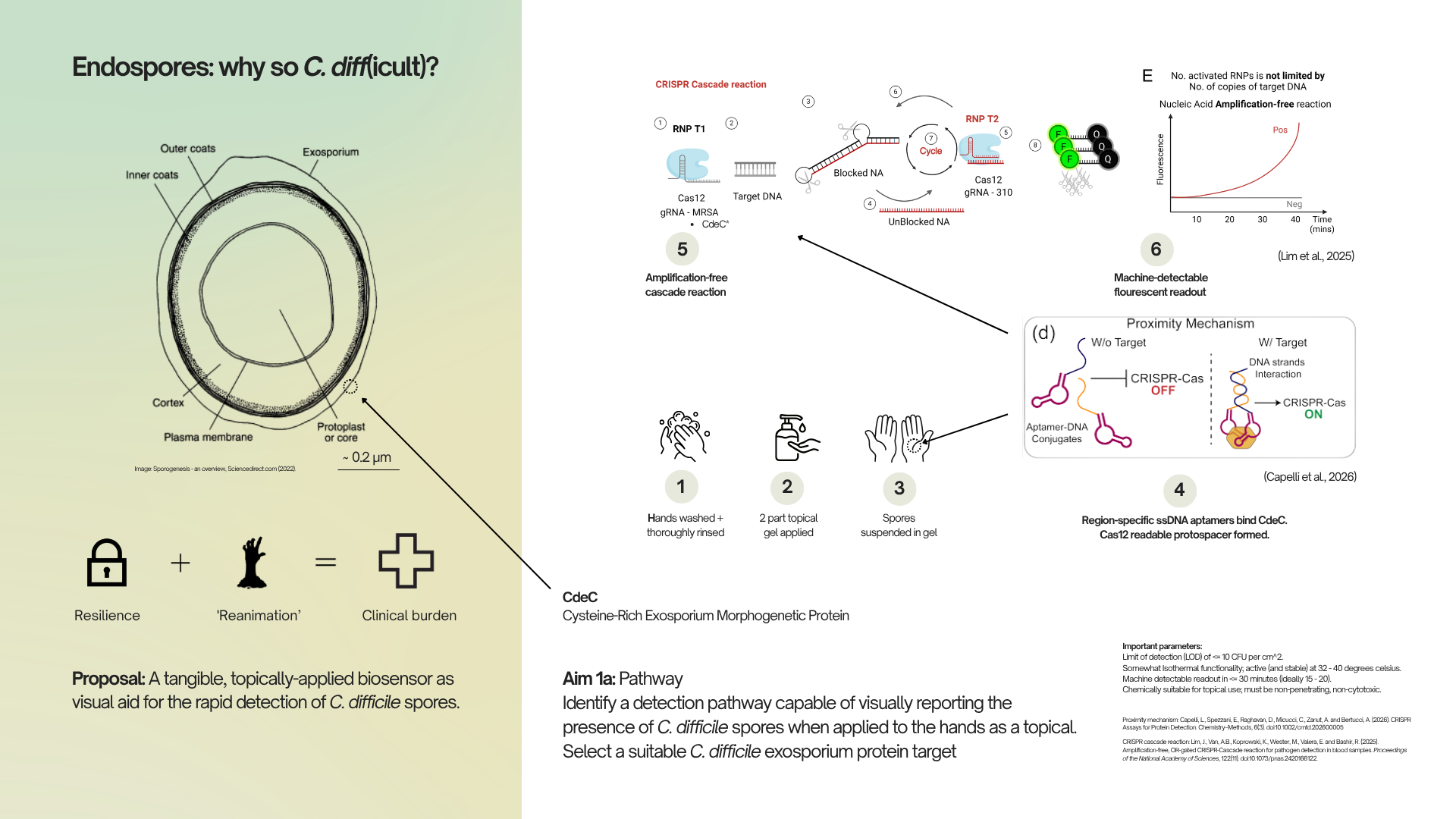
Seeming to model phenomena found frequently throughout nature, the Reaction-Diffusion equations give rise to an endless assortment of intriguing interactions. I whipped up the one above courtesy of @pmneila via Reaction-Diffusion System (Grey-Scott model).
About me
Hey there, I’m Zander!
I’m a recent industrial design graduate with a growing fascination for how synthetic biology may be harnessed as a novel manufacturing technique. I was fortunate enough to receive a taster of the world of biology through the completion of a minor in Biotechnology, having gained not only some time in the wetlab, but an unquenchable desire to culture, plate, and microscopically observe every microorganism in a 10 kilometre radius. This time around, I’m pretty thrilled to be diving into the world of synthetic biology. Lets leave no stone unturned!
First, describe a biological engineering application or tool you want to develop and why.
I’m fascinated by morphogenesis, in which the complex, three-dimensional organisation of biological entities can emerge! The possibilities of synthetically harbouring this process feel endless, however I am most intrigued by the potential of its application in radically-sustainable manufacturing.
Conventional manufacturing techniques (i.e formative plastic methods, subtractive techniques) in their archetypal incarnation contain inherent inefficacies at all stages, from the pre-production through to disposal of the manufactured artefact by the end-user. The plastic spork is an exemplar for how contemporary industrial logic constrains a product from its inception. Compatibility with standard machinery immediately informs material selection. Injection moulding techniques demand a particular design rationale, where minor features like undercuts can cause costs to balloon through added mould complexity. Instead of being repossessed wholly by organic processes at the atomic level when disposed, conventional thermoplastics degrade and colonize such exotic destinations as the mariana trench, our water supply, the human brain etc.
Organisms in the natural world appear unshackled by these constraints. I think immediately of the functionally-graded characteristics of bone, in which matter distribution is varied spatially, optimised to provide lightweight strength and support.
Next, describe one or more governance/policy goals related to ensuring that this application or tool contributes to an “ethical” future, like ensuring non-malfeasance (preventing harm).
Consistency of use:
Fair dissemination of information is necessary to permit strong, equitable discussions between distinct stakeholders (public, policymakers, technicians etc), ensuring the ethical concerns of all groups are platformed to guide accurate policymaking.
Transparency in research and application:
‘Growth’ processes should likely be strongly standardized (at least initially) to establish a safe, repeatable precedent of procedure.
Prevention of environmental contamination:
Current biowaste disposal processes should be adapted to fit novel requirements as they emerge.
Prevention of utilisation in scenarios considered universally unethical:
This one feels pretty clear: no application should cause innate suffering or distress to involved biological materials (or societies in which they are used).
a. Sound disposal practices of biomass bioproducts as industrial waste:
Current biowaste disposal processes should be adapted to fit novel requirements as they emerge.
Next, describe at least three different potential governance “actions”.
1. Implement sound genetic safeguards.
Purpose: emergency termination of cellular processes in the event of dangerous circumstances, whereby the health of a population or ecosystem is suddenly threatened.
Induced auxotrophy (metabolic or via xenonucleic biochemistry), toxin gene expression cassettes, and engineered addiction are all contemporary methods of halting cellular activity outside of a specific environment (lab). A mosaic of approaches is recommended as the most robust containment method, especially when accounting for the spontaneous occurrence of safeguard-negating mutations.
Design: continued research, testing, and update of existing safeguards. Assumptions: established safeguard techniques identified are inadequate or incompatible for this use case.
Risk of failure (or success!):
a. Strategies prove ineffective and difficult to consistently implement. Environmental contamination occurs frequently; consequences are unpredictable and catastrophic.
b. Genetic safeguards utilised are too complex or specific to permit equitable access.
2. Compose a universal convention outlining acceptable and unacceptable applications in manufacturing (and beyond), requiring strict agreement and adherence at a national level prior to industrial use.
Purpose: establish legal basis for international enforcement of manufacturing practices collectively identified as ethical and sound. Dissuade bad actors from potential misuse, encourage international collaboration, and mandate stewardship at the national scale. Unlike traditional manufacturing where inanimate materials are used, it feels like a necessity to require international collaboration where unethical practices could give rise to unpredictable, dangerous outcomes.
Design: The Biological Weapons Convention (BWC) serves as a strong example for the potential of a multilateral agreement to address and prevent misuse of biological practices at a global scale, reaching practically universal agreement across UN member states. I propose the creation of a convention following a similar framework, however with emphasis placed on proactively identifying and sanctioning acceptable and unacceptable use cases.
Assumptions: success hinges heavily upon members reaching a collective understanding. Additionally, it assumes that national governance voices are strongly aligned with the private interests of the manufacturing sector in each member state.
Risk of failure (or success!):
a. Private interests commence manufacturing via synthetic morphogenesis despite absent participation at UN level.
b. An unanimous decision is made, however coverage of convention proves inadequate, requiring frequent amendments.
3. Establish an independent watchdog for regulation of industrial activity.
Purpose: provide additional layer of security to possible corruption at both national and international scales, serving to conduct random audits of the industrial activity within member states.
