Week 6 HW: Genetic Circuits Part i
DNA Assembly
The primary component of Phusion High-Fidelity PCR Master Mix is the polymerase Phusion, an alternative to standard taq polymerase which provides upwards of 40x greater fidelity (the accuracy at which a template strand is replicated) comparative to taq. It also contains free-deoxynucleotides (bases for formation of replicated strand) and a reaction buffer (provides optimal pH conditions for Phusion polymerase activity).
Factors may include primer melting temperature, composition of reaction buffer, characteristics of target sequence, and type of polymerase. For example, primers containing higher concentrations of guanine and cytosine bases will possess a higher melting temperature due to the strength imparted by the triple hydrogen bonds of GC.
PCR involves the amplification and ‘construction’ of fragments from a provided template sequence, while restriction digests produce linear fragments from the repeated ‘truncation’ of larger fragments within a concentrated pool (via restriction enzymes).
- Both methods are commonly used in conjunction.
- Both methods produce linear double-stranded fragments.
- The result of both methods may be assayed via gel electrophoresis.
Primers with the appropriate 20-40bp overlaps must be precisely designed and validated for use prior to their use as the initiation point of PCR. This ensures the required 5’ to 3’ orientation and overlaps are present to permit the successful cloning via Gibson assembly.
Simple diffusion permits the entry of cloned plasmids into E. coli cells permitted that heat shock or electroporation techniques have been used prior, as these methods generate pores within the plasma membrane that would otherwise be impassable.
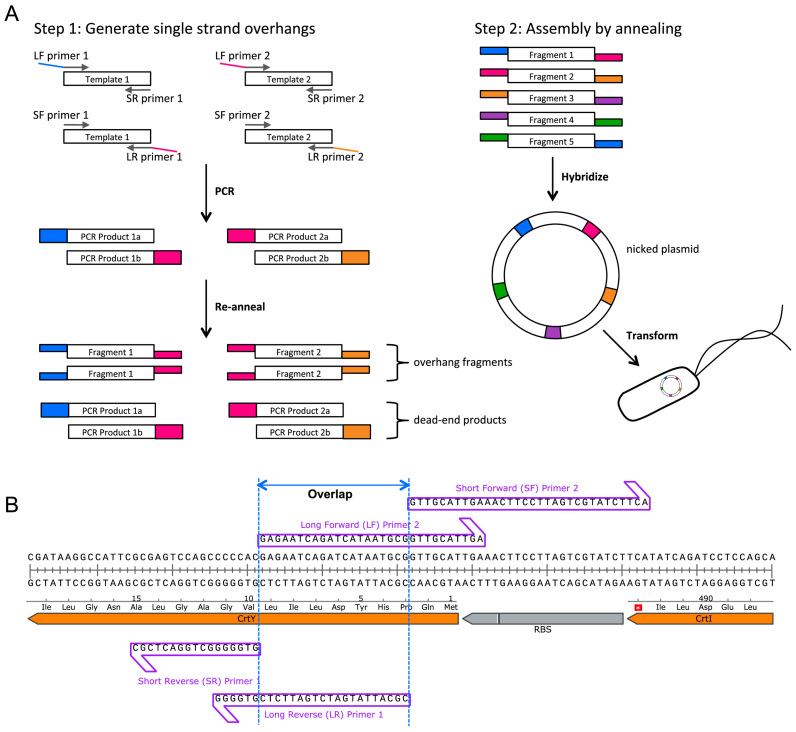
Twin-Primer assembly is an non-enzymatic, alternative technique of assembling PCR-amplified fragments into plasmids, while retaining the necessary efficacy and fidelity of conventional methodologies:
Segment the overall sequence in-silco into a series of fragments with complimentary designed overlaps between adjacent fragments, with two ‘sticky ends’ required per fragment. Overlaps are characteristically 16-20 bp long, and are selected to possess a universal melting temperature of approximately 50 C.
Amplification is then performed separately, twice per fragment, with each iteration using a different set of primer pairs. Per fragment, this produces: A, sequence with overlap to previous fragment, and B, sequence with overlap to next fragment.
