Week 5 HW: Protein Design Part ii
Part 1: Generate Binders with PepMLM
Human SOD1 sequence (without A4V):
A4V represents a point mutation within the 4th codon of the human SOD1 gene. This results in the substitution of the conventional alanine for a valine amino acid.
Mutant SOD1 sequence (with A4V):
Four experimental peptides were then generated using the PepMLM tool:
| Binder | Pseudo Perplexity |
|---|---|
| HRYYPVAVRLKE | 11.513239315710866 |
| HRYPVVAVAWKE | 14.299230176233568 |
| KRYPPVAARWKE | 20.264871722997526 |
| WLYPVAAARHKE | 20.749563629245603 |
Part 2: Evaluate Binders with AlphaFold3
| Entry | Binder | Pseudo Perplexity | ipTM | Description | Image |
|---|---|---|---|---|---|
| 🎮 | FLYRWLPSRRGG | — | 0.31 | Appears tightly surface-bound, primarily binding to the dimer interface. | 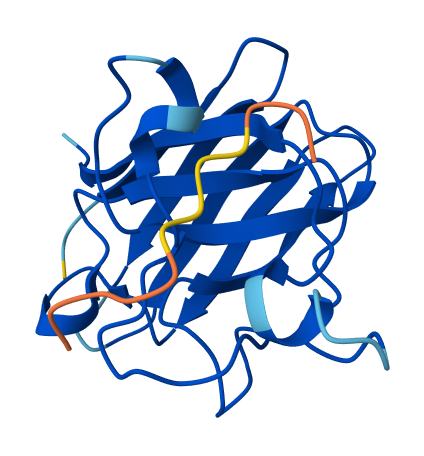 |
| 0 | HRYYPVAVRLKE | 11.513239315710866 | 0.26 | Appears weakly surface bound to the dimer interface; half the residues do not appear bound. | 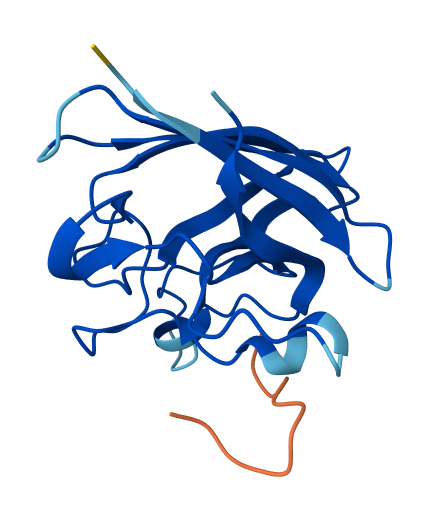 |
| 1 | HRYPVVAVAWKE | 14.299230176233568 | 0.26 | Appears weakly surface bound; similar positioning to HRYYPVAVRLKE, but reversed in that the central residues appear to bind while the flanks do not. | 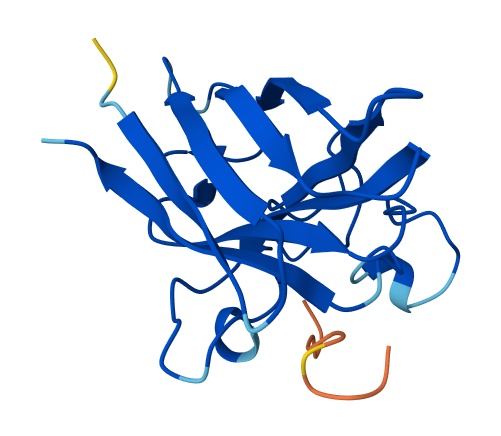 |
| 2 | KRYPPVAARWKE | 20.264871722997526 | 0.34 | Appears to bind superficially with an alpha helix at residue 56-61; also possesses its own alpha helix. In proximity to the rear of β-barrel (opposite end to N and C terminus). | 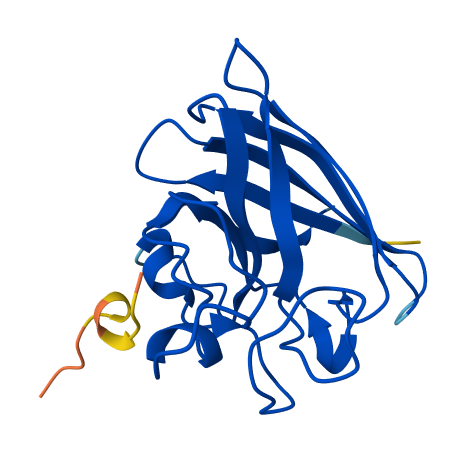 |
| 3 | WLYPVAAARHKE | 20.749563629245603 | 0.35 | Appears strongly surface bound with proximity to the rear of β-barrel. | 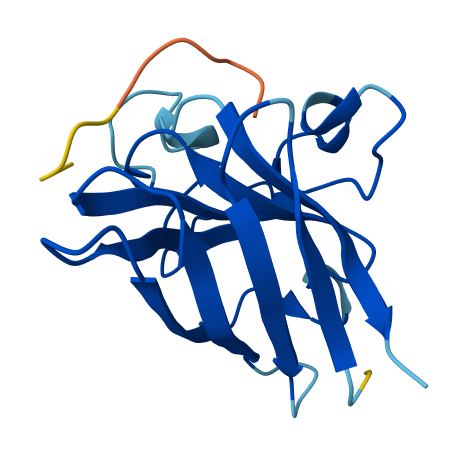 |
Compared to a control ipTM of 0.31, binders appeared tightly clustered into two distinct groups; 0.26 and 0.34~. The ipTM of binders KRYPPVAARWKE and WLYPVAAARHKE appeared to exceed the known binders score or 0.31, with 0.34 and 0.35 respectively. When considering ipTM alongside observed binding characteristics, WLYPVAAARHKE appears the most theoretically desirable of the set generated.
