Week 4 HW: Protein Design Part i
Part A: Conceptual Questions
1. How many molecules of amino acids do you take with a piece of 500 grams of meat? (on average an amino acid is ~100 Daltons)
500 grams of beef contains ≈ 112 g of protein. 1 dalton corresponds to ≈ 1.6605×10-24 gram. 1 amino acid (100 daltons) ≈ 1.6605×10−22 grams. 112 grams / 1.6605×10−22 grams ≈ 6.745×1023.
The 112 g of protein contained with 500 g of beef represents ≈ 6.745×1023 molecules of amino acid!
2. Why do humans eat beef but do not become a cow, eat fish but do not become fish?
One aspect of “cowness” could be considered the successful construction, transcription, and translation of genetic material which results in a heritable bovine phenotype; physical traits and behaviours which can be associated with cows. For this to be possible, genetic material must not only be incorporated inherently across all tissues at the cellular level, but be intact, and complemented by the correct composition of functional enzymes.
When humans consume tissues from another organism, a series of digestive enzymes effectively destroy the structure of foreign macromolecules (such as DNA), into recyclable constituent molecules (like nucleotides). Even if foreign genetic material were to remain intact inside the stomach, it would be impossible for it to be integrated across all cells at the scale required to produce observable “cowness”.
3. Why are there only 20 natural amino acids?
This is a very intriguing question by which the precise answer still remains elusive. One theory suggests that the canonical 20 were selected gradually on the basis of parsimony (extreme stinginess, or frugality), where monomers favoured provided the widest spectrum of utility for the lowest investment of energy and material (Bywater, 2018).
4. Can you make other non-natural amino acids? Design some new amino acids.
The synthesis of non-canonical amino acids (NcAA’s) is possible, with large advancements in the field made in the early 21st century. For my attempt at NcAA design, see 4.1 below.
5. Where did amino acids come from before enzymes that make them, and before life started?
Amino acids are theorised to have emerged abiotically via complex chemical processes that were facilitated by the proximity of specific chemical reactants and physical parameters (heat, electricity). It is also possible that amino acids arrived on earth from extraterrestrial sources via a meteorite(s), where other methods of synthesis may have first preluded their arrival (Kirsching, 2022).
6. If you make an α-helix using D-amino acids, what handedness (right or left) would you expect?
Amino acids can be characterised by their chirality, with pairs known as enantiomers representing their two possible configurations (L and D). This pair represents the ‘mirror-image’, non-superimposable structural quality central to chirality. In living organisms, L-aminos are almost exclusively used to construct proteins. In α-helices, this curiously results in the final protein having a D-configuration. Using D-aminos to form α-helices instead yields an overall L-configuration in the resultant structure.
7. Can you discover additional helices in proteins?
Additional types of helices have been discovered and researched, such as the 3-10 helices and Pi helices!
8. Why are most molecular helices right-handed?
Most molecular helices appear right-handed as a resultant property of their left-handed amino monomers. This D configuration is said to improve steric hindrance (reduce reactivity of specific molecules in macromolecules) and energetic stability (qualities of hydrogen bonds in backbone of a-helices are optimised).
9. Why do β-sheets tend to aggregate? What is the driving force for β-sheet aggregation?
The chemical structure of the edges of β-sheets promote aggregation, as it readily encourages the formation of hydrogen bonds with adjacent β-sheets to form larger aggregates.
4.1: Design some new amino acids.