The two products (A and B) are then combined to generate two separate versions of the same fragment however featuring unique overlapping regions.
Post mix., the PCR’ed products are denatured to generate ssDNA, before controlled cooling to generate hybrid ‘duplex intermediates’.
All duplex intermediates are then incubated collectively, permitting the annealing of overhangs from complimentary fragments, and ultimately assembling a plasmid (albeit with nicked backbone)
Upon transformation into the desired host, bacterial machinery is co-opted to phosphorylate DNA ends and ligate nicks throughout the DNA backbone, ultimately resulting in a complete plasmid.
 |
Asimov Kernel
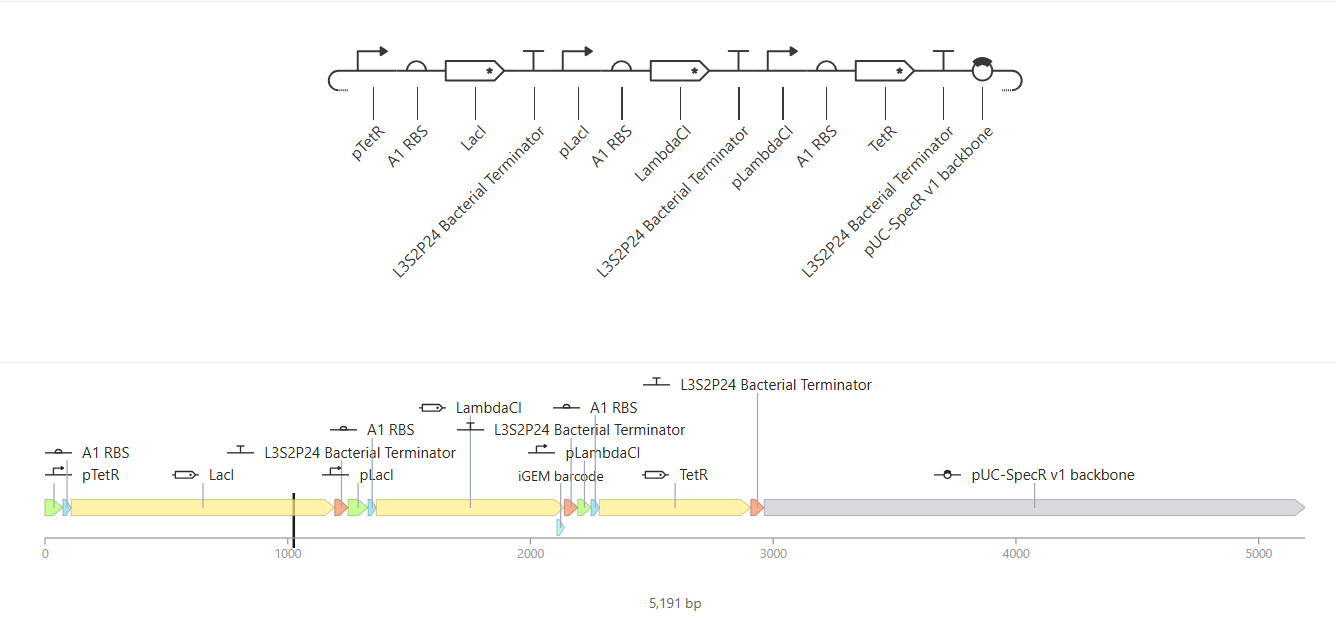
The Repressilator
 |
The Repressilator construct. Although its title is very fitting, it's still such a cool name! |
Iteration 1
- Began by referencing the Repressilator paper: source
- Unsure of the appropriate RBS to use prior to each coding region. There’s a fair few available in ‘Charecterized Bacterial Parts’ repository! Wasn’t able to locate BBa_B0034, the original RBS utilised within the construct described in the paper. Selected S1 RBS at random; lets see how it goes!
- I was able to locate and insert pSC101, the original low-copy backbone used by Elowitz & Liebler!
- Stock settings were employed in the initial simulation (E. coli, 72 hours, 10 min timestep).
- Results were not aligned with the oscillatory behaviour of the Repressilator. Protein expression appeared to peak across each gene, then remain constant.