Design: using universal convention as a basis, establish key measurable indicators as a baseline for monitoring and governance. Indicators must be specific and comprehensive, defining actions by industry which must be precisely met to guarantee compliance.
Assumptions:
a. The watchdog is sufficiently capable of avoiding corruption. Handlings are fair, just, and do not impose on the national security or privacy of member states.
b. Member states (and industry) corporate sufficiently with requests of watchdog.
Risk of failure (or success!):
a. Undetected convention violations threaten international biosecurity or violate agreements on ethical practices.
4. Implement traceability protocols as..
a. Chain Of Custody (CoC) for synthesis and distribution of manufacturing precursors.
b. Integrated genetic barcodes.
Purpose: ensure all stakeholders are accountable for their contribution to the design, production, and distribution of biological materials prior, during, and post manufacture. In the event that a ‘leak’ occurs whereby an ecosystem is threatened, traceability permits rapid response and containment, as well as identification of processes violating the convention.
Design:
a. Define ‘custody’ objects, assigning each with a distinct identifier, lifecycle, and scope of use. Establish assignable states (created, modified, transported, archived, destroyed etc.). Design custody record around requirements identified in convention.
b. Establish composition, length, and ideal location of barcode locus.
Assumptions:
a. Stakeholders adhere stringently to CoC protocol.
Risk of failure (or success!):
CoC processes are too authoritative to strongly discourage prospective stakeholders, with sound intent, from entering the market. The approach cannot achieve the required market leverage to replace the use of conventional manufacturing techniques.
Next score (from 1-3 with, 1 as the best, or n/a) each of your governance actions against your rubric of governance goals.
“Synthetic morphogenesis as a manufacturing technique”
Does the option:
Option 1
Option 2
Option 3
Option 4
Enhance Biosecurity
• By preventing incidents
1
1
1
1
• By helping respond
3
3
2
1
Foster Lab Safety
• By preventing incident
1
1
1
3
• By helping respond
3
3
2
1
Protect the environment
• By preventing incidents
1
1
1
3
• By helping respond
3
3
3
1
Other considerations
• Minimizing costs and burdens to stakeholders
2
3
3
2
• Feasibility?
1
3
3
2
• Not impede research
2
2
2
1
• Promote constructive applications
2
1
1
3
Total (lower > higher)
19
21
19
18
Last, drawing upon this scoring, describe which governance option, or combination of options, you would prioritize, and why. Outline any trade-offs you considered as well as assumptions and uncertainties.
Although likely the most politically challenging, the convention outlined in Option 2 appears central to establishing the precedent required for ethical use of the novel technology. Option 1 is almost mandatory as a guardrail in the event of industrial pollution, as built-in failsafes provide the greatest degree of protection where incidental mishaps could have dire consequences for public health.
I propose the initial priorisation of Options 1 and 2 consecutively, whereby research seeks to first establish integrated safeguards appropriate for the requirements unique to manufacturing, whilst national policymakers work collaboratively to negotiate terms of the convention. Due to somewhat uncertain ramifications of misuse, initial collaboration directly with the United Nations Office of the Secretary General will provide the expertise necessary to move forward at a national scale, where progress is likely to be more fruitful.
Principles & Practices - Weekly Homework
Reflecting on what you learnt and did in class this week, outline any ethical concerns that arose, especially any that were new to you.
The ability to control the growth of organic matter in a way that is so precise feels unprecedented in the sense that it is not dissimilar to ‘playing god’; it is clear that this gives rise to countless ethical concerns:
Concern: Could the process inadvertently give rise to conscious organisms under the right (or more so wrong) circumstances?
Action 2.
Concern: What happens if the technique is abused to grow human flesh for use in applications considered universally immoral?
Action 2, Action 3.
Concern: Could self-propagating organisms be inadvertently created and released into ecosystems?
Action 1.
Concern: Could the process create significant social upheaval amongst religious societies?
Action 2.
I will continue to strongly consider this topic throughout the coming weeks, aiming to further philosophise and describe ideas as they arise!
Preparatory Questions (Week 2)
Professor Jacobsen
Nature’s machinery for copying DNA is called polymerase. What is the error rate of polymerase? How does this compare to the length of the human genome. How does biology deal with that discrepancy?
1:10^6
Human genome = ~ 3.2 gbp; approximately one incorrect base is inserted per 1 million correct insertions.
Human biology employs several DNA proofreading and correction techniques; sense of correct geometry of base pairings, slowing catalysis when mismatch detected, and separation of mismatched primer to exonuclease site for removal etc.
How many different ways are there to code (DNA nucleotide code) for an average human protein? In practice what are some of the reasons that all of these different codes don’t work to code for the protein of interest?
Substantial degree of combinations possible mathematically (approx 10^190)
Degeneracy of genetic code (multiple distinct codons encode amino acid) means a small fraction of total combinations actually encode for protein.
Dr. LeProust
What’s the most commonly used method for oligo synthesis currently?
Phosphoramidite oligo synthesis
Why is it difficult to make oligos longer than 200nt via direct synthesis?
Small discrepancies in coupling efficiency (95% - 99%) compound exponentially beyond 200nt, reducing sequence accuracy beyond.
Why can’t you make a 2000bp gene via direct oligo synthesis?
As described above, synthesis becomes incredibly imprecise beyond about 200nt. Accumulation of errors will yield strand coding for dysfunctional characteristics.
[Using Google & Prof. Church’s slide #4] What are the 10 essential amino acids in all animals and how does this affect your view of the “Lysine Contingency”?
Arginine, histidine, methionine, isoleucine, leucine, lysine, phenylalanine, threonine, tryptophan, and valine compose the 10 essential amino acids, where arginine is described as ‘conditionally essential’ in some mammalian species, such as humans (Rychen et al., 2018).
The ‘Lysine Contingency’ describes the knock-out mutation performed in Jurassic Park (1993), in which the dinosaurs were rendered incapable of producing the essential aa Lysine (synthetic auxotrophy), requiring constant supplementation.
The organic abundance of Lysine as an essential aa effectively renders this method useless, as they would be capable of receiving it through their conventional diet. This is especially interesting when considering prospective use of synthetic auxotrophy to control the growth of a manufactured organism; how might nature creatively find a method to circumvent the integrated safeguard?
A method to arrange bacterial growth (magnetotaxis)
Manufacturing technique (key concepts / words):
Morphogenesis
Developmental biology
Tissue patterning
Hox genes
Homeoboxes
Positional information (morphogen gradient)
References
Huigang, L., Menghui, L., Xiaoli, Z., Cui, H. and Zhiming, Y. (2022). Development of and prospects for the biological weapons convention. Journal of Biosafety and Biosecurity, [online] 4(1), pp.50–53. doi:https://doi.org/10.1016/j.jobb.2021.11.003.
Johnson, M.S., Venkataram, S. and Kryazhimskiy, S. (2023). Best Practices in Designing, Sequencing, and Identifying Random DNA Barcodes. Journal of Molecular Evolution, [online] 91(3), pp.263–280. doi:https://doi.org/10.1007/s00239-022-10083-z.
Teufel, J., López Hernández, V., Greiter, A., Kampffmeyer, N., Hilbert, I., Eckerstorfer, M., Narendja, F., Heissenberger, A. and Simon, S. (2024). Strategies for Traceability to Prevent Unauthorised GMOs (Including NGTs) in the EU: State of the Art and Possible Alternative Approaches. Foods, [online] 13(3), p.369. doi:https://doi.org/10.3390/foods13030369.
Trotsyuk, A.A., Waeiss, Q., Bhatia, R.T., Aponte, B.J., Heffernan, I.M.L., Madgavkar, D., Felder, R.M., Lehmann, L.S., Palmer, M.J., Greely, H., Wald, R., Goetz, L., Trengove, M., Vandersluis, R., Lin, H., Cho, M.K., Altman, R.B., Endy, D., Relman, D.A. and Levi, M. (2024). Toward a framework for risk mitigation of potential misuse of artificial intelligence in biomedical research. Nature Machine Intelligence, [online] 6(12), pp.1435–1442. doi:https://doi.org/10.1038/s42256-024-00926-3.
Wang, F. and Zhang, W. (2019). Synthetic biology: Recent progress, biosafety and biosecurity concerns, and possible solutions. Journal of Biosafety and Biosecurity, [online] 1(1), pp.22–30. doi:https://doi.org/10.1016/j.jobb.2018.12.003.
Week 2 HW: DNA Read / Write / Edit
Part 0: Basics of Gel Electrophoresis
Attend Bootcamp:
Part 1: Benchling & In-silico Gel Art
Left: practice in-silico digestion featuring base sequence (Lambda) and restriction enzymes as specified.
Right: in-silico digestion representing a three-subunit amino polypeptide! To achieve the central grouping of bands I desired, I first asked chatGPT to recommend a short base sequence, then referenced the linear map in Benchling to iterate with endonucleases which produced cuts at the positions needed to make bp lengths which would match an outline I sketched.