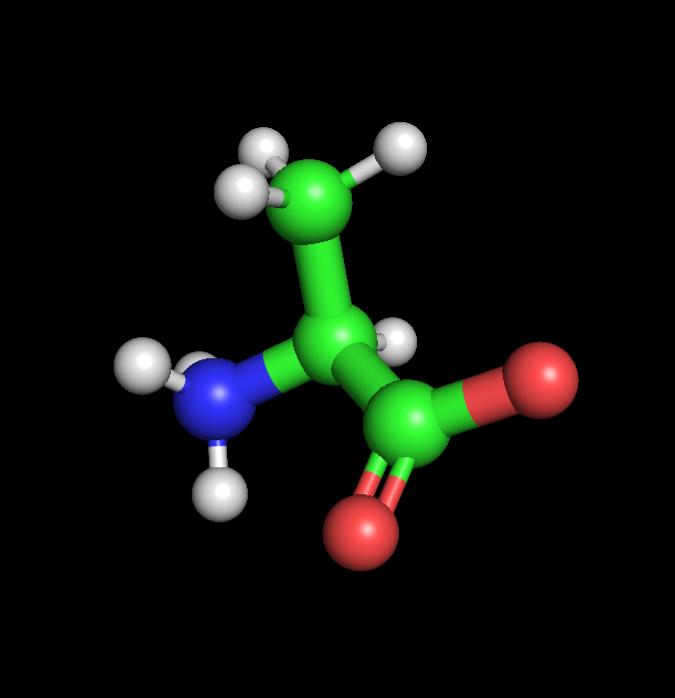 | 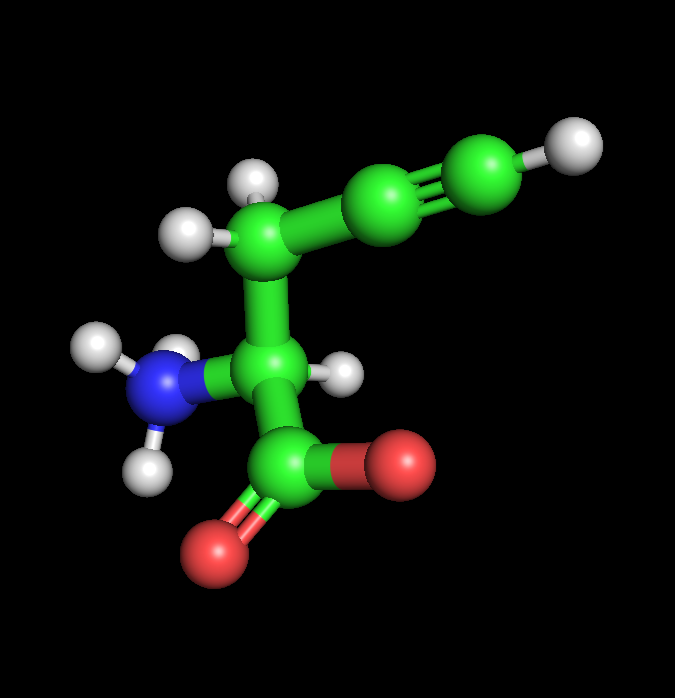 | 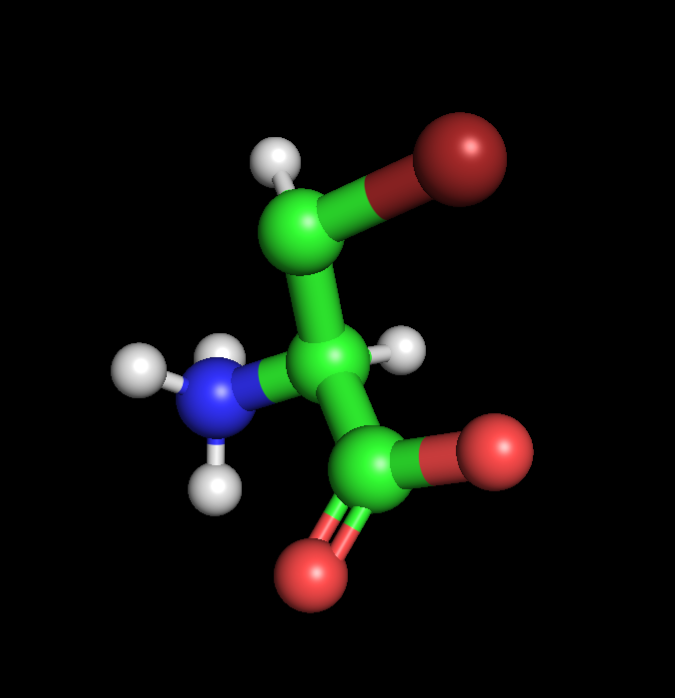 |
| Alanine | Propargyl-valine | Halogenated-valine |
I found and followed a short tutorial online detailing how Pymol can be used to quickly design NcAA models, starting with Valine as per the demonstration. With an extremely limited background in chemistry, I used chatGPT to aid me in selecting and positioning functional groups. I decided to create 2 NcAA’s, one optimised for click-reactivity (Acetylene) and photo-reactivity (Bromine), via the separate addition of acetylene and bromine to the methyl group of each Alanine respectively. ChatGPT prompts:
Part B: Protein Analysis and Visualization
Many bacterial species are capable of cell-to-cell communication as a response to changes in localised population density. This system is known as quorum sensing.
1. Briefly describe the protein you selected and why you selected it.
I have selected LuxR, a transcriptional activator, and one of the first proteins (alongside LuxL) to be identified as integral in a QS pathway. LuxR works with LuxL in the prokaryote Vibrio fischeri to permit bioluminescence once a particular population density is reached. QS pathways are particularly interesting to me as I would like to understand how they may be leveraged to enable external control of a bacterial population’s behaviour at the cellular level via controlled chemical signalling.
