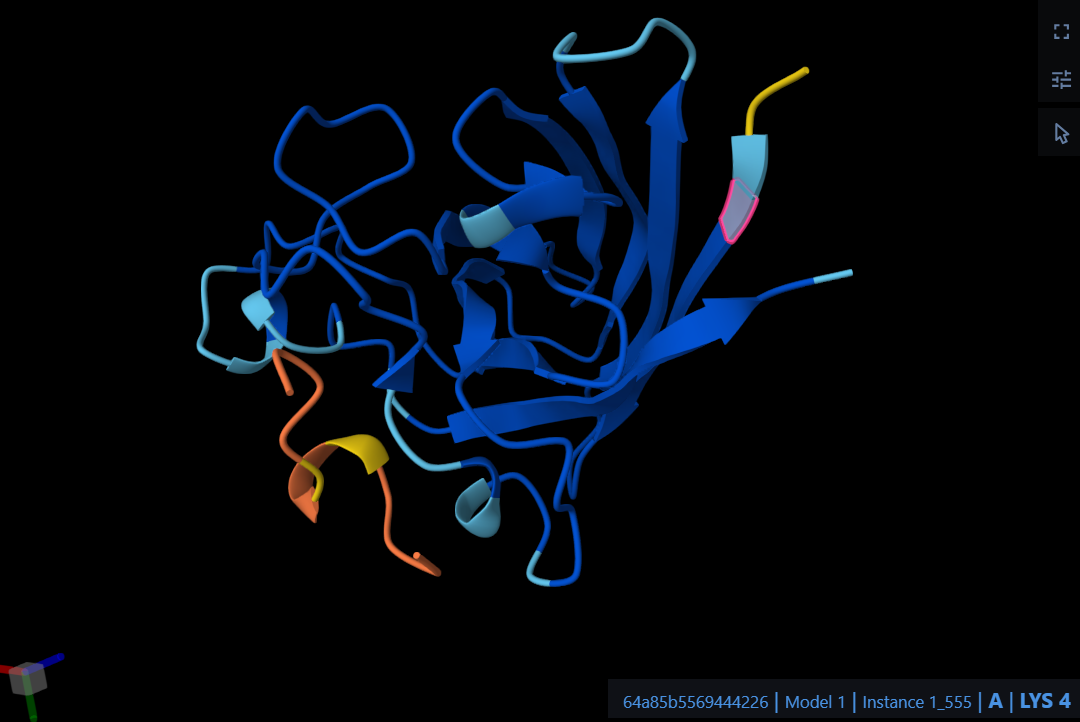
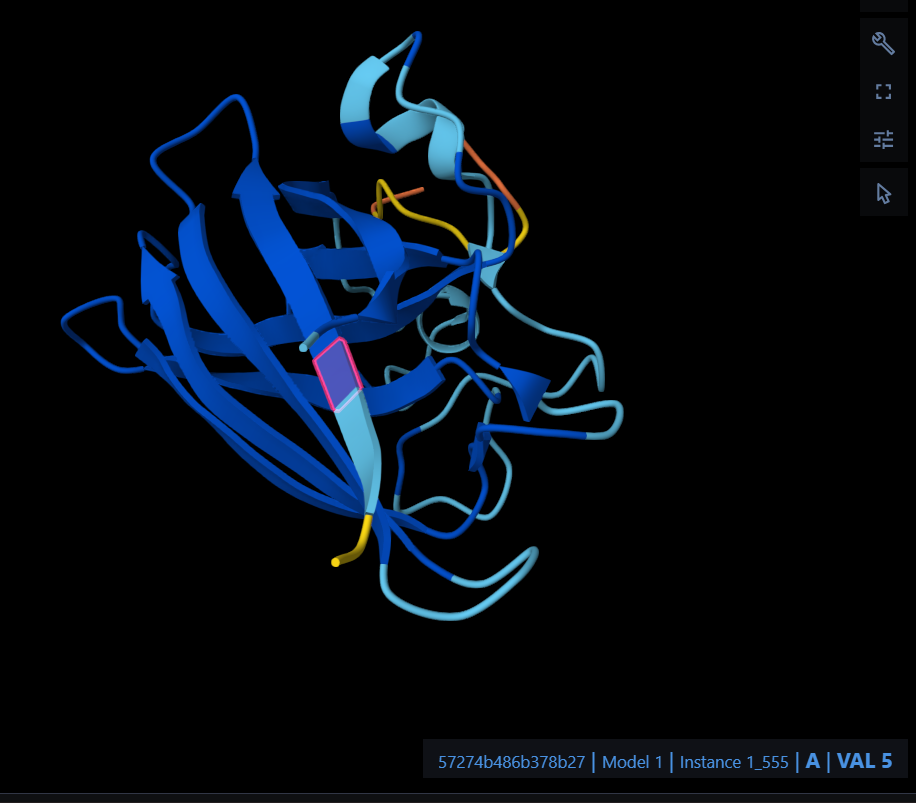