Part 2: Gel Art - Restriction Digests and Gel Electrophoresis
No wetlab access. Given access in the coming months, I’ll definitely attempt to create my artwork given the required endonucleases + base sequence are accessible!
Part 3: DNA Design Challenge
3.1. Choose your protein.
From Magnetospirillum magneticum (AMB-1): HtrA/DegP family protease MamE
Magnetotaxic bacteria possess the highly curious ability to form organised, magnetic ‘inclusions’ in structures composed of ‘magnetosomes’. Magnetotaxis as a behavioural characteristic is particularly advantageous in aquatic environments, where detection of the earth’s magnetic field permits spatial sense in the water column as a method to locate nutrient-rich microenvironments. In the model organism Magnetospirillum magneticum AMB-1 (AMB-1), MamE protease is central to the biomineralization process required for magnetic crystal formation within magnetosomes.
I selected this protein as I find this behaviour incredibly enchanting! Its role in both magnetoreception and the biomineralization process is fascinating, and promises many intriguing applications if harnessed.
>reverse translation of sp|Q2W8Q8|MAME_PARM1 Magnetosome formation protease MamE OS=Paramagnetospirillum magneticum (strain ATCC 700264 / AMB-1) OX=342108 GN=mamE PE=1 SV=1 to a 2184 base sequence of most likely codons.
atggcgatgtttaacggcgatgtggaagatggcggccgcggcgatgcgagctgcggcaaagatctgaaacgctatctgatgctgatgggcgtggtggcgctggtggtgctgtttggcgcgtttatttatcgccagagcagcggcggcctgcgcctgggcgcgatgctggaacagatgggccgcggcaccggcccggcggtgaacgtgccggtgcagcagggcggcccgagcgcggcggtgaacccggcgatgagcgtgccggcgggcgcgcgcgtggcgccgccgagcgcggcgggcgcgattgcgaccatgccgccgatggtggattttggcccggcgccgattggcgcgggcggcccgtttagcagcgtggtgaccctgctgcgcaacagcgtggtggcggtgaccgcgagcagcgcgaacggccaggcgatgccggatccgctgggcctggcgaacccggatggcctgccgcattttgcgaacccggcgacccgcagcgtggaaaacattggcaccggcgtgattgtgcgcaacgatggctttattgtgaccaactatcatgtggtgcgcggcgcgaacagcgtgtttgtgaccgtgcaggatgatgtgggcagcacccgctatagcgcggaaattattaaaatggatgaagcgctggatctggcgctgctgaaagtggcgccgaaaaccccgctgaccgcggcggtgctgggcgatagcgatggcgtgcaggtggcggatgaagtgattgcgattggcaccccgtttggcctggatatgaccgtgagccgcggcattattagcgcgaaacgcaaaagcatggtgattgaaggcgtgacccatagcaacctgctgcagaccgatgcggcgattaaccagggcaacagcggcggcccgctggtgattagcaacggcaccgtggtgggcattaacaccgcgatttataccccgaacggcgcgtttgcgggcattggctttgcggtgccgagcaaccaggcgcgcctgtttattctggatgaagtgggctggctgccgaccagcaccgcggaaggcgcgagcatgggcctggtggcgatgcagcgcccgatgggcggcggcgtgggcgcggcgggcccggcgatttttgcgggcacccgcgcgccgcataccgatggccgccagaacatggattgcaccacctgccatgatctgattccggcgggcaacggccgcccggcgccgatgatgccgattgcggcgccgattccgccgccgccgattccgatgggcgcggtgagcccgcataccgatggccgccagaacatgaactgcgcgaactgccatcagatgctgggcggcgcggcgccgattgcggcgccgggcctgggcggcggcgcgtatcgctttgcgcagccgccgggcagcctggcgattaacattcagggcccgcgcggcggccagagcaccgcggcgggcaccggccgcgtgaccctgctgggcgcggcgctgaccccgatgagccagcgcctgggcgcgcagaccggcgtgccggtgggccgcggcgtgtttattagcggcgtgaccccgaacaccccggcggcgaccgcgggcctgcgcccgggcgatgtgctgctgaaagtggatggccgcccggtgcgcctgccggaagaagtgagcgcgattatggtggaaatgcatgcgggccgcagcgtgcgcctgggcgtgctgcgcgatggcgatgtgcgcaacatgaccctggtggcgggcccggcgggcctggcggcggcggcggtgcaggcgccggcgattgcggatatggcgcagccgccgatgggcggcatggcgccgaccgcgccgggcatggtggcggtgccgggcggcccggcggtgatgccgaaaccgccgaccgaatttaactggctgggcatggaaattgaaacctttcaggcgccgcgcccgattaccggcgtgccgggcgcggtgccggtgccgggcgcgaaaggcgcgcaggtggcggaagtgctggtgggcagccgcgcggcggtggcgggcctgcaggcgaacgatctgattctggaagtgaacaaccgcccggtggcgggcccggcgcgcctggatgcggcgattaaaggcgcgaccaacgcgggccagcagattctgctgaaagtgaaccgcaacggccaggaattttggattgtgctg
3.3. Codon optimization.
When engineering the genome of one organism to express a foreign gene, codon optimization increases the chances that the translated sequence will result in the correct, functional protein product. Although the central dogma is inherent to all lifeforms, each class of organism will possess a ‘codon bias’ by which the particular codon (of which there may be multiple for one amino acid; degeneracy) will be represented by a comparatively higher concentration of tRNA molecules.
Vibrio natriegens is a gram negative, marine-dwelling prokaryote understood to possess one of the highest growth rates of any organism. This trait would be highly desirable for manufacturing applications where controlled biomineralisation could be harnessed to create solid artefacts in a short timeframe.
As V. Natriegens is not so forth a model organism and was thus omitted as an option from available online tools, I retrieved the codon usage table for Vibrio natriegens from the Codon Usage Database and used the BioInfromatics Reverse Translation Tool to directly generate the most likely to succeed, non-degenerate DNA sequence. This instead required the input of the AA sequence previously retrieved in 3.1.
>reverse translation of sp|Q2W8Q8|MAME_PARM1 Magnetosome formation protease MamE OS=Paramagnetospirillum magneticum (strain ATCC 700264 / AMB-1) OX=342108 GN=mamE PE=1 SV=1 to a 2184 base sequence of most likely codons.
atggcaatgtttaacggtgatgtagaagatggtggtcgtggtgatgcatcatgtggtaaagatcttaaacgttaccttatgcttatgggtgtagtagcacttgtagtactttttggtgcatttatctaccgtcaatcatcaggtggtcttcgtcttggtgcaatgcttgaacaaatgggtcgtggtacaggtccagcagtaaacgtaccagtacaacaaggtggtccatcagcagcagtaaacccagcaatgtcagtaccagcaggtgcacgtgtagcaccaccatcagcagcaggtgcaatcgcaacaatgccaccaatggtagattttggtccagcaccaatcggtgcaggtggtccattttcatcagtagtaacacttcttcgtaactcagtagtagcagtaacagcatcatcagcaaacggtcaagcaatgccagatccacttggtcttgcaaacccagatggtcttccacattttgcaaacccagcaacacgttcagtagaaaacatcggtacaggtgtaatcgtacgtaacgatggttttatcgtaacaaactaccatgtagtacgtggtgcaaactcagtatttgtaacagtacaagatgatgtaggttcaacacgttactcagcagaaatcatcaaaatggatgaagcacttgatcttgcacttcttaaagtagcaccaaaaacaccacttacagcagcagtacttggtgattcagatggtgtacaagtagcagatgaagtaatcgcaatcggtacaccatttggtcttgatatgacagtatcacgtggtatcatctcagcaaaacgtaaatcaatggtaatcgaaggtgtaacacattcaaaccttcttcaaacagatgcagcaatcaaccaaggtaactcaggtggtccacttgtaatctcaaacggtacagtagtaggtatcaacacagcaatctacacaccaaacggtgcatttgcaggtatcggttttgcagtaccatcaaaccaagcacgtctttttatccttgatgaagtaggttggcttccaacatcaacagcagaaggtgcatcaatgggtcttgtagcaatgcaacgtccaatgggtggtggtgtaggtgcagcaggtccagcaatctttgcaggtacacgtgcaccacatacagatggtcgtcaaaacatggattgtacaacatgtcatgatcttatcccagcaggtaacggtcgtccagcaccaatgatgccaatcgcagcaccaatcccaccaccaccaatcccaatgggtgcagtatcaccacatacagatggtcgtcaaaacatgaactgtgcaaactgtcatcaaatgcttggtggtgcagcaccaatcgcagcaccaggtcttggtggtggtgcataccgttttgcacaaccaccaggttcacttgcaatcaacatccaaggtccacgtggtggtcaatcaacagcagcaggtacaggtcgtgtaacacttcttggtgcagcacttacaccaatgtcacaacgtcttggtgcacaaacaggtgtaccagtaggtcgtggtgtatttatctcaggtgtaacaccaaacacaccagcagcaacagcaggtcttcgtccaggtgatgtacttcttaaagtagatggtcgtccagtacgtcttccagaagaagtatcagcaatcatggtagaaatgcatgcaggtcgttcagtacgtcttggtgtacttcgtgatggtgatgtacgtaacatgacacttgtacaggtccagcaggtcttgcagcagcagcagtacaagcaccagcaatcgcagatatggcacaaccaccaatgggtggtatggcaccaacagcaccaggtatggtagcagtaccaggtggtccagcagtaatgccaaaaccaccaacagaatttaactggcttggtatggaaatcgaaacatttcaagcaccacgtccaatcacaggtgtaccaggtgcagtaccagtaccaggtgcaaaaggtgcacaagtagcagaagtacttgtaggttcacgtgcagcagtagcaggtcttcaagcaaacgatcttatccttgaagtaaacaaccgtccagtagcaggtccagcacgtcttgatgcagcaatcaaaggtgcaacaaacgcaggtcaacaaatccttcttaaagtaaaccgtaacggtcaagaattttggatcgtactt
For HW consistency, I additionally optimised the ‘most likely codon’ MamE sequence for expression in Escherichia Coli (E. Coli) via the tool at Vectorbuilder:
The resulting Codon Adaption Index (CAI) for E. Coli fortunately still appeared to fall within a suitable range of <0.8.