2. Identify the amino acid sequence of your protein.
LuxR has a length of 250 amino acids. The most common is Isoleucine (I), which appears 28 times. Using BLAST, LuxR appeared to have approx. 186 possible homologs after using the E value to cull the total pool of 250 matches. The protein is also the title for the family it is contained within: LuxR-type transcriptional regulators.
3. Identify the structure page of your protein in RCSB.
I couldn’t find a non ML-resolved match for the LuxR sequence from V. fischeri, which was intriguing considering it was supposedly the first organism where this protein was identified.
I did however find a direct match that was resolved with Alphafold DB on 2021-12-09 with a global model confidence of 88.61%. I couldn’t determine if there were additional molecules in the structure. There was also a homolog called 7AMT resolved though x-ray diffraction, which was deposited on 2020-10-09 with an excellent resolution of 2.60 Å. Both proteins belong to the transcription classification family.
4. Open the structure of your protein in any 3D molecule visualization software.
I continued with 7AMT as it was readily available in the correct format from PDB.
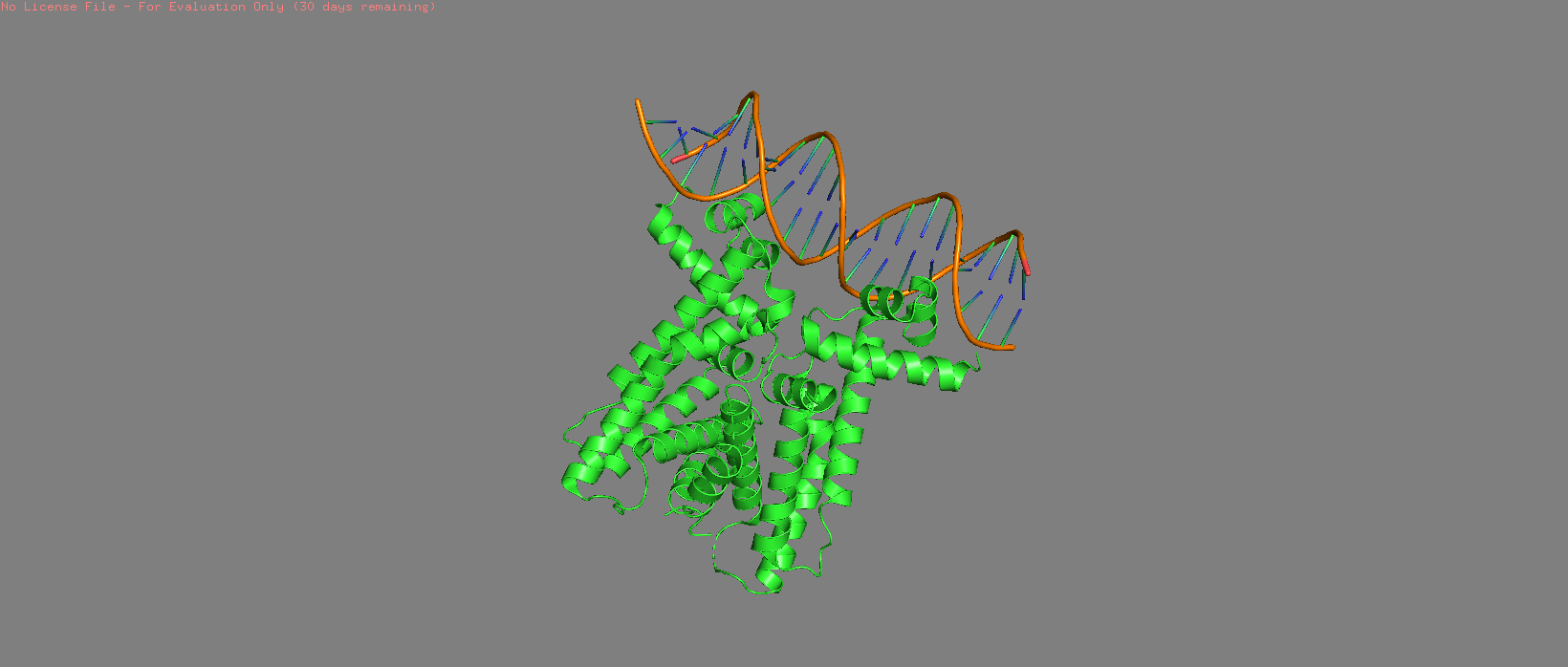 Cartoon |  Cylinder |  Stick + Sphere |
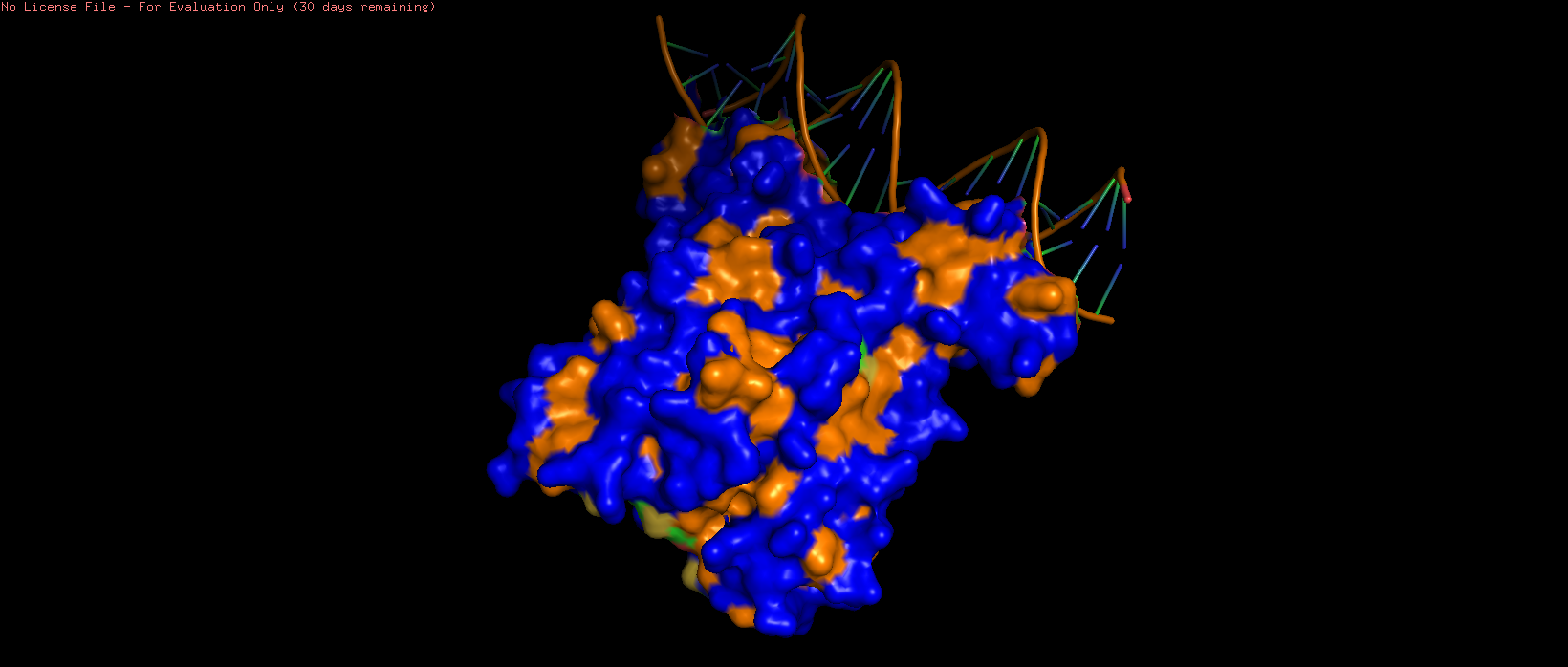 | 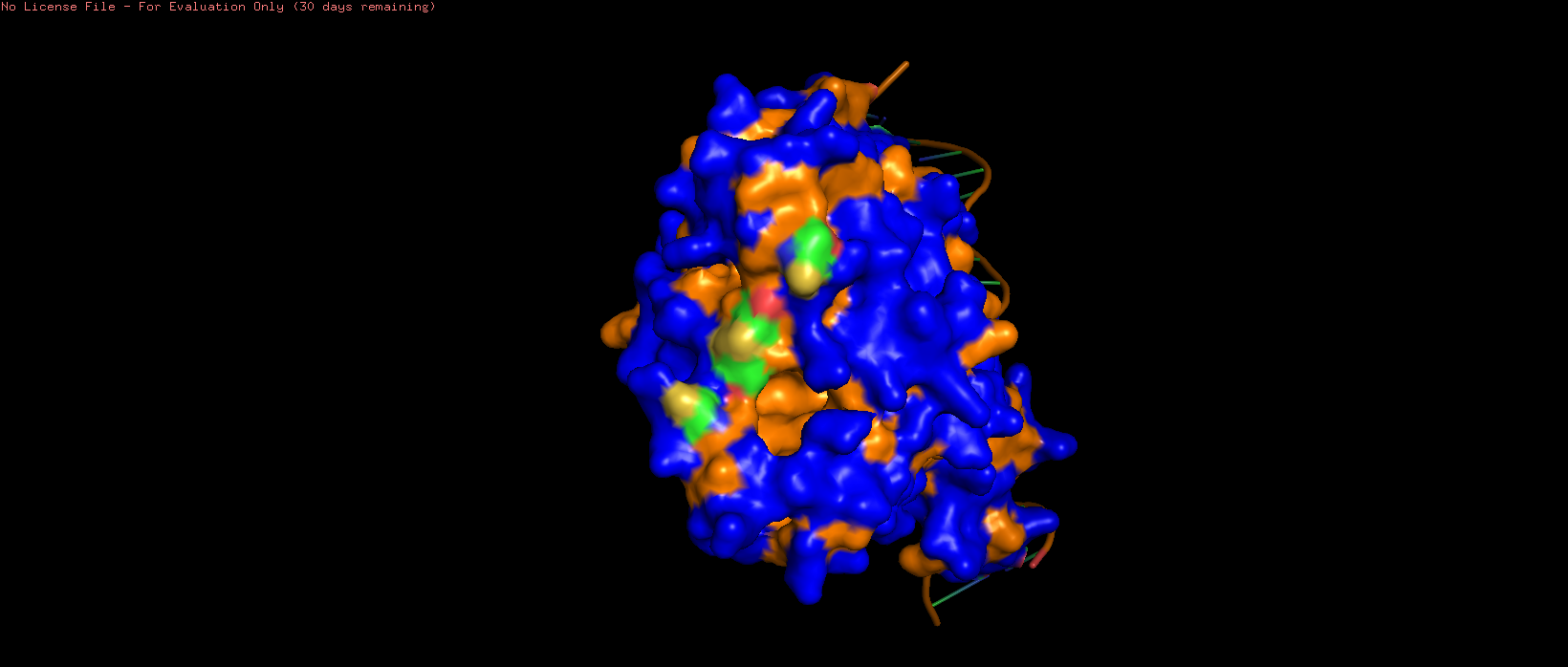 | 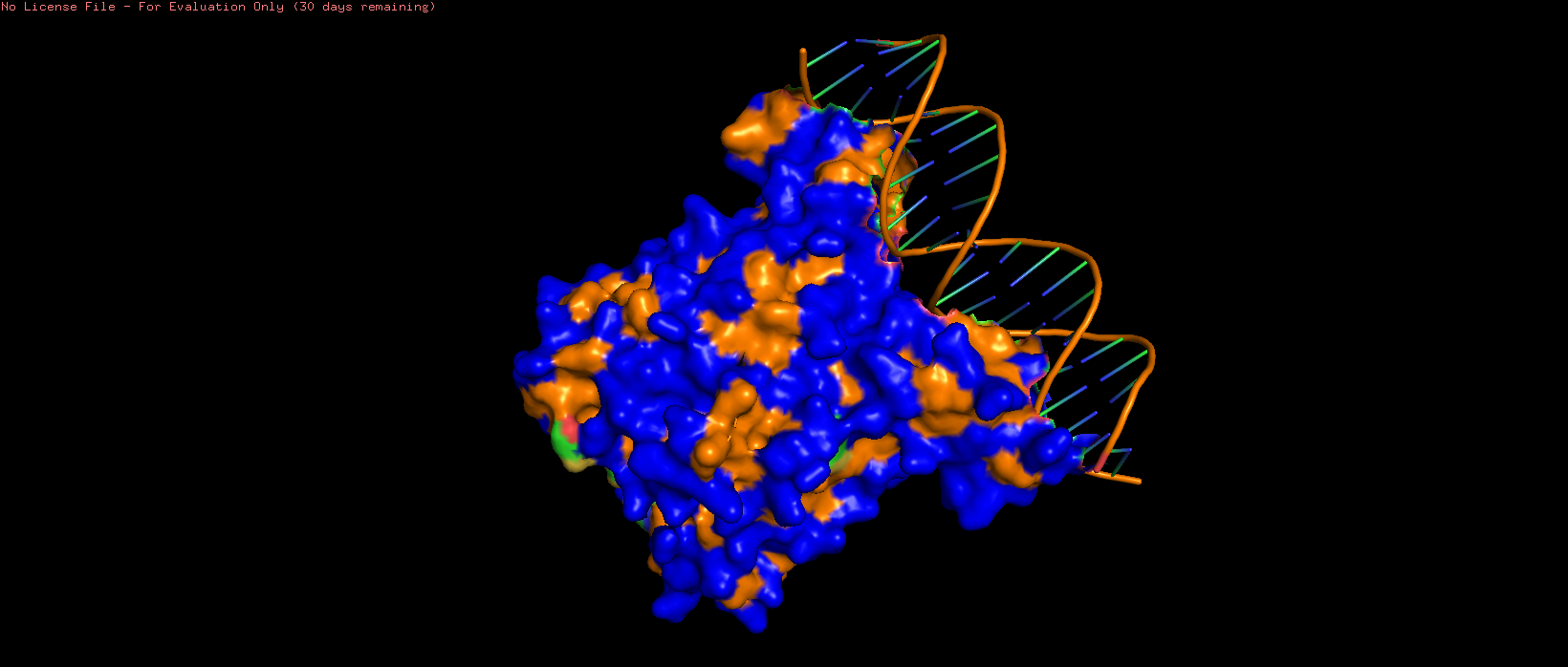 |
| 7AMT surface structure visualised with hydrophilic (blue) and hydrophobic (orange) regions highlighted. Several pits can be observed, particularly on the side opposite to the DNA binding region. | ||
The surface residues are more commonly hydrophilic, with internal, ‘hidden’ residues appearing to be mainly hydrophobic.