Part 3: Evaluate Properties of Generated Peptides in the PeptiVerse
| Binder | Prediction Summary |
|---|---|
| HRYYPVAVRLKE | 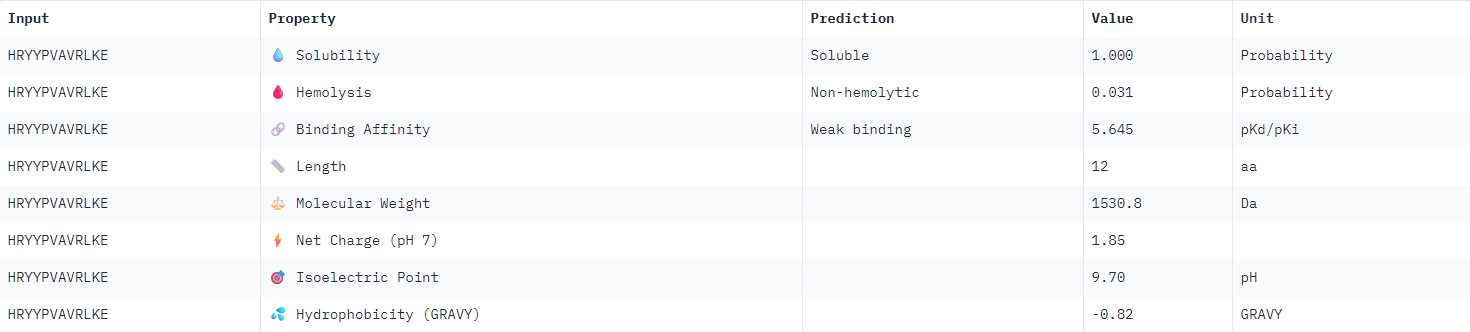 |
| HRYPVVAVAWKE | 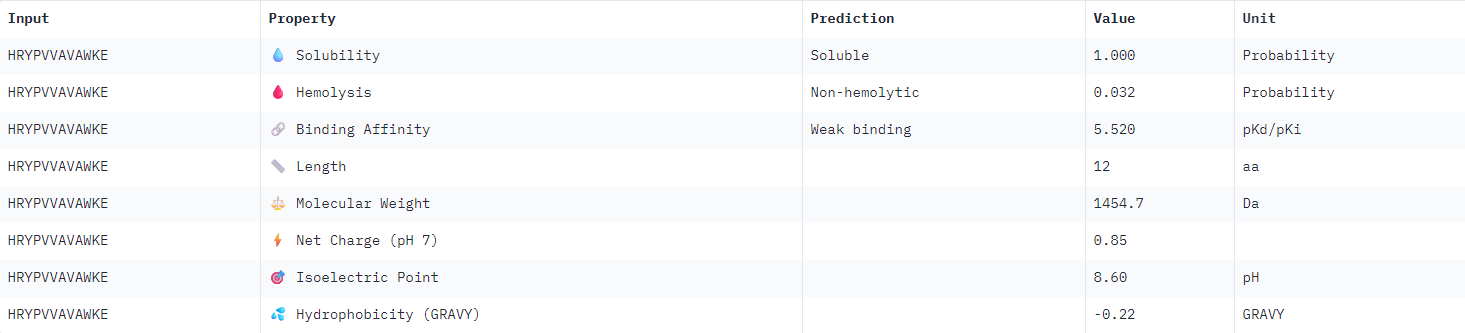 |
| KRYPPVAARWKE | 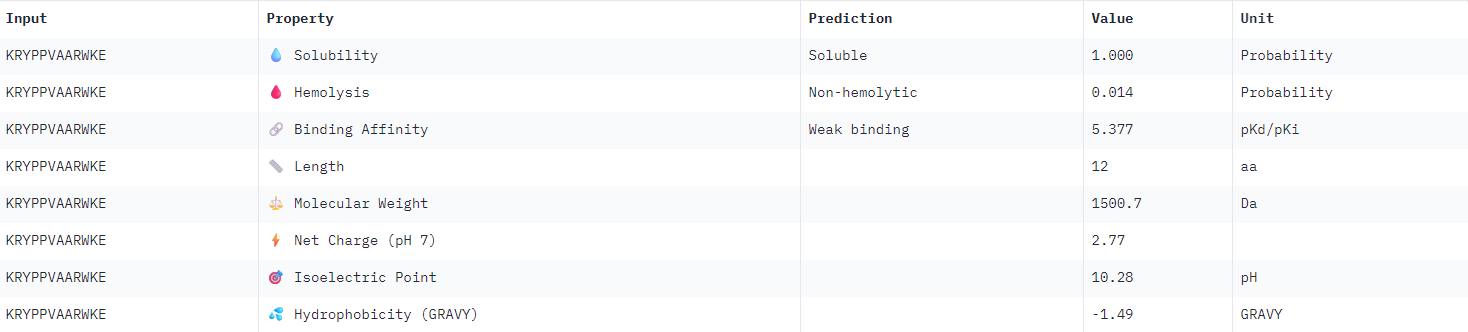 |
| WLYPVAAARHKE | 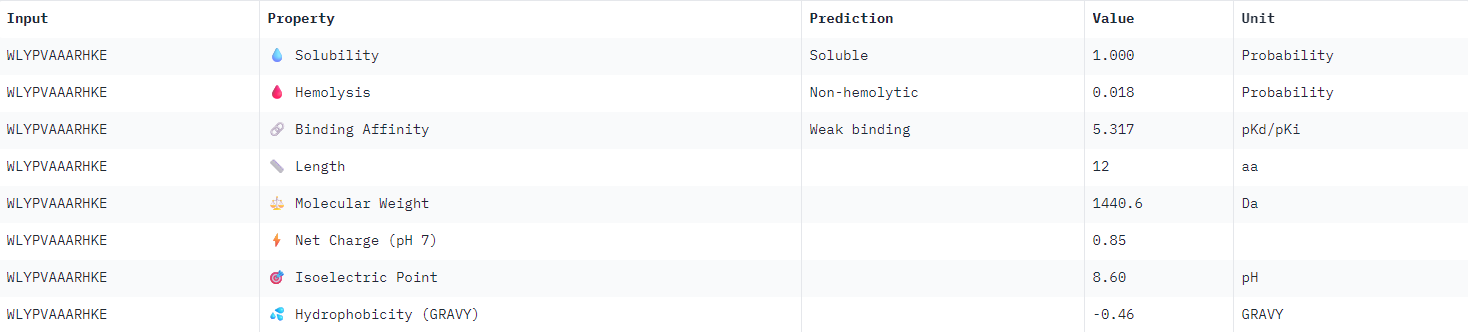 |
All binder candidates were predicted to have weak binding affinity, even those that appeared visually in AlphaFold to be potentially strong candidates. I was not able to discern any strong patterns between Peptiverse results and previous ipTM predictions. The most promising binder from the previous section (WLYPVAAARHKE) intriguingly appeared the second least hydrophobic, and was predicted to have the weakest binding affinity.
I would select KRYPPVAARWKE to advance for further examination given the considerations outlined above. It possesses the highest predicted solubility, while remaining the second strongest candidate identified in the previous section.
Part 4: Generate Optimized Peptides with moPPIt
I provided the A4V mutant SOD1 sequence and specified motif 50-70 for binding, as this region appeared promising as a target from previous observations in AlphaFold. Objective weights for Affinity and Motif selections were kept default (1).
| Rank | Binder | Predicted Affinity |
|---|---|---|
| 1 | AWWEYVWWWWCV | 8.3275 |
| 2 | GYYGCYGAVYYY | 8.3254 |
| 3 | CTSCCYVGWCWW | 8.2258 |
| 4 | FAWYWPCYWYYR | 8.2060 |
| 5 | YCVYCYDAYVWW | 8.1153 |