3.4. You have a sequence! Now what?
The mamE sequence could first be integrated in-vitro into a recombinant plasmid, then transformed into V. Natriegen cells via electroporation, by which plasmids enter the cell through the creation of temporary pores in its membrane. The recombinant plasmids should then be transcribed and translated as normal by the appropriate enzymes within the cytosol. Electroporation is advantageous as is both efficient and relatively fast.
3.5. Bonus: How does it work in nature/biological systems?
In prokaryotes such as V. Natriegen, polycistronic transcription for a single mRNA molecule to be translated into different proteins. This is achieved via the inclusion of separate, distinct START and STOP codons, creating separate regions along the same mRNA molecule which are thus translated into distinct proteins.
I attempted to align the flow of information from DNA through polypeptide via the stacking of my three produced Benchling sequences manually. Unfortunately begins to break down in alignment, but accurate for the first three codons:
ChatGPT used to format correct alignment:
Part 4: Prepare a Twist DNA Synthesis Order
This process was really fascinating! I would love to learn more about the different inclusions along the way, and of their functionality + importance.
5.1 DNA Read(i) What DNA would you want to sequence (e.g., read) and why?
It would be intriguing to sequence morphogen-encoding genes (such as the HOX group) in species which have so far not been included. I would love to compare the base composition of different morphogen-encoding genes across distinct species to better understand the mechanisms behind different types of tissue patterning, and perhaps discover more conserved sequences.
(ii) In lecture, a variety of sequencing technologies were mentioned. What technology or technologies would you use to perform sequencing on your DNA and why?
I would select some combination of Single Molecule (third-generation; sequences individual bases directly without prior amplification) and Fluorescent 3D (other; distant in characteristics) to map genes of interest. Fluorescent 3D could be used to first determine the spatial concentration of suspected morphogen-encoding genes in a given tissue, then use Single Molecule to determine finer structural details. The capacity for Single Molecule to produce very long reads is desirable where developmental regions may be large and complex.
Fluorescent 3D:
Input: Hybridization target probe pre-prepared based on theorized gene of interest (GOI). Tissue segment of interest pre-prepared on slide.
Base calling method: iterative cycles of images detect each base as ‘spot in tissue’; corresponds to a particular gene identity.
Output: spatial coordinates for gene expression + cells it is expressed by.
Single Molecule:
Input: cells identified to contain GOI previous step are isolated and cultured, then lysed before separating and purifying DNA. Construction (oligonucleotide synthesis) and addition of nanopore sequencing adapters. Segments loaded into flow cell.
Base calling method: individual DNA molecules pulled through nanopore by sequencing adapter, disrupting baseline ionic current of the detector which is detected and recorded by device. ‘Trace signals’ correspond to a particular base.
Output: Long-read sequences (LRS)
5.2 DNA Write(i) What DNA would you want to synthesize (e.g., write) and why?
Artificially constructing a cluster of genes which allows for relatively precise, customisable control of tissue patterning would be highly appealing. Think programmable osteablasts, where the functional grading of bone-like structures can be controlled to produce an organic car chassis.
(ii) What technology or technologies would you use to perform this DNA synthesis and why?
Multiplexed Oligo Synthesis followed by Gibson Assembly appears a feasible option for the construction of large genes. Many sub-80 bp oligos can first be synthesised in parallel as a means of error-rate limitation, then combined via Gibson Assembly to construct the entire gene. In regards to morphogenic genes, both high precision and accuracy are required as the resulting phenotype is likely highly sensitive to accurate sequence composition.
Multiplexed Oligo Synthesis (in situ, via inkjet printing)
Essential steps: computational design of desired DNA sequence, followed by division into 60-80 bp oligos with overlapping regions. Microarray pre-prepared with contact primer at each oligo synthesis site; single added sequentially in cycles at each site adhering to phosphamidite chemistry process. At termination length, cleaved, retrieved, and amplified.
Limitations: error rate increases as sequence grows larger. Overlapping requirements of gibson assembly requires more cycles to be performed.
Gibson assembly
Essential steps: Addition of exonuclease to prepared oligonucleotides to create sticky overhangs. Segments hybridise at designed complementary overlapping regions. DNA polymerase introduced to integrate missing nt’s at gaps. Ligase seals gaps throughout the incomplete backbone.
Limitations: efficiency decreases as quantity of oligo fragments increases. Precise design of overlapping regions essential.
5.3 DNA Edit(i) What DNA would you want to edit and why?
It would be ideal to edit the genomes of more agricultural crops to introduce robust autofertilisation traits via symbiosis with microorganisms, for use in both developed and developing nations alike. In industrialised agriculture, external fertilisation is responsible for damaging pollution events throughout adjacent ecosystems. In developing regions where soil viability is poor, endemic low-yields resulting from limited fertilizer access drive perilous consequences. As the most abundant atmospheric gas, fixation of nitrogen directly from where directly where it is required makes significant sense.
(ii) What technology or technologies would you use to perform these DNA edits and why?
CRISPR/Cas9 currently appears the most desirable method for performing DNA modifications within plant embryo cells, particularly due to its efficiency and precision.
Preparation + inputs: Design gRNA to match target sequence within plant embryo; incorporate into suitable vector alongside Cas9 gene.
Limitations: Cuts at sequences similar to DNA target in embryo possible, requiring careful planning and validation post-edit. Editing efficiency dependent on accessibility of target gene.
References
Jerlie Mhay Matres, H., Hilscher, J., Datta, A., Armario-Nájera, V., Baysal, C., He, W., Huang, X., Zhu, C., Valizadeh-Kamran, R., Trijatmiko, K.R., Capell, T., Christou, P., Stoger, E. and Slamet-Loedin, I.H. (2021). Genome editing in cereal crops: an overview. Transgenic Research, 30(4), pp.461–498. https://doi.org/10.1007/s11248-021-00259-6
Lee, H.H., Ostrov, N., Wong, B.G., Gold, M.A., Khalil, A.S. and Church, G.M. (2016). Vibrio natriegens, a new genomic powerhouse. https://doi.org/10.1101/058487
Quinlan, A., Murat, D., Vali, H. and Komeili, A. (2011). The HtrA/DegP family protease MamE is a bifunctional protein with roles in magnetosome protein localization and magnetite biomineralization. Molecular Microbiology, 80(4), pp.1075–1087. https://doi.org/10.1111/j.1365-2958.2011.07631.x
Wan, J., Ji, R., Liu, J., Ma, K., Pan, Y. and Lin, W. (2024). Biomineralization in magnetotactic bacteria: From diversity to molecular discovery-based applications. Cell Reports, 43(12), p.114995. https://doi.org/10.1016/j.celrep.2024.114995
Weinstock, M.T., Hesek, E.D., Wilson, C.M. and Gibson, D.G. (2016). Vibrio natriegens as a fast-growing host for molecular biology. Nature Methods, 13(10), pp.849–851. https://doi.org/10.1038/nmeth.3970
Week 3 HW: Lab Automation
Part 1: OT-2 Automation
This week involved the plating and growth of a design created via the controlled aspiration of fluorescent Escherichia Coli (E. coli) through the OT-2 virtual platform.
Left: the result! P(eace)CR seemed a fitting name.
After some brief iteration, I settled on an 8-bit interpretation of the classic ‘dove with olive branch’, a relatively universal symbol of peace. As DesignerCells only has access to sfGFP and mRFP1 fluorescent recombinants, this design provided a suitable compromise to the design limitations. I will include the final outcome pending the lab is successful (fingers crossed!).