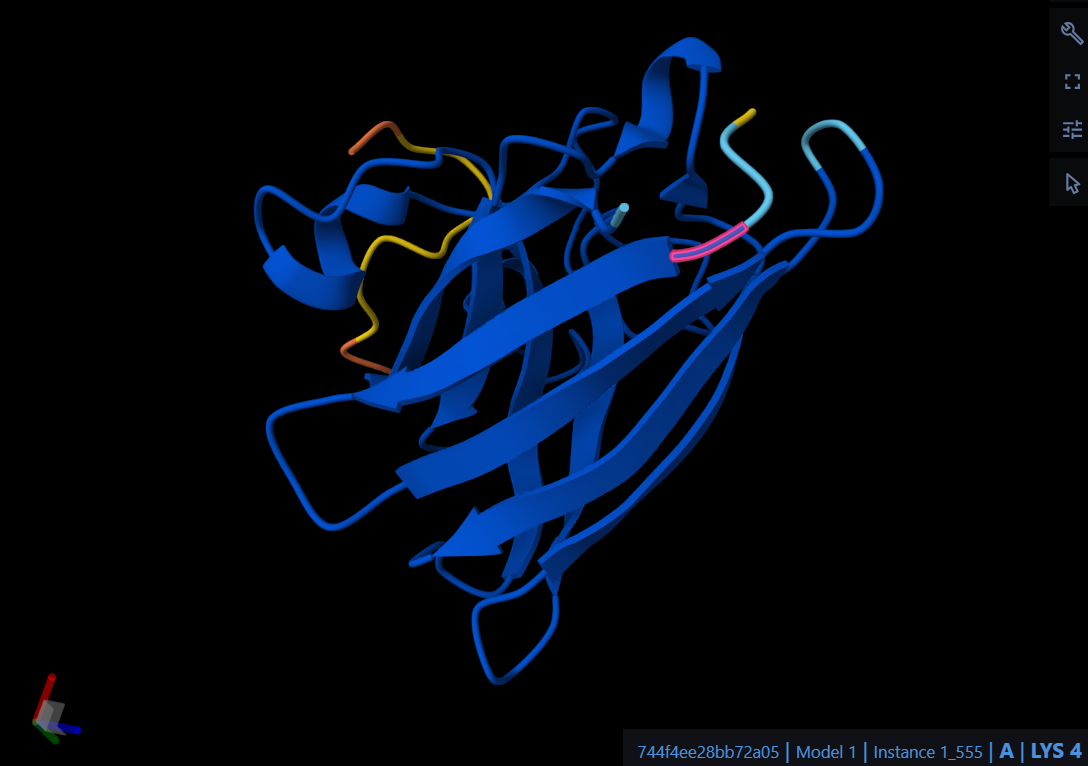
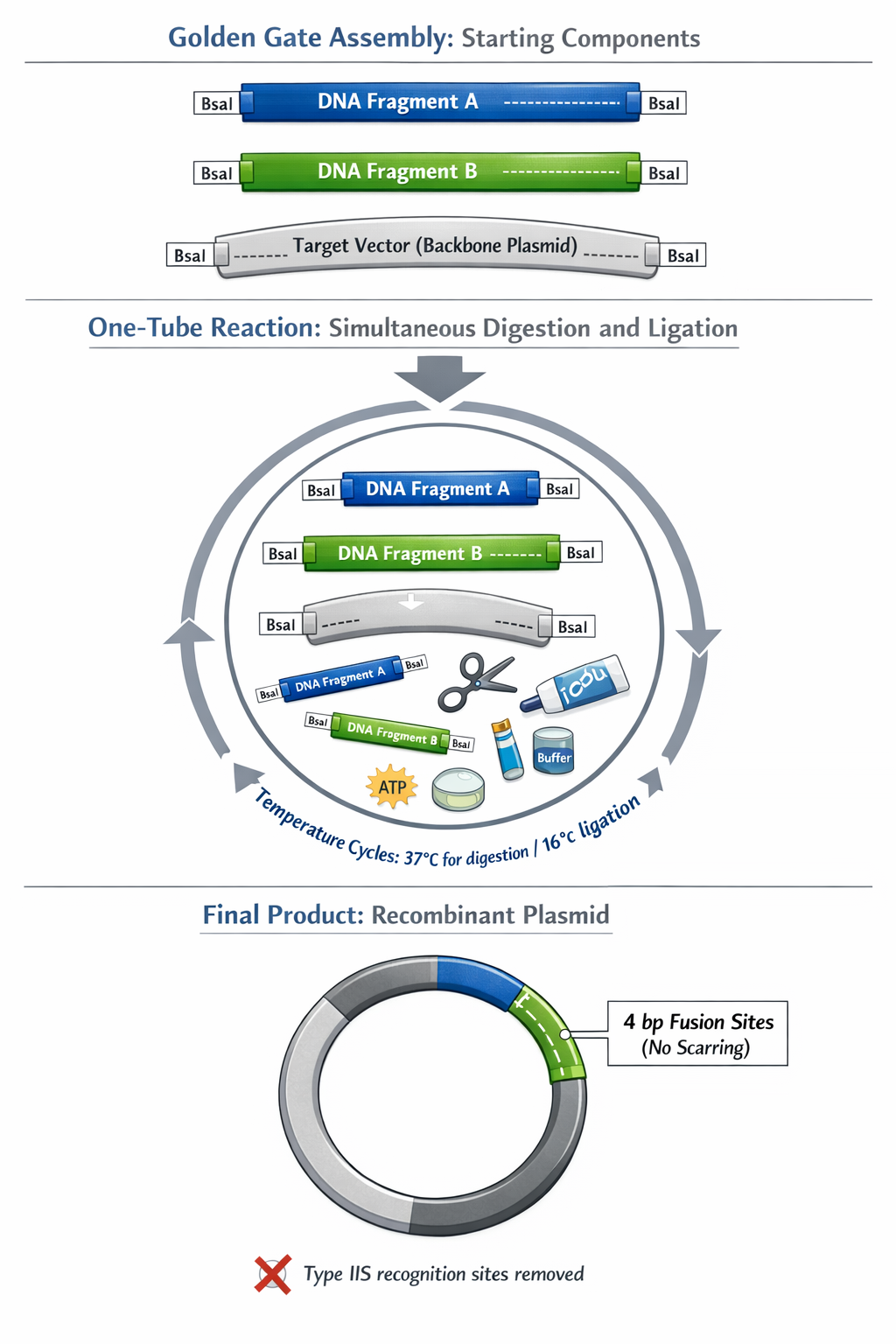