| 6 | DGDCRYCLHCCW | 8.1107 |
| 7 | AVYCYYVCRNWW | 7.9624 |
| 8 | GSEYWWYWWHYT | 7.7270 |
| 9 | MVAGIWVWWVAR | 7.3000 |
| 10 | AYYTRVHWPCVW | 6.9906 |
Unlike PepMLM, moPPIt is intended to bias the generation of binders towards specific residue indices, providing a finer degree of control given particular clinical (or functional) objectives. Intriguingly, they appear much more varied in residue composition compared to the PepMLM run.
Conducting a series of ligand-binding assays would be the best approach for identifying binders for advancement into clinical trials. The equilibrium dissociation constant KD is the universal standard for confirming ligand-target binding affinity; I would employ surface plasmon resonance (SPR) as the final method of filtering candidates via KD for selection.
I used AlphaFold to predict the binding location and ipTM of the top moPPit binder as I was curious to compare with the earlier outcome. Curiously, it formed quite a large alpha helix:
 |
| AWWEYVWWWWCV (PD: 8.3275) |
Part C: Final Project: L-Protein Mutants
Please refer to the group final project.
References
Hahn, D., Bayly, C., Boby, M.L., Bruce Macdonald, H., Chodera, J., Gapsys, V., Mey, A., Mobley, D., Perez Benito, L., Schindler, C., Tresadern, G. and Warren, G. (2022). Best Practices for Constructing, Preparing, and Evaluating Protein-Ligand Binding Affinity Benchmarks [Article v1.0]. Living Journal of Computational Molecular Science, [online] 4(1), p.1497. doi:10.33011/livecoms.4.1.1497.