Iteration 2
- Realised almost immediately upon inspection of the repressilator in the bacterial samples folder that I had failed to include terminators!
- Retained pSC101 and S1 RBS.
- Stock settings (E. coli, 72 hours, 10 min timestep).
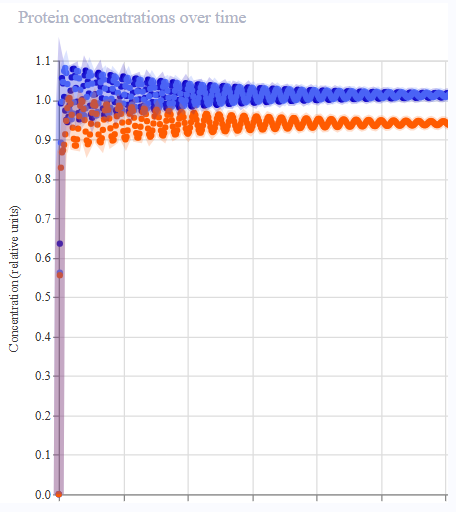
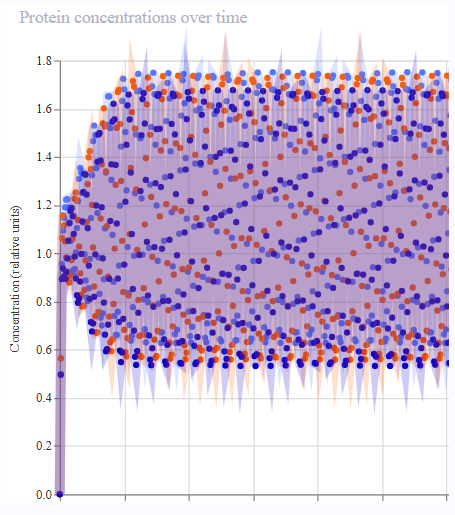
- Results were much more reminiscent of reference! Oscillatory expression behaviour appeared to ‘tighten’, with the period each gene was expressed narrowing throughout the simulation.
Iteration 3
- Finally adjusted RBS to A1 and backbone to pUC-SpecR v1 (as featured within the reference Repressilator)
- Promoters still appear in slightly different locations to reference, however the overall architecture appears to match that of Elowitz & Liebler.
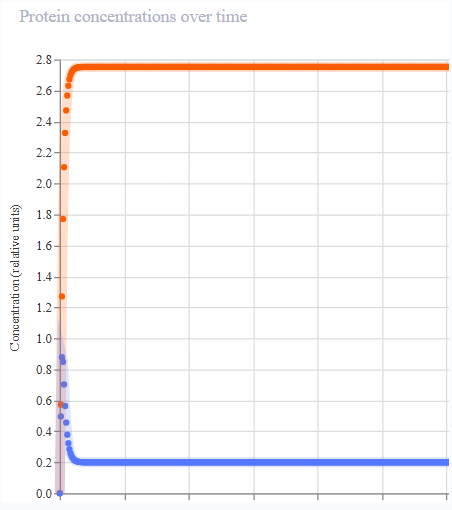
- Results were again significantly different; actually less relevant than those obtained in iteration 2!
- Definitely suspicious that the layout of the promoters is altering the outcome.
Iteration 4
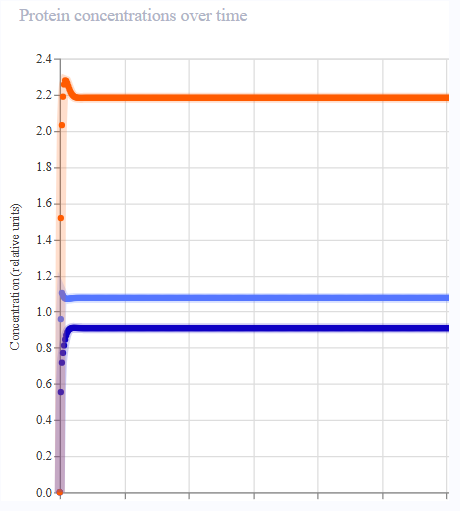
- Precisely copied the layout of the reference construct.