ChatGPT was used in conjunction with the CoLabs reference code to create the final script for the OT-2 liquid handling platform:
fromopentronsimporttypesmetadata={# see https://docs.opentrons.com/v2/tutorial.html#tutorial-metadata'author':'Zander Morris','protocolName':'P(eace)CR','description':'Prints the P(eace)CR graphic via the set of data points.','source':'HTGAA 2026 Opentrons Lab','apiLevel':'2.20'}################################################################################# Robot deck setup constants - don't change these##############################################################################TIP_RACK_DECK_SLOT=9COLORS_DECK_SLOT=6AGAR_DECK_SLOT=5PIPETTE_STARTING_TIP_WELL='A1'well_colors={'A1':'Red','B1':'Green','C1':'Orange'}sfgfp_points=[(-26.4,21.6),(-26.4,19.2),(-24,19.2),(-26.4,16.8),(-24,16.8),(-21.6,16.8),(-14.4,16.8),(-26.4,14.4),(-24,14.4),(-21.6,14.4),(-16.8,14.4),(-14.4,14.4),(-21.6,12),(-19.2,12),(-16.8,12),(-14.4,12),(-24,9.6),(-21.6,9.6),(-19.2,9.6),(-16.8,9.6),(-26.4,7.2),(-24,7.2),(-19.2,7.2),(-16.8,4.8),(-21.6,2.4),(-19.2,2.4),(-16.8,2.4),(-19.2,0),(-16.8,0),(-14.4,0),(-14.4,-2.4),(-14.4,-4.8),(-14.4,-7.2),(-12,-9.6),(-12,-12),(-9.6,-14.4),(-9.6,-16.8),(-9.6,-19.2)]mrfp1_points=[(12,26.4),(7.2,24),(9.6,24),(12,24),(26.4,24),(4.8,21.6),(7.2,21.6),(9.6,21.6),(12,21.6),(24,21.6),(26.4,21.6),(2.4,19.2),(4.8,19.2),(9.6,19.2),(12,19.2),(24,19.2),(26.4,19.2),(0,16.8),(9.6,16.8),(12,16.8),(21.6,16.8),(24,16.8),(26.4,16.8),(0,14.4),(9.6,14.4),(12,14.4),(19.2,14.4),(21.6,14.4),(24,14.4),(26.4,14.4),(0,12),(9.6,12),(12,12),(14.4,12),(16.8,12),(21.6,12),(24,12),(26.4,12),(0,9.6),(7.2,9.6),(9.6,9.6),(12,9.6),(21.6,9.6),(24,9.6),(26.4,9.6),(-9.6,7.2),(-7.2,7.2),(0,7.2),(7.2,7.2),(9.6,7.2),(21.6,7.2),(24,7.2),(-12,4.8),(-9.6,4.8),(-4.8,4.8),(-2.4,4.8),(0,4.8),(4.8,4.8),(21.6,4.8),(24,4.8),(-14.4,2.4),(-12,2.4),(-2.4,2.4),(0,2.4),(2.4,2.4),(21.6,2.4),(-9.6,0),(-7.2,0),(16.8,0),(19.2,0),(21.6,0),(-7.2,-2.4),(-4.8,-2.4),(14.4,-2.4),(16.8,-2.4),(19.2,-2.4),(-7.2,-4.8),(-4.8,-4.8),(7.2,-4.8),(9.6,-4.8),(12,-4.8),(14.4,-4.8),(16.8,-4.8),(-4.8,-7.2),(-2.4,-7.2),(12,-7.2),(-4.8,-9.6),(-2.4,-9.6),(0,-9.6),(14.4,-9.6),(16.8,-9.6),(19.2,-9.6),(-2.4,-12),(0,-12),(2.4,-12),(4.8,-12),(19.2,-12),(21.6,-12),(0,-14.4),(2.4,-14.4),(4.8,-14.4),(7.2,-14.4),(9.6,-14.4),(12,-14.4),(19.2,-14.4),(21.6,-14.4),(24,-14.4),(26.4,-14.4),(28.8,-14.4),(14.4,-16.8),(19.2,-16.8),(21.6,-16.8),(24,-16.8),(26.4,-16.8),(28.8,-16.8),(16.8,-19.2),(19.2,-19.2),(21.6,-19.2),(24,-19.2),(19.2,-21.6),(21.6,-21.6),(19.2,-24)]defrun(protocol):################################################################################# Load labware, modules and pipettes############################################################################### Tipstips_20ul=protocol.load_labware('opentrons_96_tiprack_20ul',TIP_RACK_DECK_SLOT,'Opentrons 20uL Tips')# Pipettespipette_20ul=protocol.load_instrument("p20_single_gen2","right",[tips_20ul])# Modulestemperature_module=protocol.load_module('temperature module gen2',COLORS_DECK_SLOT)# Temperature Module Platetemperature_plate=temperature_module.load_labware('opentrons_96_aluminumblock_generic_pcr_strip_200ul','Cold Plate')# Choose where to take the colors fromcolor_plate=temperature_plate# Agar Plateagar_plate=protocol.load_labware('htgaa_agar_plate',AGAR_DECK_SLOT,'Agar Plate')## TA MUST CALIBRATE EACH PLATE!# Get the top-center of the plate, make sure the plate was calibrated before running thiscenter_location=agar_plate['A1'].top()pipette_20ul.starting_tip=tips_20ul.well(PIPETTE_STARTING_TIP_WELL)################################################################################# Patterning#################################################################################### Helper functions for this lab#### pass this e.g. 'Red' and get back a Location which can be passed to aspirate()deflocation_of_color(color_string):forwell,colorinwell_colors.items():ifcolor.lower()==color_string.lower():returncolor_plate[well]raiseValueError(f"No well found with color {color_string}")# For this lab, instead of calling pipette.dispense(1, loc) use this: dispense_and_detach(pipette, 1, loc)defdispense_and_detach(pipette,volume,location):"""
Move laterally 5mm above the plate (to avoid smearing a drop); then drop down to the plate,
dispense, move back up 5mm to detach drop, and stay high to be ready for next lateral move.
5mm because a 4uL drop is 2mm diameter; and a 2deg tilt in the agar pour is >3mm difference across a plate.
"""assert(isinstance(volume,(int,float)))above_location=location.move(types.Point(z=location.point.z+5))# 5mm abovepipette.move_to(above_location)# Go to 5mm above the dispensing locationpipette.dispense(volume,location)# Go straight downwards and dispensepipette.move_to(above_location)# Go straight up to detach drop and stay high###### YOUR CODE HERE to create your design#### -----------------------------# Printing parameters# -----------------------------VOL_PER_DOT=0.75# Keep aspirates comfortably below 20uL for accuracy/safetyMAX_ASPIRATE_UL=18.0MAX_BATCH_DOTS=int(MAX_ASPIRATE_UL//VOL_PER_DOT)# 18.0 // 0.75 = 24# Choose where on Z you actually want to dispense.# Start conservative: 0 means "at agar_plate['A1'].top() plane".# If your drops need to touch the agar more, try -0.5 or -1.0 after testing.DISPENSE_DZ=2defpoint_location_from_center(dx,dy,dz=DISPENSE_DZ):# Offsets are in mmreturncenter_location.move(types.Point(x=dx,y=dy,z=dz))defprint_points(points,color_name):pipette_20ul.pick_up_tip()i=0whilei<len(points):batch=points[i:i+MAX_BATCH_DOTS]batch_volume=len(batch)*VOL_PER_DOT# Pull enough dye for this batchpipette_20ul.aspirate(batch_volume,location_of_color(color_name))# Dispense each dotfor(dx,dy)inbatch:loc=point_location_from_center(dx,dy)dispense_and_detach(pipette_20ul,VOL_PER_DOT,loc)i+=MAX_BATCH_DOTSpipette_20ul.drop_tip()# -----------------------------# Print your two datasets# -----------------------------print_points(sfgfp_points,"Green")print_points(mrfp1_points,"Red")
Prompts were primarily as follows. Small corrections and confirmations were omitted for clarity:
I want to take a set of predefined x and y points and allow my them to be interpreted by Opentrons OT-2 via a Python script, so that the actuator can move to each point and dispense a set volume of liquid. How do setup a grid and allow the OT-2 to move to each location?
[code inserted]. This is the code I have so far. How can I get it to navigate between the points?
0.75 ul per dot please.
I can’t get my points list to correctly define?
Okay starting over. this is my code so far. what should I add precisely to get it to work?
[series of errors corrected via feedback and correction loop with ChatGPT]
Final Result
Thank you TA’s for carrying out the labwork and sharing the magnificent photos! Very pleased with the result.
Part 2: Find and describe a published paper that utilizes the Opentrons or an automation tool to achieve novel biological applications.