Part C: Using ML-Based Protein Design Tools
C.1: Protein Language Modeling
I decided to progress with 7AMT as its prior resolvement through x-ray diffraction makes it a good candidate for comparison and testing with the provided ML tools!
1. Deep Mutational Scans
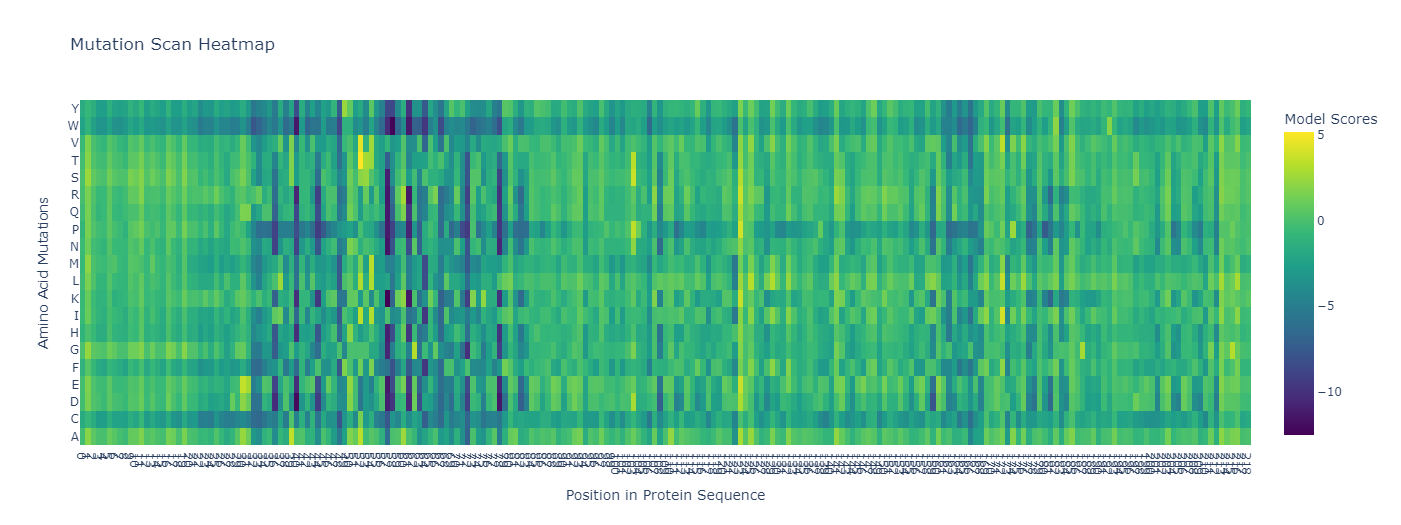 |
7AMT: Unsupervised Deep Mutational Scan. |
Substitution for Tryptophan, proline, and cystine across most residues appears to have a negative effect. Residues 50-55~ appear highly conserved, as mutations here appear problematic for functionality.