- Results matched the reference.
 |  |  |  |
| Iteration 1 | Iteration 2 | Iteration 3 | Iteration 4 |
Custom Constructs
Construct A
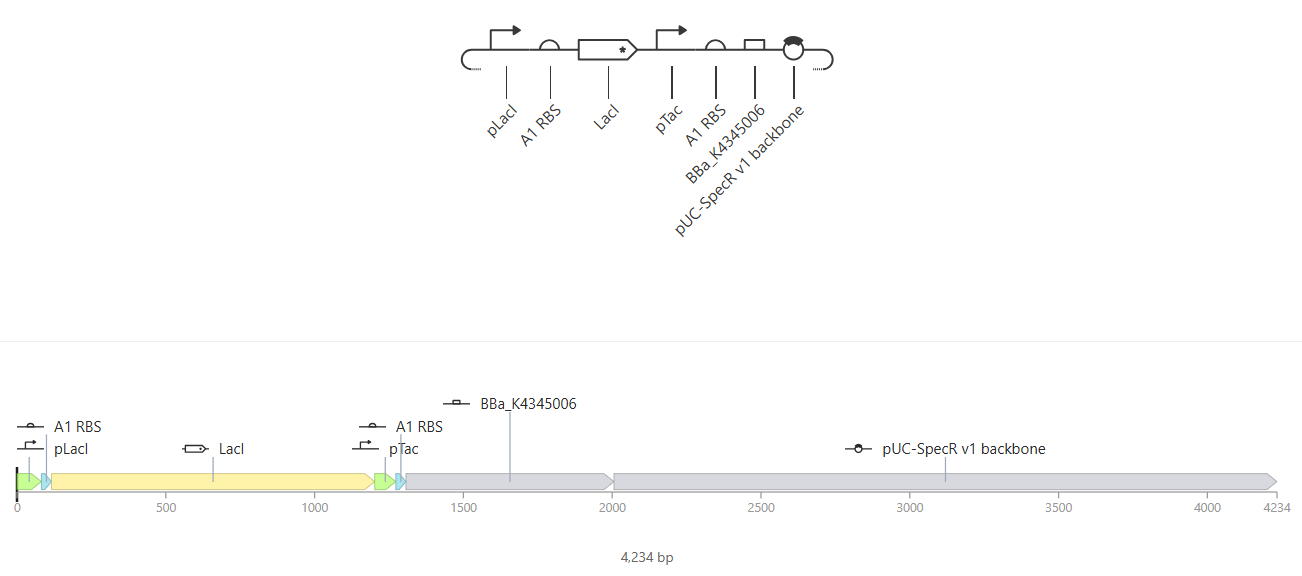 |
Construct A |
I wanted to understand how the pTac promoter could be used in conjunction with IPTG ligands to control expression of a specific gene. In construct A, the intention is that the transcription of LacL produces a repressor preventing the transcription of mKate, and hence its translation to yield the fluorescent reporter protein.
Simulation 1
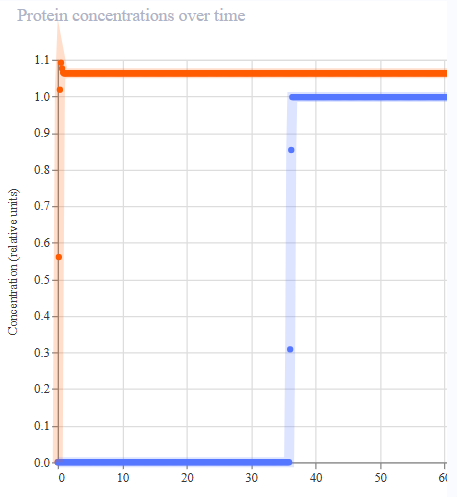
- Settings: E. coli, 72 hours, 10 min intervals. Ligand (IPTG) introduced at max concentration at 36 hours.
