Week 2 HW: DNA Read / Write / Edit
Part 0: Basics of Gel Electrophoresis
Attend Bootcamp:
Part 1: Benchling & In-silico Gel Art
 | 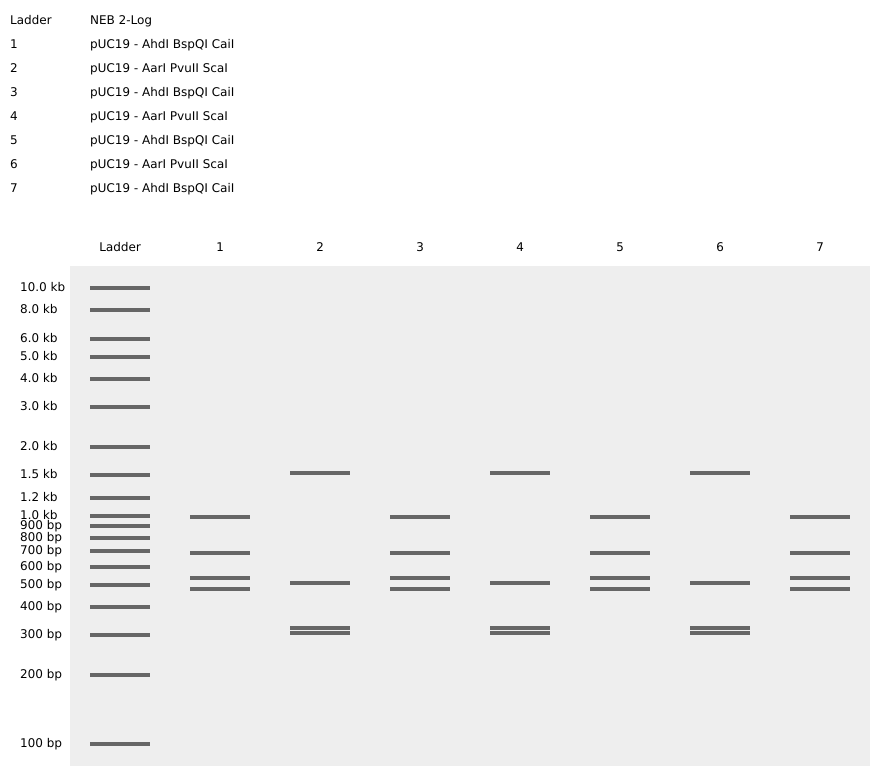 |
Left: practice in-silico digestion featuring base sequence (Lambda) and restriction enzymes as specified.
Right: in-silico digestion representing a three-subunit amino polypeptide! To achieve the central grouping of bands I desired, I first asked chatGPT to recommend a short base sequence, then referenced the linear map in Benchling to iterate with endonucleases which produced cuts at the positions needed to make bp lengths which would match an outline I sketched.
Part 2: Gel Art - Restriction Digests and Gel Electrophoresis
No wetlab access. Given access in the coming months, I’ll definitely attempt to create my artwork given the required endonucleases + base sequence are accessible!
Part 3: DNA Design Challenge
3.1. Choose your protein.
From Magnetospirillum magneticum (AMB-1): HtrA/DegP family protease MamE
Magnetotaxic bacteria possess the highly curious ability to form organised, magnetic ‘inclusions’ in structures composed of ‘magnetosomes’. Magnetotaxis as a behavioural characteristic is particularly advantageous in aquatic environments, where detection of the earth’s magnetic field permits spatial sense in the water column as a method to locate nutrient-rich microenvironments. In the model organism Magnetospirillum magneticum AMB-1 (AMB-1), MamE protease is central to the biomineralization process required for magnetic crystal formation within magnetosomes.
I selected this protein as I find this behaviour incredibly enchanting! Its role in both magnetoreception and the biomineralization process is fascinating, and promises many intriguing applications if harnessed.
3.2. Reverse Translate: Protein (amino acid) sequence to DNA (nucleotide) sequence.
MamE protease gene, reverse-translated via Genecorner:
3.3. Codon optimization.
When engineering the genome of one organism to express a foreign gene, codon optimization increases the chances that the translated sequence will result in the correct, functional protein product. Although the central dogma is inherent to all lifeforms, each class of organism will possess a ‘codon bias’ by which the particular codon (of which there may be multiple for one amino acid; degeneracy) will be represented by a comparatively higher concentration of tRNA molecules.