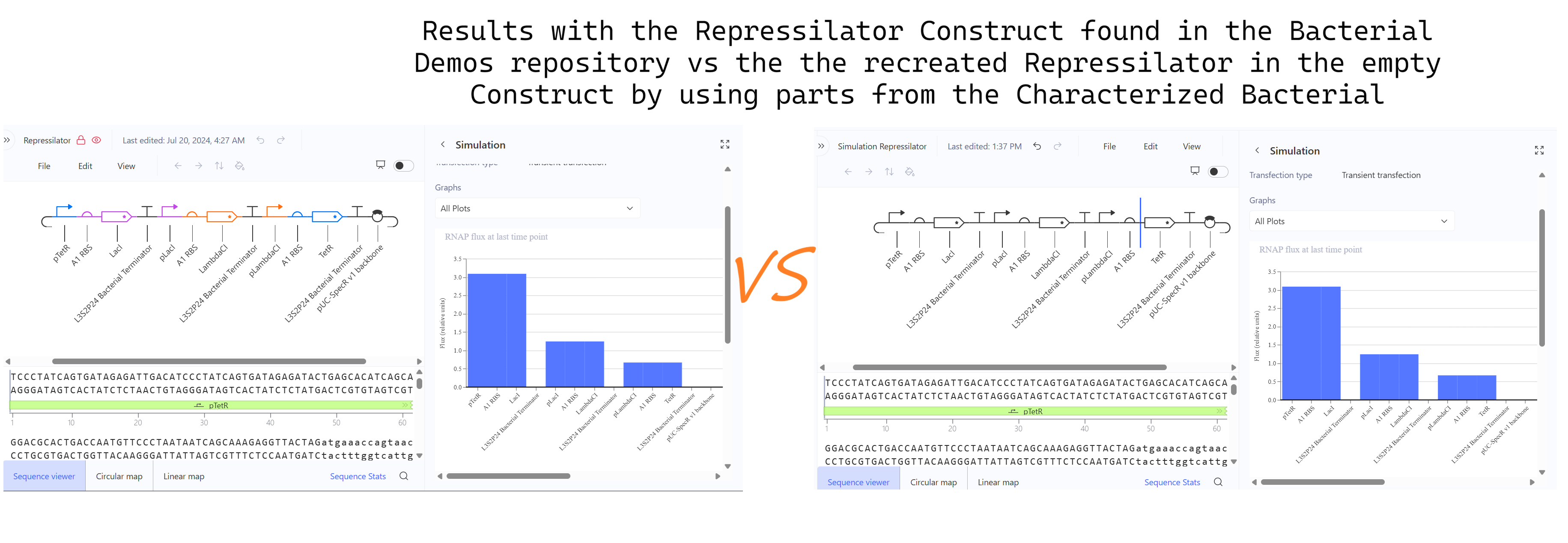
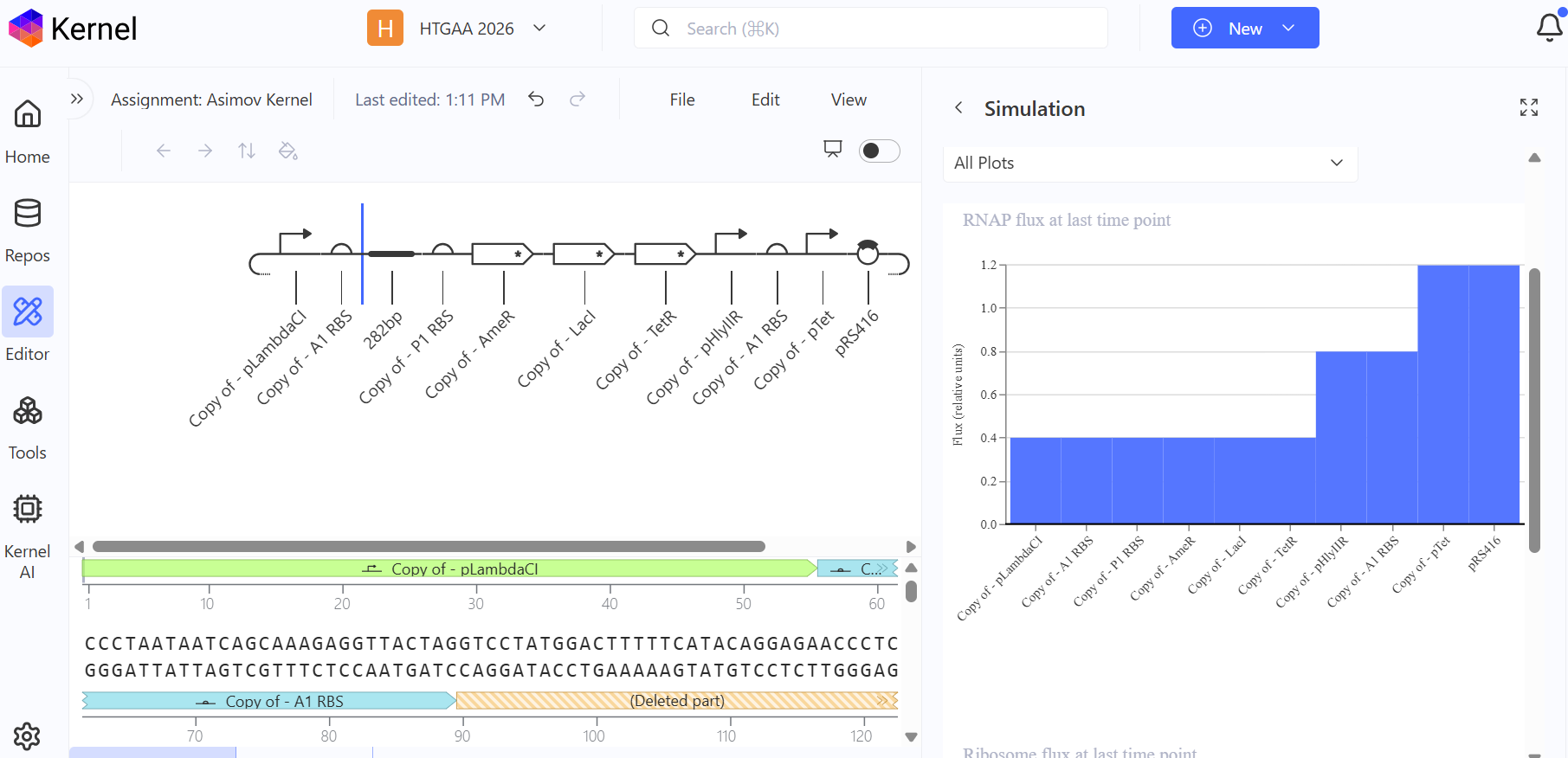