- Appeared to work first try! LacL was transcribed at a constant rate, repressing the activity of the pTac promoter. At 36 hours, the introduction of IPTG functioned as a structural analog to allolactose, inhibiting the activity of the repressor and permitting the transcription of mKate.
 | 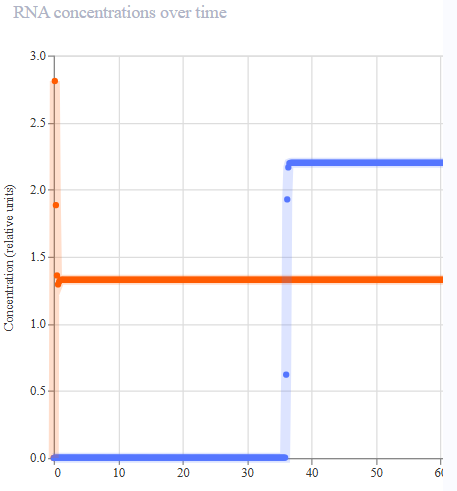 | 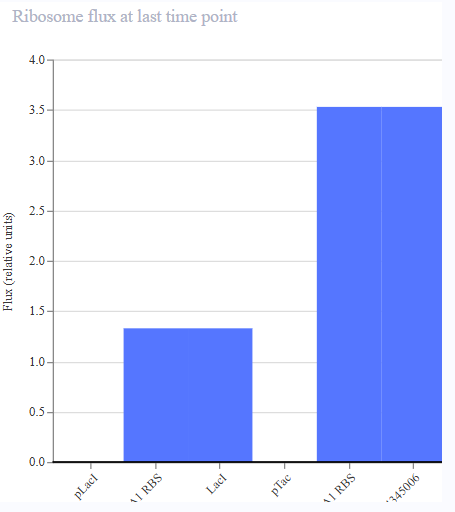 | 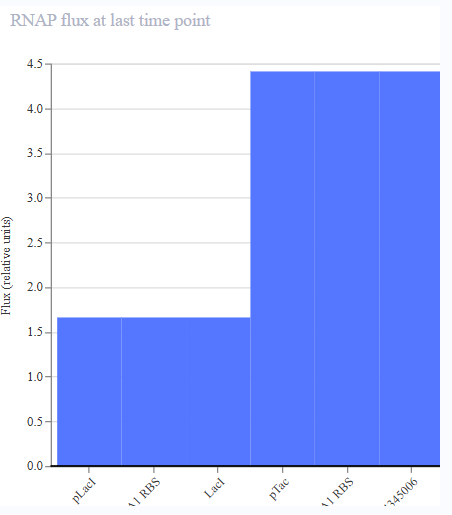 |
| Protien Concentrations | RNA Concentrations | Ribosome Flux | RNAP FLux |
Construct B