2. Latent Space Analysis
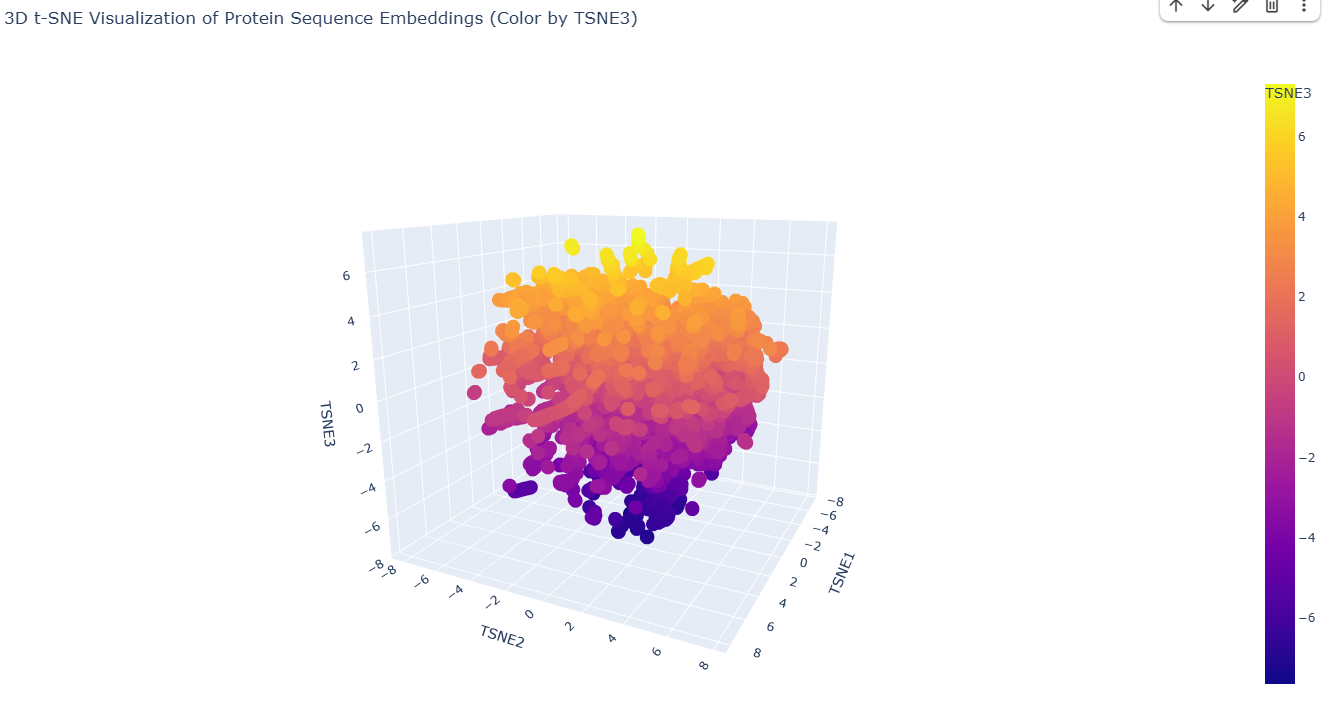 |
7AMT: Latent Space Analysis. |
The visualisation generated is free from distinct neighbourhoods, instead appearing relatively smooth and continuous between residues. I was unfortunately unable to embed and visualise 7AMT, however I hope to return to this beyond the timeline of the course!
C.2: Protein Folding
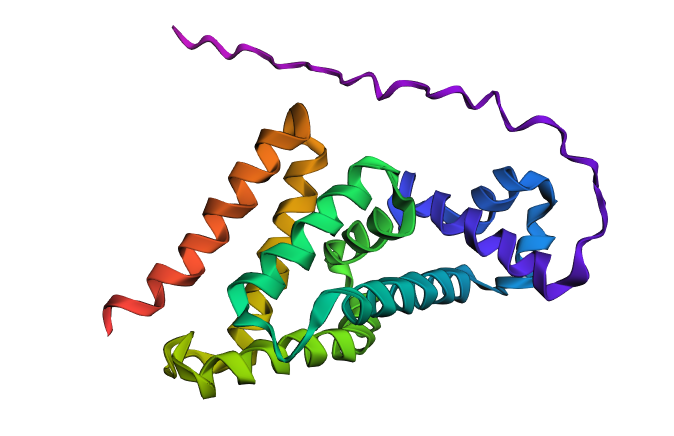 | 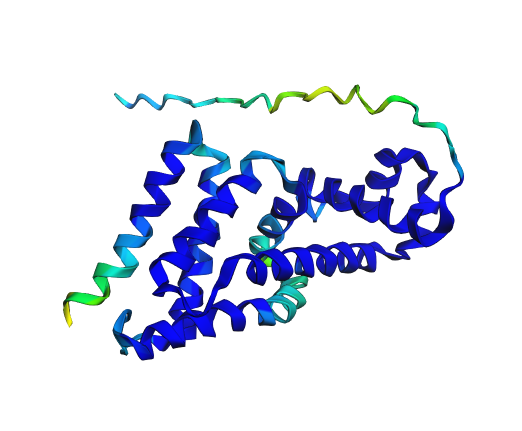 |
| 7AMT ESMfold 2: rainbow for clarity. | 7AMT ESMfold 1: colour gradient representing confidence of resolution. |
While somewhat reminiscent, folding of 7AMT in ESMfold did not perfectly match the original coordinates retrieved from UniProt. The structure appeared highly susceptible to change upon alteration of the original sequence.