Vibrio natriegens is a gram negative, marine-dwelling prokaryote understood to possess one of the highest growth rates of any organism. This trait would be highly desirable for manufacturing applications where controlled biomineralisation could be harnessed to create solid artefacts in a short timeframe.
As V. Natriegens is not so forth a model organism and was thus omitted as an option from available online tools, I retrieved the codon usage table for Vibrio natriegens from the Codon Usage Database and used the BioInfromatics Reverse Translation Tool to directly generate the most likely to succeed, non-degenerate DNA sequence. This instead required the input of the AA sequence previously retrieved in 3.1.
For HW consistency, I additionally optimised the ‘most likely codon’ MamE sequence for expression in Escherichia Coli (E. Coli) via the tool at Vectorbuilder:

The resulting Codon Adaption Index (CAI) for E. Coli fortunately still appeared to fall within a suitable range of <0.8.
3.4. You have a sequence! Now what?
The mamE sequence could first be integrated in-vitro into a recombinant plasmid, then transformed into V. Natriegen cells via electroporation, by which plasmids enter the cell through the creation of temporary pores in its membrane. The recombinant plasmids should then be transcribed and translated as normal by the appropriate enzymes within the cytosol. Electroporation is advantageous as is both efficient and relatively fast.
3.5. Bonus: How does it work in nature/biological systems?
In prokaryotes such as V. Natriegen, polycistronic transcription for a single mRNA molecule to be translated into different proteins. This is achieved via the inclusion of separate, distinct START and STOP codons, creating separate regions along the same mRNA molecule which are thus translated into distinct proteins.
I attempted to align the flow of information from DNA through polypeptide via the stacking of my three produced Benchling sequences manually. Unfortunately begins to break down in alignment, but accurate for the first three codons:
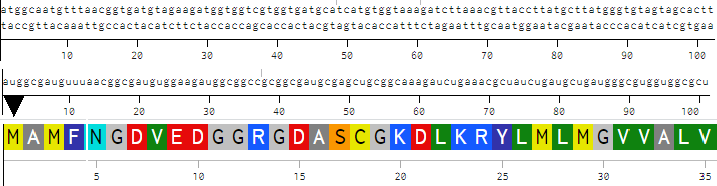
ChatGPT used to format correct alignment:

Part 4: Prepare a Twist DNA Synthesis Order
This process was really fascinating! I would love to learn more about the different inclusions along the way, and of their functionality + importance.
Review my plasmid here.
Part 5: DNA Read/Write/Edit
5.1 DNA Read (i) What DNA would you want to sequence (e.g., read) and why?
It would be intriguing to sequence morphogen-encoding genes (such as the HOX group) in species which have so far not been included. I would love to compare the base composition of different morphogen-encoding genes across distinct species to better understand the mechanisms behind different types of tissue patterning, and perhaps discover more conserved sequences.
(ii) In lecture, a variety of sequencing technologies were mentioned. What technology or technologies would you use to perform sequencing on your DNA and why?
I would select some combination of Single Molecule (third-generation; sequences individual bases directly without prior amplification) and Fluorescent 3D (other; distant in characteristics) to map genes of interest. Fluorescent 3D could be used to first determine the spatial concentration of suspected morphogen-encoding genes in a given tissue, then use Single Molecule to determine finer structural details. The capacity for Single Molecule to produce very long reads is desirable where developmental regions may be large and complex.
Fluorescent 3D:
Input: Hybridization target probe pre-prepared based on theorized gene of interest (GOI). Tissue segment of interest pre-prepared on slide.
Base calling method: iterative cycles of images detect each base as ‘spot in tissue’; corresponds to a particular gene identity.
Output: spatial coordinates for gene expression + cells it is expressed by.
Single Molecule:
Input: cells identified to contain GOI previous step are isolated and cultured, then lysed before separating and purifying DNA. Construction (oligonucleotide synthesis) and addition of nanopore sequencing adapters. Segments loaded into flow cell.
Base calling method: individual DNA molecules pulled through nanopore by sequencing adapter, disrupting baseline ionic current of the detector which is detected and recorded by device. ‘Trace signals’ correspond to a particular base.