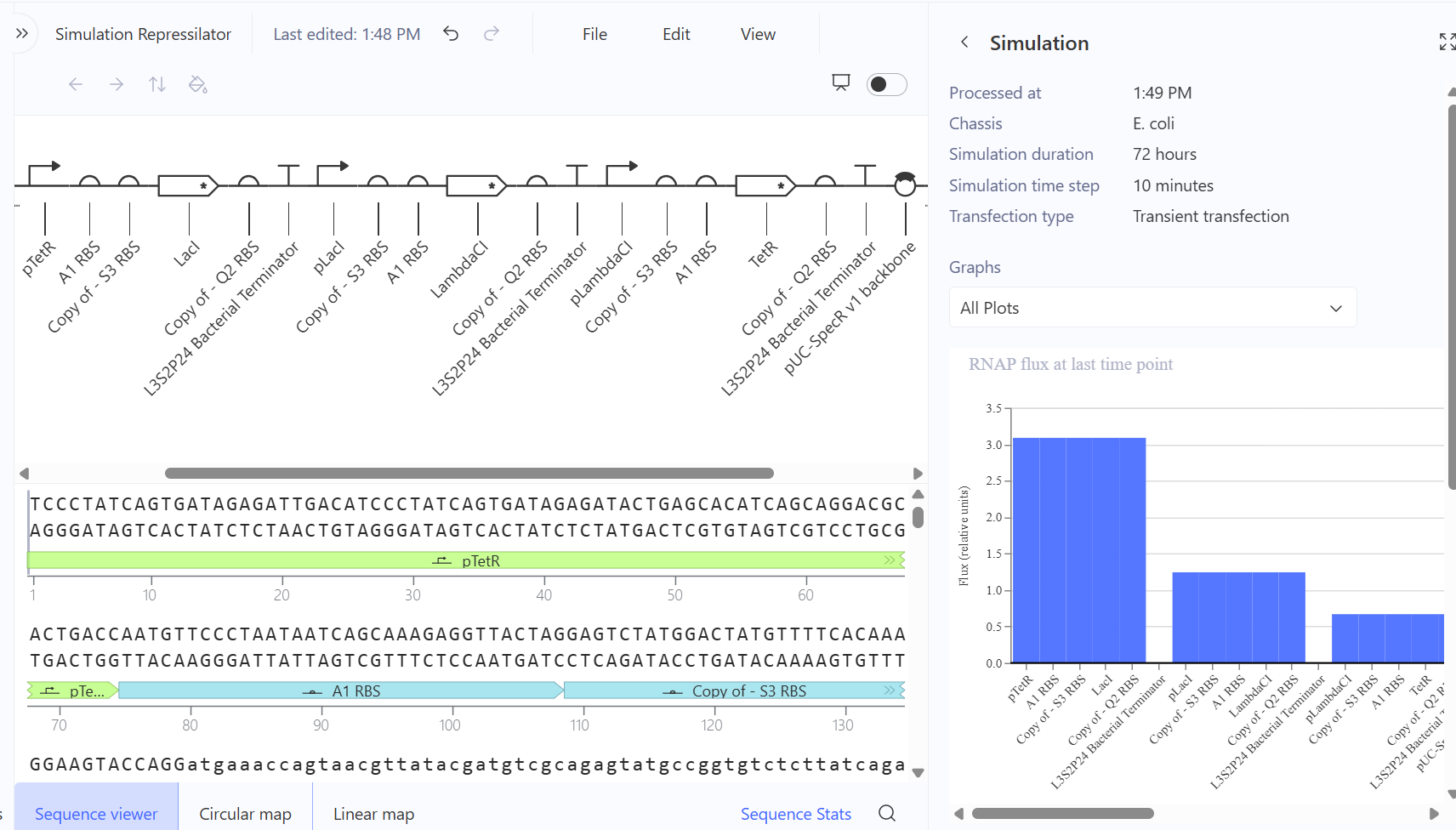
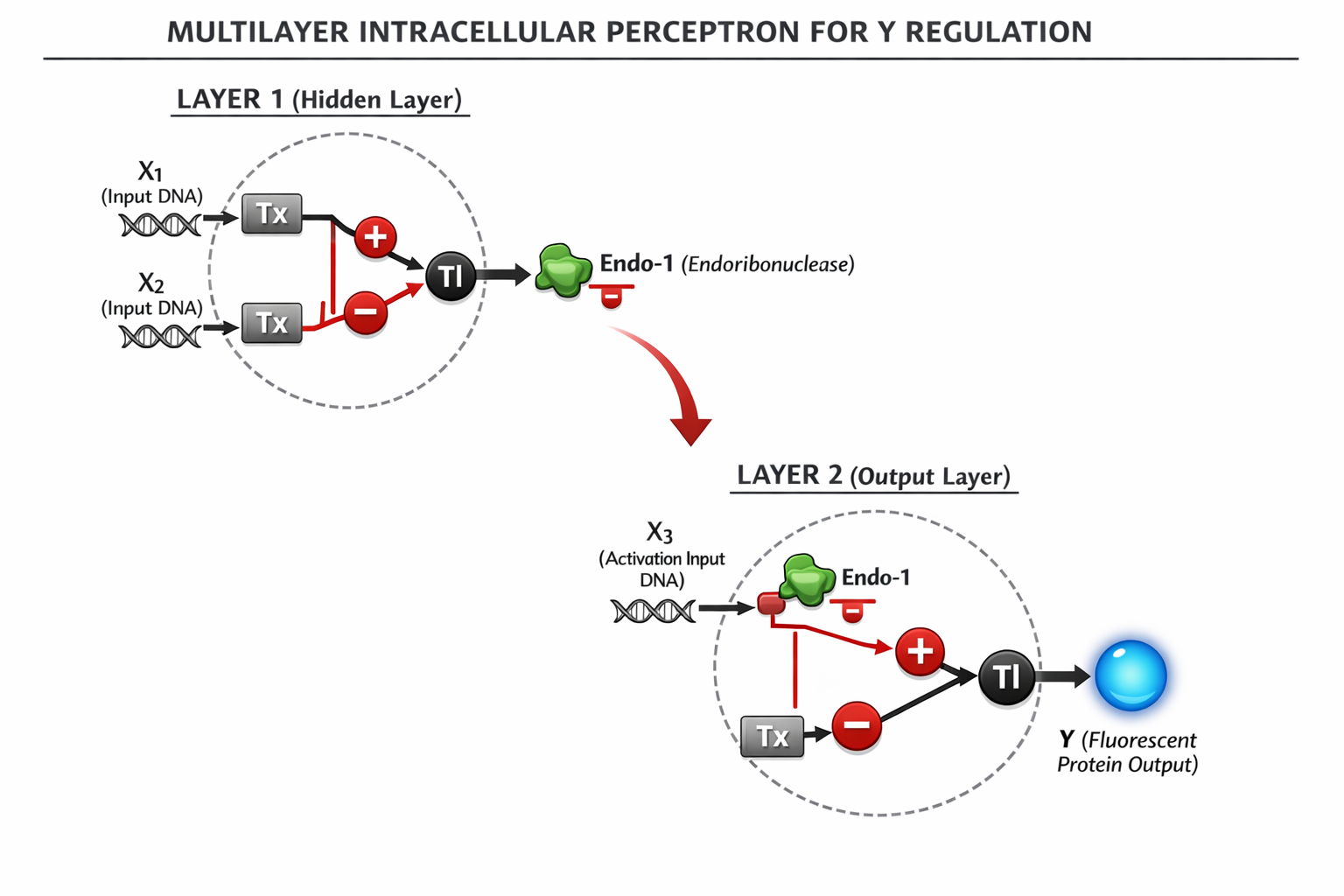