C.3: Protein Generation
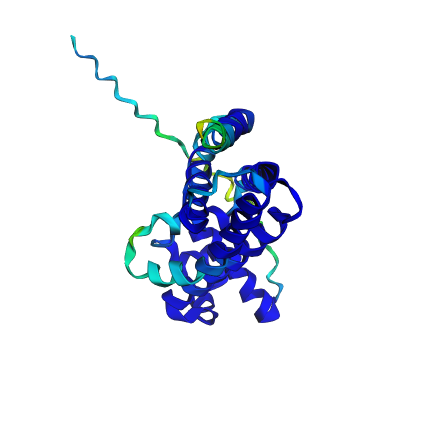 | 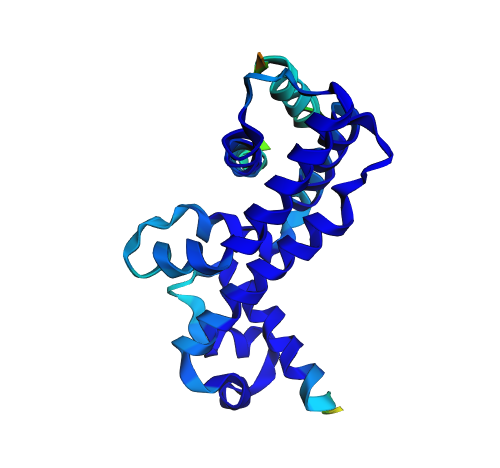 |
| Original structure (confidence) | Predicted structure (confidence) |
Intriguingly, the reverse folding processes did appear to yield quite an accurate prediction compared to ESMfold, as indicated by the dark blue colouring of the structure on the right. A quick visualisation in ESMfold yielded a similar result.
Part D. Group Brainstorm on Bacteriophage Engineering
Group formed, initial brainstorm session conducted:
Please refer to the group final project section for our brainstorming session writeup.
References
Foodstandards.gov.au. (2026). Australian Food Composition Database. [online] Available at: https://afcd.foodstandards.gov.au/fooddetails.aspx?PFKID=F000678 [Accessed 6 Mar. 2026].
Bywater, R.P. (2018). Why twenty amino acid residue types suffice(d) to support all living systems. PLoS ONE, 13(10), pp.e0204883–e0204883. https://doi.org/10.1371/journal.pone.0204883
Cooley, R.B., Arp, D.J. and Karplus, P.A. (2010). Evolutionary origin of a secondary structure: π-helices as cryptic but widespread insertional variations of α-helices that enhance protein functionality. Journal of Molecular Biology, 404(2), pp.232–246. https://doi.org/10.1016/j.jmb.2010.09.034
Doig, A.J. (2017). Frozen, but no accident – why the 20 standard amino acids were selected. The FEBS Journal, 284(9), pp.1296–1305. https://doi.org/10.1111/febs.13982
Kirschning, A. (2022). On the evolutionary history of the twenty encoded amino acids. Chemistry – A European Journal, 28(55). https://doi.org/10.1002/chem.202201419
Munier, R. and Cohen, G.N. (1956). Incorporation d’analogues structuraux d’aminoacides dans les protéines bactériennes. Biochimica et Biophysica Acta, 21(3), pp.592–593. https://doi.org/10.1016/0006-3002(56)90207-4
Oliveira, N.M., Foster, K.R. and Durham, W.M. (2016). Single-cell twitching chemotaxis in developing biofilms. Proceedings of the National Academy of Sciences, 113(23), pp.6532–6537. https://doi.org/10.1073/pnas.1600760113
Soto-Aceves, M.P., Diggle, S.P. and Greenberg, E.P. (2023). Microbial primer: LuxR-LuxI quorum sensing. Microbiology, 169(9). https://doi.org/10.1099/mic.0.001343