Output: Long-read sequences (LRS)
5.2 DNA Write (i) What DNA would you want to synthesize (e.g., write) and why?
Artificially constructing a cluster of genes which allows for relatively precise, customisable control of tissue patterning would be highly appealing. Think programmable osteablasts, where the functional grading of bone-like structures can be controlled to produce an organic car chassis.
(ii) What technology or technologies would you use to perform this DNA synthesis and why?
Multiplexed Oligo Synthesis followed by Gibson Assembly appears a feasible option for the construction of large genes. Many sub-80 bp oligos can first be synthesised in parallel as a means of error-rate limitation, then combined via Gibson Assembly to construct the entire gene. In regards to morphogenic genes, both high precision and accuracy are required as the resulting phenotype is likely highly sensitive to accurate sequence composition.
Multiplexed Oligo Synthesis (in situ, via inkjet printing)
Essential steps: computational design of desired DNA sequence, followed by division into 60-80 bp oligos with overlapping regions. Microarray pre-prepared with contact primer at each oligo synthesis site; single added sequentially in cycles at each site adhering to phosphamidite chemistry process. At termination length, cleaved, retrieved, and amplified.
Limitations: error rate increases as sequence grows larger. Overlapping requirements of gibson assembly requires more cycles to be performed.
Gibson assembly
Essential steps: Addition of exonuclease to prepared oligonucleotides to create sticky overhangs. Segments hybridise at designed complementary overlapping regions. DNA polymerase introduced to integrate missing nt’s at gaps. Ligase seals gaps throughout the incomplete backbone.
Limitations: efficiency decreases as quantity of oligo fragments increases. Precise design of overlapping regions essential.
5.3 DNA Edit (i) What DNA would you want to edit and why?
It would be ideal to edit the genomes of more agricultural crops to introduce robust autofertilisation traits via symbiosis with microorganisms, for use in both developed and developing nations alike. In industrialised agriculture, external fertilisation is responsible for damaging pollution events throughout adjacent ecosystems. In developing regions where soil viability is poor, endemic low-yields resulting from limited fertilizer access drive perilous consequences. As the most abundant atmospheric gas, fixation of nitrogen directly from where directly where it is required makes significant sense.
(ii) What technology or technologies would you use to perform these DNA edits and why?
CRISPR/Cas9 currently appears the most desirable method for performing DNA modifications within plant embryo cells, particularly due to its efficiency and precision.
Preparation + inputs: Design gRNA to match target sequence within plant embryo; incorporate into suitable vector alongside Cas9 gene.
Limitations: Cuts at sequences similar to DNA target in embryo possible, requiring careful planning and validation post-edit. Editing efficiency dependent on accessibility of target gene.
References
Jerlie Mhay Matres, H., Hilscher, J., Datta, A., Armario-Nájera, V., Baysal, C., He, W., Huang, X., Zhu, C., Valizadeh-Kamran, R., Trijatmiko, K.R., Capell, T., Christou, P., Stoger, E. and Slamet-Loedin, I.H. (2021). Genome editing in cereal crops: an overview. Transgenic Research, 30(4), pp.461–498. https://doi.org/10.1007/s11248-021-00259-6
Lee, H.H., Ostrov, N., Wong, B.G., Gold, M.A., Khalil, A.S. and Church, G.M. (2016). Vibrio natriegens, a new genomic powerhouse. https://doi.org/10.1101/058487
Quinlan, A., Murat, D., Vali, H. and Komeili, A. (2011). The HtrA/DegP family protease MamE is a bifunctional protein with roles in magnetosome protein localization and magnetite biomineralization. Molecular Microbiology, 80(4), pp.1075–1087. https://doi.org/10.1111/j.1365-2958.2011.07631.x
Wan, J., Ji, R., Liu, J., Ma, K., Pan, Y. and Lin, W. (2024). Biomineralization in magnetotactic bacteria: From diversity to molecular discovery-based applications. Cell Reports, 43(12), p.114995. https://doi.org/10.1016/j.celrep.2024.114995
Weinstock, M.T., Hesek, E.D., Wilson, C.M. and Gibson, D.G. (2016). Vibrio natriegens as a fast-growing host for molecular biology. Nature Methods, 13(10), pp.849–851. https://doi.org/10.1038/nmeth.3970