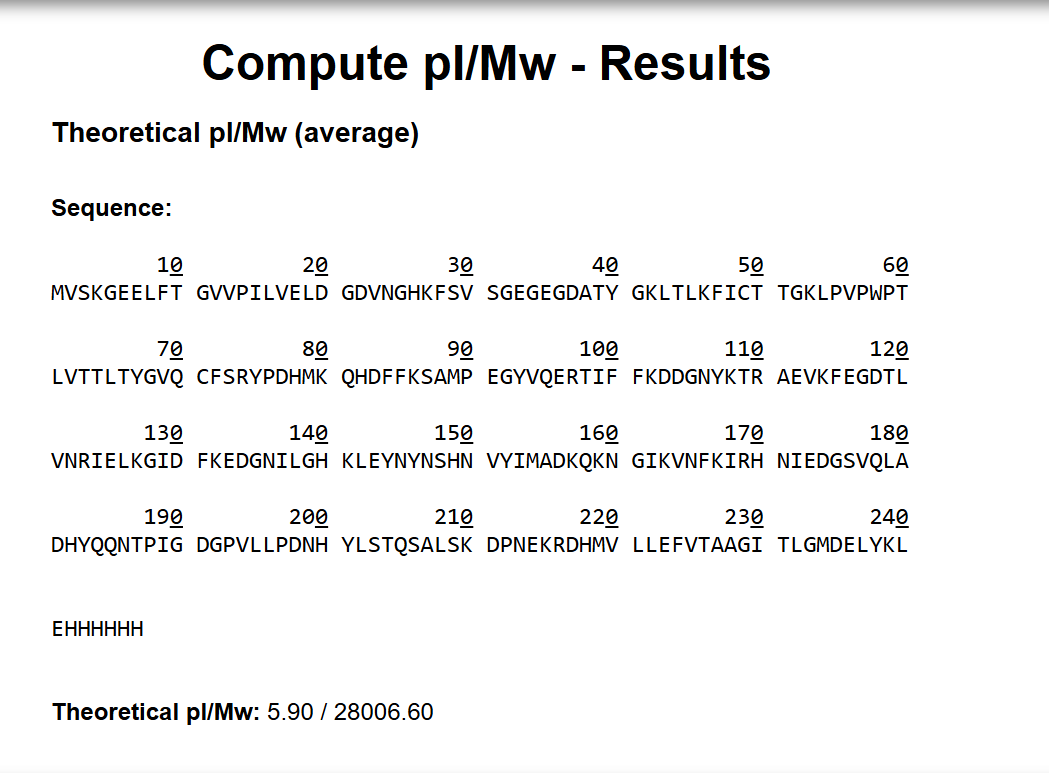

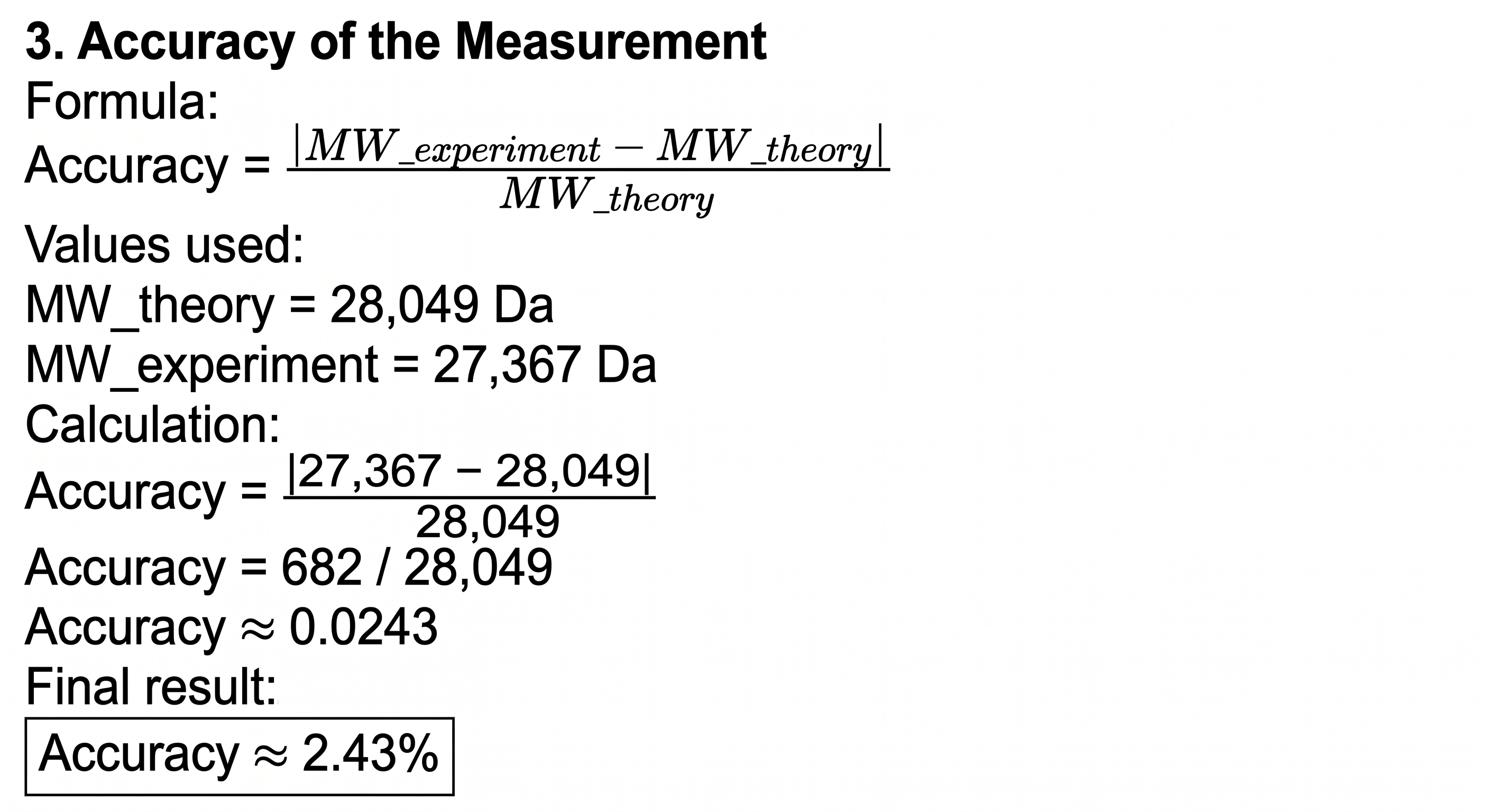