This paper details the use of the OT-2 as a means to automate various processes and assays associated with the culture of patient-derived organoids. Conventionally a process performed manually, the scaffold-supported platform for orgonoid-based tissue (SPOT) method is automated as a possible solution to improve throughput and scalability. This approach provides a possible solution to the limited conventional scalability of SPOT. As SPOT is a method of drug discovery, specifically in the development of therapeutics personalized to the specific tissue of a patient’s tumour, increasing the speed and volume at which this process may be performed via OT-2 lab automation is a highly promising application of lab-automation technology.
Part 3: Write a description about what you intend to do with automation tools for your final project.
I want to investigate the process of controlling the spatial formation of a biofilm (or the biomineralisation process) across the surface of a morphable 3D artifact (scaffold).
Although I am still unsure of the precise outcome, I would like to explore the possibility of integrating automation at two points:
At culture production: to perform the recombinant process, automating the transformation process to facilitate the uptake of engineered plasmids via a desirable prokaryotic host.
Within physical tool: to serve as a repeatbly programmable platform for biofilm formation.
I will work to crystalise the precise integration of automation methods in the coming weeks.
Cao, R., Li, N.T., Latour, S., Cadavid, J.L., Tan, C.M., Forman, A., Jackson, H.W. and McGuigan, A.P. (2023). An Automation Workflow for High‐Throughput Manufacturing and Analysis of Scaffold‐Supported 3D Tissue Arrays. Advanced Healthcare Materials, [online] 12(19). doi:https://doi.org/10.1002/adhm.202202422.
1. How many molecules of amino acids do you take with a piece of 500 grams of meat? (on average an amino acid is ~100 Daltons)
500 grams of beef contains ≈ 112 g of protein.
1 dalton corresponds to ≈ 1.6605×10-24 gram.
1 amino acid (100 daltons) ≈ 1.6605×10−22 grams.
112 grams / 1.6605×10−22 grams ≈ 6.745×1023.
The 112 g of protein contained with 500 g of beef represents ≈ 6.745×1023 molecules of amino acid!
2. Why do humans eat beef but do not become a cow, eat fish but do not become fish?
One aspect of “cowness” could be considered the successful construction, transcription, and translation of genetic material which results in a heritable bovine phenotype; physical traits and behaviours which can be associated with cows. For this to be possible, genetic material must not only be incorporated inherently across all tissues at the cellular level, but be intact, and complemented by the correct composition of functional enzymes.
When humans consume tissues from another organism, a series of digestive enzymes effectively destroy the structure of foreign macromolecules (such as DNA), into recyclable constituent molecules (like nucleotides). Even if foreign genetic material were to remain intact inside the stomach, it would be impossible for it to be integrated across all cells at the scale required to produce observable “cowness”.
3. Why are there only 20 natural amino acids?
This is a very intriguing question by which the precise answer still remains elusive. One theory suggests that the canonical 20 were selected gradually on the basis of parsimony (extreme stinginess, or frugality), where monomers favoured provided the widest spectrum of utility for the lowest investment of energy and material (Bywater, 2018).
4. Can you make other non-natural amino acids? Design some new amino acids.
The synthesis of non-canonical amino acids (NcAA’s) is possible, with large advancements in the field made in the early 21st century. For my attempt at NcAA design, see 4.1 below.
5. Where did amino acids come from before enzymes that make them, and before life started?
Amino acids are theorised to have emerged abiotically via complex chemical processes that were facilitated by the proximity of specific chemical reactants and physical parameters (heat, electricity). It is also possible that amino acids arrived on earth from extraterrestrial sources via a meteorite(s), where other methods of synthesis may have first preluded their arrival (Kirsching, 2022).
6. If you make an α-helix using D-amino acids, what handedness (right or left) would you expect?
Amino acids can be characterised by their chirality, with pairs known as enantiomers representing their two possible configurations (L and D). This pair represents the ‘mirror-image’, non-superimposable structural quality central to chirality. In living organisms, L-aminos are almost exclusively used to construct proteins. In α-helices, this curiously results in the final protein having a D-configuration. Using D-aminos to form α-helices instead yields an overall L-configuration in the resultant structure.
7. Can you discover additional helices in proteins?
Additional types of helices have been discovered and researched, such as the 3-10 helices and Pi helices!
8. Why are most molecular helices right-handed?
Most molecular helices appear right-handed as a resultant property of their left-handed amino monomers. This D configuration is said to improve steric hindrance (reduce reactivity of specific molecules in macromolecules) and energetic stability (qualities of hydrogen bonds in backbone of a-helices are optimised).
9. Why do β-sheets tend to aggregate? What is the driving force for β-sheet aggregation?
The chemical structure of the edges of β-sheets promote aggregation, as it readily encourages the formation of hydrogen bonds with adjacent β-sheets to form larger aggregates.
4.1: Design some new amino acids.
Alanine
Propargyl-valine
Halogenated-valine
I found and followed a short tutorial online detailing how Pymol can be used to quickly design NcAA models, starting with Valine as per the demonstration. With an extremely limited background in chemistry, I used chatGPT to aid me in selecting and positioning functional groups. I decided to create 2 NcAA’s, one optimised for click-reactivity (Acetylene) and photo-reactivity (Bromine), via the separate addition of acetylene and bromine to the methyl group of each Alanine respectively. ChatGPT prompts:
I would like to take this valine and make a non-canonical amino acid by adding different functional groups. which spots should I add groups onto, and what can they be?
I would like to edit for photo-reactivity and click-reactivity. What groups do I add, and what part of the image represents the methyl group?
[Showed me a series of possible combinations.]
These are the only fragments I have available.
[I provided the options available under the ‘fragment’ tab in PyMOL, which then allowed it to narrow down the additions.]
[Two final options I narrowed down two based on desirable characteristics]
[Propargyl-valine = Acetylene addition = click-reactivity]
[Halogenated-valine = bromine addition = photo-reactivity]
Part B: Protein Analysis and Visualization
Many bacterial species are capable of cell-to-cell communication as a response to changes in localised population density. This system is known as quorum sensing.
1. Briefly describe the protein you selected and why you selected it.
I have selected LuxR, a transcriptional activator, and one of the first proteins (alongside LuxL) to be identified as integral in a QS pathway. LuxR works with LuxL in the prokaryote Vibrio fischeri to permit bioluminescence once a particular population density is reached. QS pathways are particularly interesting to me as I would like to understand how they may be leveraged to enable external control of a bacterial population’s behaviour at the cellular level via controlled chemical signalling.
2. Identify the amino acid sequence of your protein.
LuxR has a length of 250 amino acids. The most common is Isoleucine (I), which appears 28 times. Using BLAST, LuxR appeared to have approx. 186 possible homologs after using the E value to cull the total pool of 250 matches. The protein is also the title for the family it is contained within: LuxR-type transcriptional regulators.
3. Identify the structure page of your protein in RCSB.
I couldn’t find a non ML-resolved match for the LuxR sequence from V. fischeri, which was intriguing considering it was supposedly the first organism where this protein was identified.
I did however find a direct match that was resolved with Alphafold DB on 2021-12-09 with a global model confidence of 88.61%. I couldn’t determine if there were additional molecules in the structure. There was also a homolog called 7AMT resolved though x-ray diffraction, which was deposited on 2020-10-09 with an excellent resolution of 2.60 Å. Both proteins belong to the transcription classification family.
4. Open the structure of your protein in any 3D molecule visualization software.
I continued with 7AMT as it was readily available in the correct format from PDB.
Cartoon
Cylinder
Stick + Sphere
7AMT surface structure visualised with hydrophilic (blue) and hydrophobic (orange) regions highlighted. Several pits can be observed, particularly on the side opposite to the DNA binding region.
The surface residues are more commonly hydrophilic, with internal, ‘hidden’ residues appearing to be mainly hydrophobic.
Part C: Using ML-Based Protein Design Tools
C.1: Protein Language Modeling
I decided to progress with 7AMT as its prior resolvement through x-ray diffraction makes it a good candidate for comparison and testing with the provided ML tools!
1. Deep Mutational Scans
7AMT: Unsupervised Deep Mutational Scan.
Substitution for Tryptophan, proline, and cystine across most residues appears to have a negative effect. Residues 50-55~ appear highly conserved, as mutations here appear problematic for functionality.
2. Latent Space Analysis
7AMT: Latent Space Analysis.
The visualisation generated is free from distinct neighbourhoods, instead appearing relatively smooth and continuous between residues. I was unfortunately unable to embed and visualise 7AMT, however I hope to return to this beyond the timeline of the course!
C.2: Protein Folding
7AMT ESMfold 2: rainbow for clarity.
7AMT ESMfold 1: colour gradient representing confidence of resolution.
While somewhat reminiscent, folding of 7AMT in ESMfold did not perfectly match the original coordinates retrieved from UniProt. The structure appeared highly susceptible to change upon alteration of the original sequence.