Attempt at a pseudo AND gate. Both L-Arabinose and IPTG must be present for each component to be expressed. Here, we imagine mOrange and GFP must both be expressed as precursors in the formation of an output.
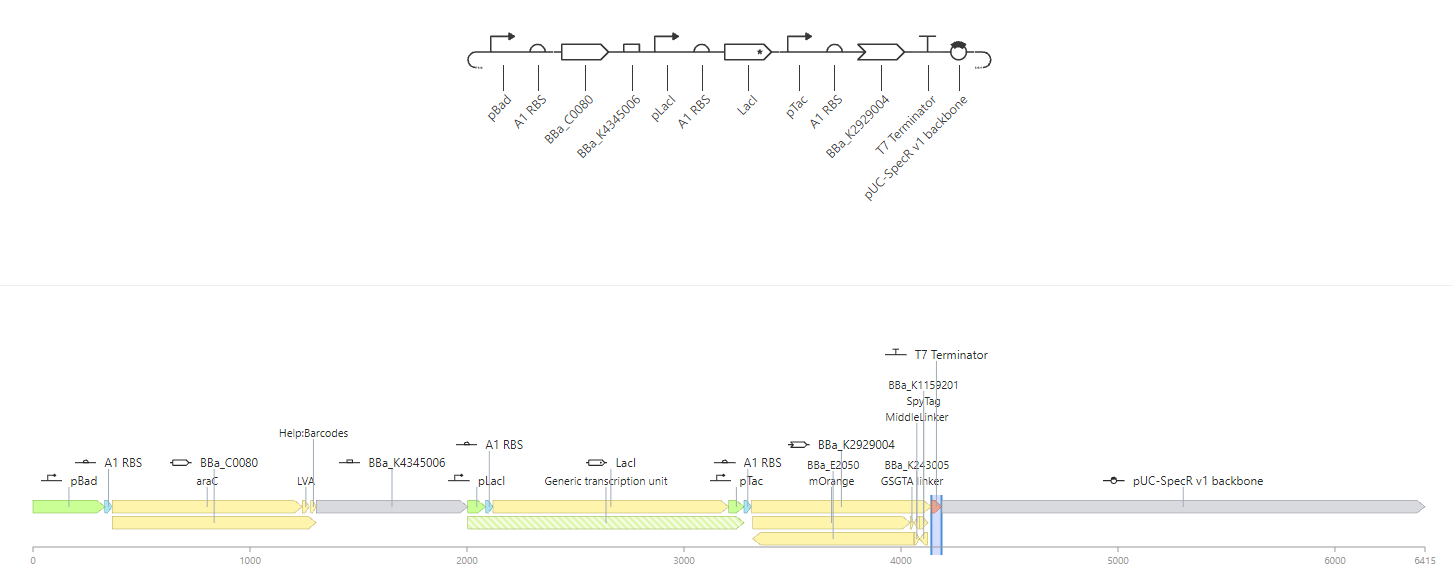 |
Construct B |
Construct C
Attempt at NOT gate. mOrange expressed in the absence of L-arabinose, and ceases in its presence.
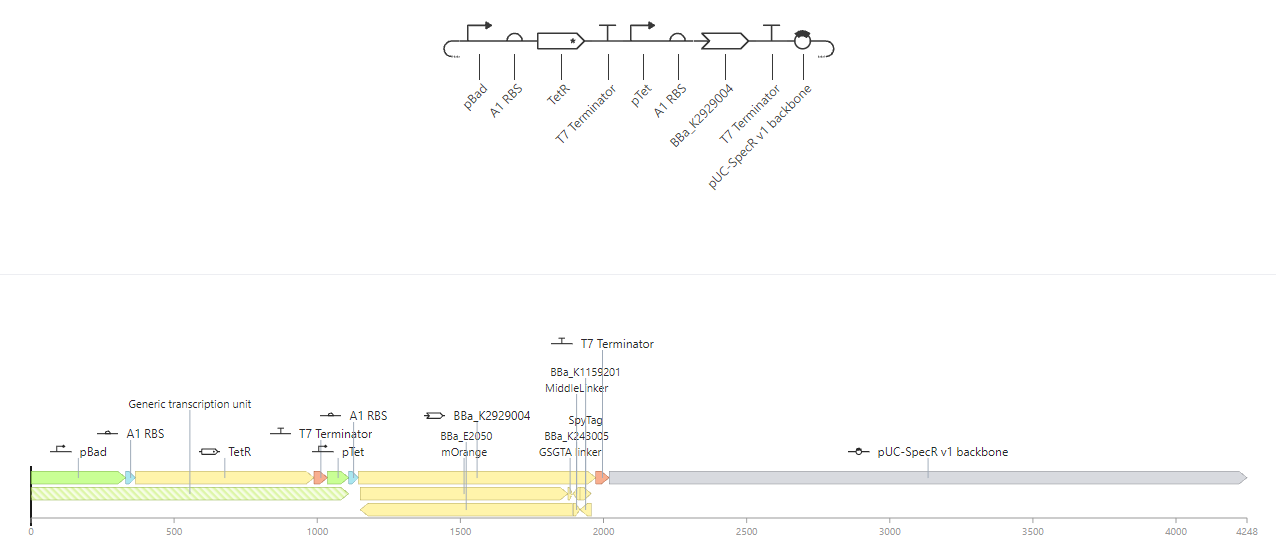 |
Construct C |
References
Elowitz, M.B. and Leibler, S. (2000). A synthetic oscillatory network of transcriptional regulators. Nature, [online] 403(6767), pp.335–338. doi:10.1038/35002125.