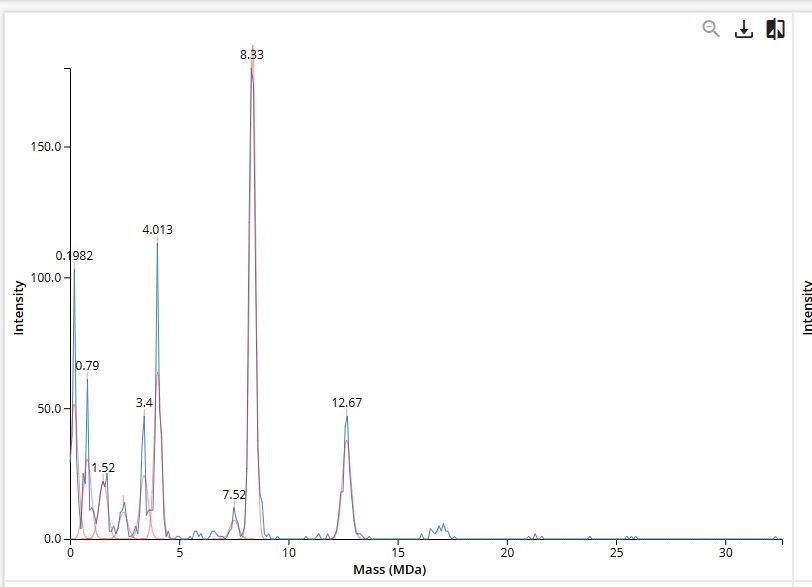

{kind=link}
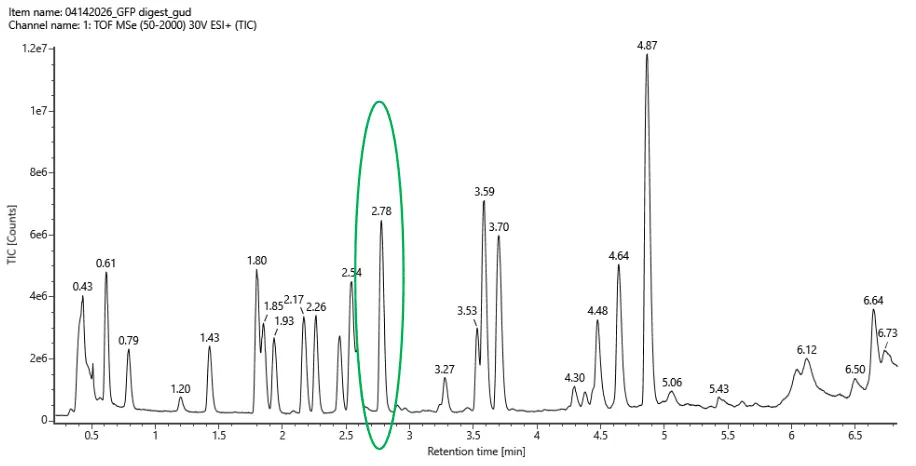