C.3: Protein Generation
Original structure (confidence)
Predicted structure (confidence)
Intriguingly, the reverse folding processes did appear to yield quite an accurate prediction compared to ESMfold, as indicated by the dark blue colouring of the structure on the right. A quick visualisation in ESMfold yielded a similar result.
Part D. Group Brainstorm on Bacteriophage Engineering
Group formed, initial brainstorm session conducted:
Please refer to the group final project section for our brainstorming session writeup.
Bywater, R.P. (2018). Why twenty amino acid residue types suffice(d) to support all living systems. PLoS ONE, 13(10), pp.e0204883–e0204883. https://doi.org/10.1371/journal.pone.0204883
Cooley, R.B., Arp, D.J. and Karplus, P.A. (2010). Evolutionary origin of a secondary structure: π-helices as cryptic but widespread insertional variations of α-helices that enhance protein functionality. Journal of Molecular Biology, 404(2), pp.232–246. https://doi.org/10.1016/j.jmb.2010.09.034
Doig, A.J. (2017). Frozen, but no accident – why the 20 standard amino acids were selected. The FEBS Journal, 284(9), pp.1296–1305. https://doi.org/10.1111/febs.13982
Kirschning, A. (2022). On the evolutionary history of the twenty encoded amino acids. Chemistry – A European Journal, 28(55). https://doi.org/10.1002/chem.202201419
Munier, R. and Cohen, G.N. (1956). Incorporation d’analogues structuraux d’aminoacides dans les protéines bactériennes. Biochimica et Biophysica Acta, 21(3), pp.592–593. https://doi.org/10.1016/0006-3002(56)90207-4
Oliveira, N.M., Foster, K.R. and Durham, W.M. (2016). Single-cell twitching chemotaxis in developing biofilms. Proceedings of the National Academy of Sciences, 113(23), pp.6532–6537. https://doi.org/10.1073/pnas.1600760113
Soto-Aceves, M.P., Diggle, S.P. and Greenberg, E.P. (2023). Microbial primer: LuxR-LuxI quorum sensing. Microbiology, 169(9). https://doi.org/10.1099/mic.0.001343
A4V represents a point mutation within the 4th codon of the human SOD1 gene. This results in the substitution of the conventional alanine for a valine amino acid.
Four experimental peptides were then generated using the PepMLM tool:
Binder
Pseudo Perplexity
HRYYPVAVRLKE
11.513239315710866
HRYPVVAVAWKE
14.299230176233568
KRYPPVAARWKE
20.264871722997526
WLYPVAAARHKE
20.749563629245603
Part 2: Evaluate Binders with AlphaFold3
Entry
Binder
Pseudo Perplexity
ipTM
Description
Image
🎮
FLYRWLPSRRGG
—
0.31
Appears tightly surface-bound, primarily binding to the dimer interface.
0
HRYYPVAVRLKE
11.513239315710866
0.26
Appears weakly surface bound to the dimer interface; half the residues do not appear bound.
1
HRYPVVAVAWKE
14.299230176233568
0.26
Appears weakly surface bound; similar positioning to HRYYPVAVRLKE, but reversed in that the central residues appear to bind while the flanks do not.
2
KRYPPVAARWKE
20.264871722997526
0.34
Appears to bind superficially with an alpha helix at residue 56-61; also possesses its own alpha helix. In proximity to the rear of β-barrel (opposite end to N and C terminus).
3
WLYPVAAARHKE
20.749563629245603
0.35
Appears strongly surface bound with proximity to the rear of β-barrel.
Compared to a control ipTM of 0.31, binders appeared tightly clustered into two distinct groups; 0.26 and 0.34~. The ipTM of binders KRYPPVAARWKE and WLYPVAAARHKE appeared to exceed the known binders score or 0.31, with 0.34 and 0.35 respectively. When considering ipTM alongside observed binding characteristics, WLYPVAAARHKE appears the most theoretically desirable of the set generated.
Part 3: Evaluate Properties of Generated Peptides in the PeptiVerse
Binder
Prediction Summary
HRYYPVAVRLKE
HRYPVVAVAWKE
KRYPPVAARWKE
WLYPVAAARHKE
All binder candidates were predicted to have weak binding affinity, even those that appeared visually in AlphaFold to be potentially strong candidates. I was not able to discern any strong patterns between Peptiverse results and previous ipTM predictions. The most promising binder from the previous section (WLYPVAAARHKE) intriguingly appeared the second least hydrophobic, and was predicted to have the weakest binding affinity.
I would select KRYPPVAARWKE to advance for further examination given the considerations outlined above. It possesses the highest predicted solubility, while remaining the second strongest candidate identified in the previous section.
Part 4: Generate Optimized Peptides with moPPIt
I provided the A4V mutant SOD1 sequence and specified motif 50-70 for binding, as this region appeared promising as a target from previous observations in AlphaFold. Objective weights for Affinity and Motif selections were kept default (1).
Rank
Binder
Predicted Affinity
1
AWWEYVWWWWCV
8.3275
2
GYYGCYGAVYYY
8.3254
3
CTSCCYVGWCWW
8.2258
4
FAWYWPCYWYYR
8.2060
5
YCVYCYDAYVWW
8.1153
6
DGDCRYCLHCCW
8.1107
7
AVYCYYVCRNWW
7.9624
8
GSEYWWYWWHYT
7.7270
9
MVAGIWVWWVAR
7.3000
10
AYYTRVHWPCVW
6.9906
Unlike PepMLM, moPPIt is intended to bias the generation of binders towards specific residue indices, providing a finer degree of control given particular clinical (or functional) objectives. Intriguingly, they appear much more varied in residue composition compared to the PepMLM run.
Conducting a series of ligand-binding assays would be the best approach for identifying binders for advancement into clinical trials. The equilibrium dissociation constant KD is the universal standard for confirming ligand-target binding affinity; I would employ surface plasmon resonance (SPR) as the final method of filtering candidates via KD for selection.
I used AlphaFold to predict the binding location and ipTM of the top moPPit binder as I was curious to compare with the earlier outcome. Curiously, it formed quite a large alpha helix:
Hahn, D., Bayly, C., Boby, M.L., Bruce Macdonald, H., Chodera, J., Gapsys, V., Mey, A., Mobley, D., Perez Benito, L., Schindler, C., Tresadern, G. and Warren, G. (2022). Best Practices for Constructing, Preparing, and Evaluating Protein-Ligand Binding Affinity Benchmarks [Article v1.0]. Living Journal of Computational Molecular Science, [online] 4(1), p.1497. doi:10.33011/livecoms.4.1.1497.
Week 6 HW: Genetic Circuits Part i
DNA Assembly
The primary component of Phusion High-Fidelity PCR Master Mix is the polymerase Phusion, an alternative to standard taq polymerase which provides upwards of 40x greater fidelity (the accuracy at which a template strand is replicated) comparative to taq. It also contains free-deoxynucleotides (bases for formation of replicated strand) and a reaction buffer (provides optimal pH conditions for Phusion polymerase activity).
Factors may include primer melting temperature, composition of reaction buffer, characteristics of target sequence, and type of polymerase. For example, primers containing higher concentrations of guanine and cytosine bases will possess a higher melting temperature due to the strength imparted by the triple hydrogen bonds of GC.
PCR involves the amplification and ‘construction’ of fragments from a provided template sequence, while restriction digests produce linear fragments from the repeated ‘truncation’ of larger fragments within a concentrated pool (via restriction enzymes).
Both methods are commonly used in conjunction.
Both methods produce linear double-stranded fragments.
The result of both methods may be assayed via gel electrophoresis.
Primers with the appropriate 20-40bp overlaps must be precisely designed and validated for use prior to their use as the initiation point of PCR. This ensures the required 5’ to 3’ orientation and overlaps are present to permit the successful cloning via Gibson assembly.
Simple diffusion permits the entry of cloned plasmids into E. coli cells permitted that heat shock or electroporation techniques have been used prior, as these methods generate pores within the plasma membrane that would otherwise be impassable.
Twin-Primer assembly is an non-enzymatic, alternative technique of assembling PCR-amplified fragments into plasmids, while retaining the necessary efficacy and fidelity of conventional methodologies:
Segment the overall sequence in-silco into a series of fragments with complimentary designed overlaps between adjacent fragments, with two ‘sticky ends’ required per fragment. Overlaps are characteristically 16-20 bp long, and are selected to possess a universal melting temperature of approximately 50 C.
Amplification is then performed separately, twice per fragment, with each iteration using a different set of primer pairs. Per fragment, this produces: A, sequence with overlap to previous fragment, and B, sequence with overlap to next fragment.
The two products (A and B) are then combined to generate two separate versions of the same fragment however featuring unique overlapping regions.
Post mix., the PCR’ed products are denatured to generate ssDNA, before controlled cooling to generate hybrid ‘duplex intermediates’.