{kind=link}
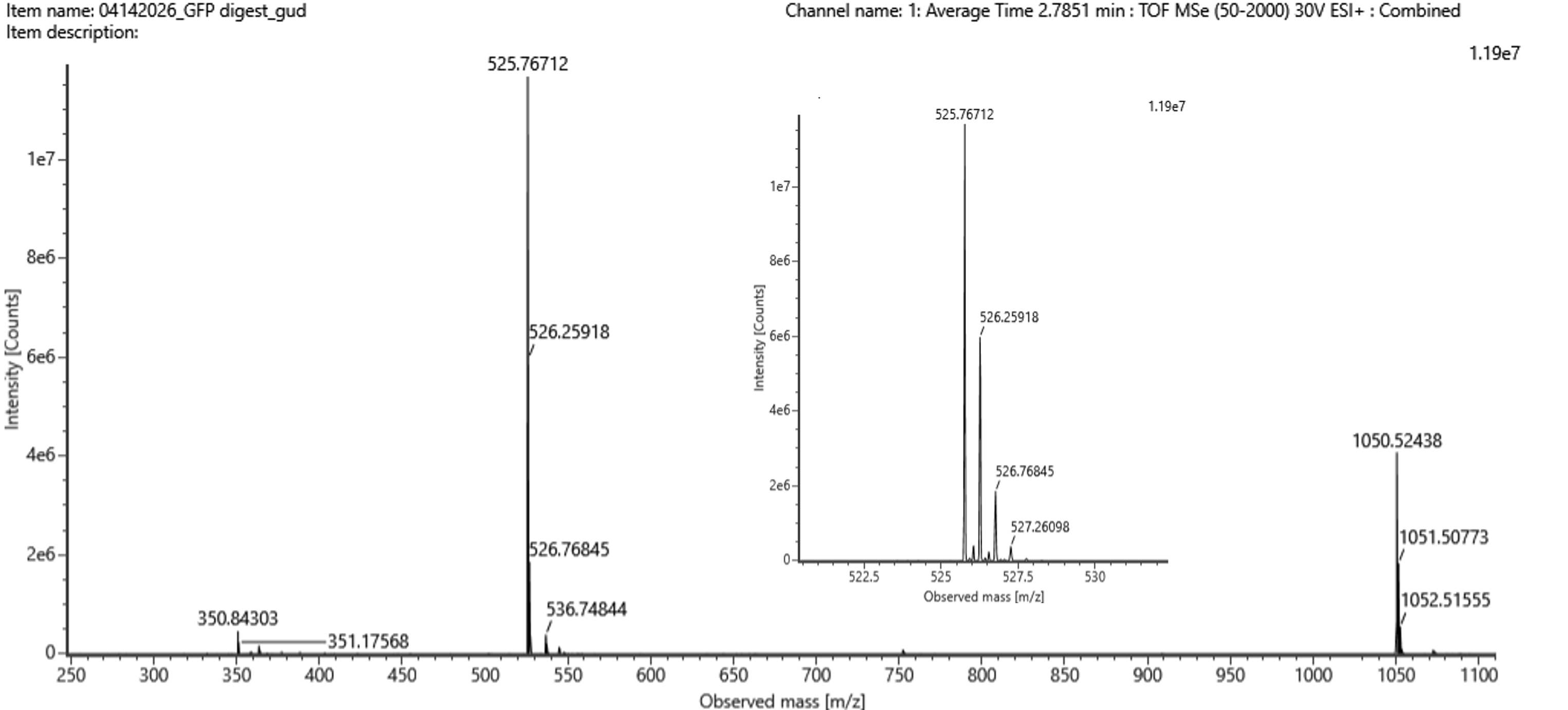{kind=link}
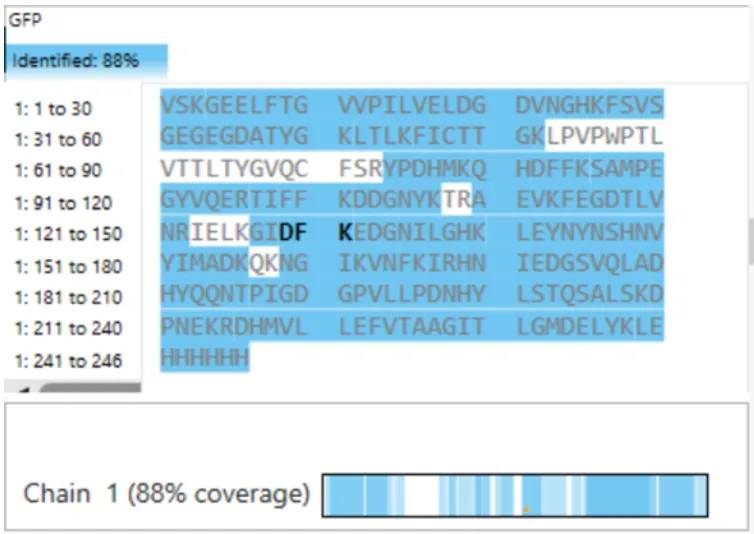{kind=link}
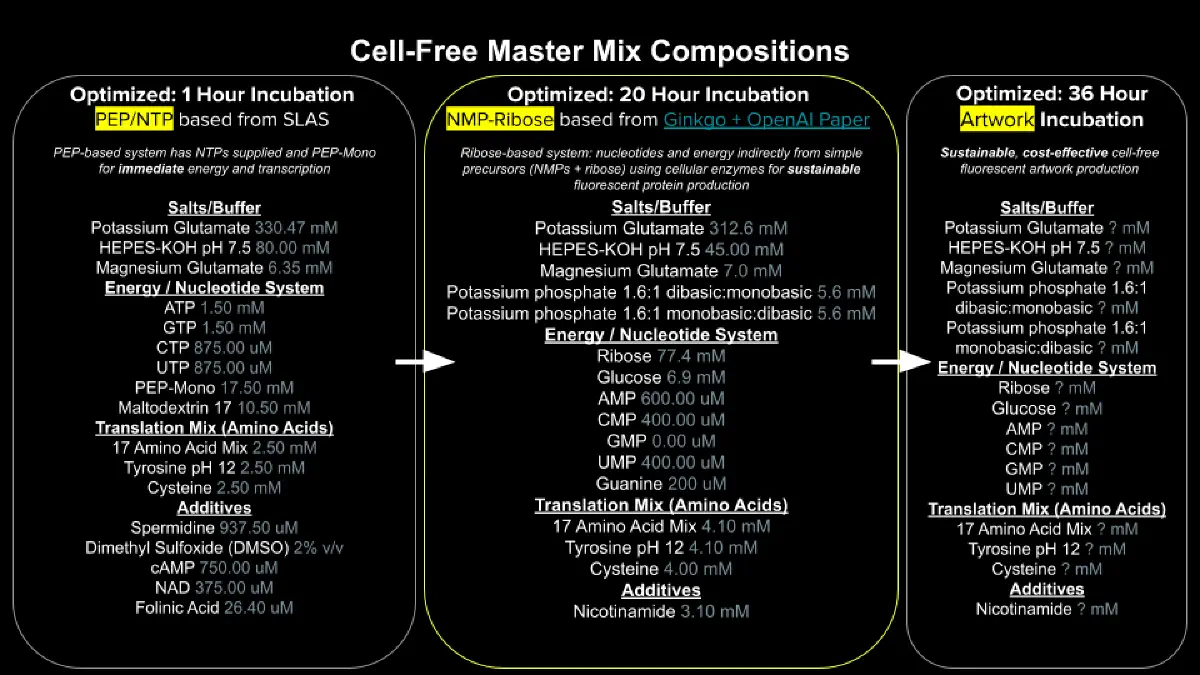{kind=link}