All duplex intermediates are then incubated collectively, permitting the annealing of overhangs from complimentary fragments, and ultimately assembling a plasmid (albeit with nicked backbone)
Upon transformation into the desired host, bacterial machinery is co-opted to phosphorylate DNA ends and ligate nicks throughout the DNA backbone, ultimately resulting in a complete plasmid.
The Repressilator construct. Although its title is very fitting, it's still such a cool name!
Iteration 1
Began by referencing the Repressilator paper: source
Unsure of the appropriate RBS to use prior to each coding region. There’s a fair few available in ‘Charecterized Bacterial Parts’ repository! Wasn’t able to locate BBa_B0034, the original RBS utilised within the construct described in the paper. Selected S1 RBS at random; lets see how it goes!
I was able to locate and insert pSC101, the original low-copy backbone used by Elowitz & Liebler!
Stock settings were employed in the initial simulation (E. coli, 72 hours, 10 min timestep).
Results were not aligned with the oscillatory behaviour of the Repressilator. Protein expression appeared to peak across each gene, then remain constant.
Iteration 2
Realised almost immediately upon inspection of the repressilator in the bacterial samples folder that I had failed to include terminators!
Retained pSC101 and S1 RBS.
Stock settings (E. coli, 72 hours, 10 min timestep).
Results were much more reminiscent of reference! Oscillatory expression behaviour appeared to ‘tighten’, with the period each gene was expressed narrowing throughout the simulation.
Iteration 3
Finally adjusted RBS to A1 and backbone to pUC-SpecR v1 (as featured within the reference Repressilator)
Promoters still appear in slightly different locations to reference, however the overall architecture appears to match that of Elowitz & Liebler.
Results were again significantly different; actually less relevant than those obtained in iteration 2!
Definitely suspicious that the layout of the promoters is altering the outcome.
Iteration 4
Precisely copied the layout of the reference construct.
Results matched the reference.
Iteration 1
Iteration 2
Iteration 3
Iteration 4
Custom Constructs
Construct A
Construct A
I wanted to understand how the pTac promoter could be used in conjunction with IPTG ligands to control expression of a specific gene. In construct A, the intention is that the transcription of LacL produces a repressor preventing the transcription of mKate, and hence its translation to yield the fluorescent reporter protein.
Simulation 1
Settings: E. coli, 72 hours, 10 min intervals. Ligand (IPTG) introduced at max concentration at 36 hours.
Appeared to work first try! LacL was transcribed at a constant rate, repressing the activity of the pTac promoter. At 36 hours, the introduction of IPTG functioned as a structural analog to allolactose, inhibiting the activity of the repressor and permitting the transcription of mKate.
Protien Concentrations
RNA Concentrations
Ribosome Flux
RNAP FLux
Construct B
Attempt at a pseudo AND gate. Both L-Arabinose and IPTG must be present for each component to be expressed. Here, we imagine mOrange and GFP must both be expressed as precursors in the formation of an output.
Construct B
Construct C
Attempt at NOT gate. mOrange expressed in the absence of L-arabinose, and ceases in its presence.
Construct C
References
Elowitz, M.B. and Leibler, S. (2000). A synthetic oscillatory network of transcriptional regulators. Nature, [online] 403(6767), pp.335–338. doi:10.1038/35002125.
Week 7 HW: Genetic Circuits Part ii
Intracellular Artificial Neural Networks (IANNs)
What advantages do IANNs have over traditional genetic circuits, whose input/output behaviors are Boolean functions?
Cellular processes are inherently continuous, with a multitude of intermeshed processes with inputs and outputs that are mutually significant. It is often difficult, if not impossible, to effectively manipulate such an environment through a boolean system alone.
IANN’s allow for genetic circuits to be programmed which respond effectively to ‘analog’ signals, whereby the rules of gene expression (when, and how much) may be governed by molecular gradients across two or three separate inputs.
Describe a useful application for an IANN; include a detailed description of input/output behavior, as well as any limitations an IANN might face to achieve your goal.
Quorum sensing is the process by which bacterial cells within a shared environment may communicate as a means to regulate population-wide behaviours, typically in response to unique population thresholds. Let’s consider a theoretical case for biomaterial formation, where we have first programmed and cultured a non-pathogenic bacterial species to function as an efficient biomineralisation unit.
To permit a degree of programmability to the duration and localisation of biomineralisation behaviour across each individual bacterium, an IANN may be employed to modify and expand the capabilities of the quorum-sensing process. If we have established that the process is the most efficient across a particular population-nutriant ratio, we will seek to construct a lower and upper threshold to provide a degree of redundancy for use in environments heterogeneous to the laboratory. This is analogous to the band-pass model described in the lecture.
Inputs
Auto-inducer A: produced only when population values exceed the lower threshold bias.
Auto-repressor B: produced only when the population values exceed the upper threshold bias (determined by training IANN on culture growing within chemostat under several different ratios; the goal is to coax the population into maintaining a stationary-like phase as the only conditions under which biomineralisation occurs.
Outputs
Low A low B: no biomineralisation.
Medium A low B: biomineralisation
High A High B: no biomineralization
Recognising when a particular population density has been exceeded could prove difficult, and is likely to manifest at different points dependent on energy source. Several separate ligands are likely to be responsible for regulating growth under different conditions; careful construction of the host genome would need to be required prior to developing IANN to limit ‘noise’.
Draw a diagram for an intracellular multilayer perceptron where layer 1 outputs an endoribonuclease that regulates a fluorescent protein output in layer 2.
Multilayer perceptron: X1 input reduces endonuclease production and thus permits translation of Y1BFP in layer 2.
Fungal Materials
What are some examples of existing fungal materials and what are they used for? What are their advantages and disadvantages over traditional counterparts?
Applications of filamentous fungal materials are quite diverse due to the varied mechanical properties of both rigid and flexible denominations; construction elements and biodegradable packaging respectively are two excellent examples. Mycelium-based materials often surpass virtually any conventional material where true biodegradability is desirable, as they may be completely decomposed and reintegrated by organic lifeforms without requiring additional processing, and often without industrial composting. Properties such as heat resilience, insulation, and rigidity are highly customisable. Resultingly, the materials are also susceptible to premature decay, moisture, and contamination. Long-term durability is also limited in applications where exposure to friction or mechanical stresses is common.
What might you want to genetically engineer fungi to do and why? What are the advantages of doing synthetic biology in fungi as opposed to bacteria?
I would be interested to explore how the structural properties of fungal growth could be controlled internally to produce highly functionally-graded materials. It would also be quite fascinating to explore the communicative-element of mycelial networks; could a network be constructed which permitted messages to be sent and received in a couple hours across, say, the local park?
The diverse structural and metabolic features of fungi make them an excellent vehicle for synthetic biology in areas such as bioremediation and, of course, biomaterials. Genetically, they share a closer resemblance to humans than plants and prokaryotic bacteria, which is intriguing in its own right. They are comparatively more rugged and resilient to environmental stressors than bacteria, which may be useful in an experimental context.
References
Hinneburg, H., Gu, S. and Naseri, G. (2025). Fungal Innovations—Advancing Sustainable Materials, Genetics, and Applications for Industry. Journal of Fungi, [online] 11(10), p.721. doi:10.3390/jof11100721.
Moreno-Gámez, S., Hochberg, M.E. and van Doorn, G.S. (2023). Quorum sensing as a mechanism to harness the wisdom of the crowds. Nature Communications, [online] 14(1). doi:10.1038/s41467-023-37950-7.
Week 9 HW: Cell-Free Systems
General homework questions
Explain the main advantages of cell-free protein synthesis over traditional in vivo methods, specifically in terms of flexibility and control over experimental variables.
Comparative to traditional methods, cell free systems:
Permit the incorporation of non-canonical amino acids (NCAA’s) for extended protein functionality not observed in nature.
Permit the synthesis and collection of difficult to express proteins, such as trans-membrane proteins.
Allow the immediate use of linear fragments in solution. This offers high flexibility for projects involving prototyping at frequent intervals; no need for plasmid construction and subsequent cloning.
Permit greater control of matrix composition during biochemical processes. Proceed with crude extracts for high-throughput, inexpensive studies, or calibrate its composition (such as in PURE) to examine or promote specific processes.
Cell-free is uniquely superior for use in environments which do not support cell production techniques, such as in materials! i.e a cell-free system may be inactivated by lyophilization then subsequently reactivated upon rehydration.
Describe the main components of a cell-free expression system and explain the role of each component.
Engineered sequences for translation; typically plasmids, but also possible with linear fragments. The core information for what is to be expressed.
Cellular lysate containing required molecular machinery for Tx/Tl (ribosomes, RNA polymerase etc). Permits the expression of the engineered sequences.
Source of energy-rich compounds and nucleosides. Provide free energy for enzymes in the reaction to do work, as well as necessary substrates for building mRNA.
Buffering compounds: stabilise reactions and maintain consistent pH.
Why is energy provision regeneration critical in cell-free systems? Describe a method you could use to ensure continuous ATP supply in your cell-free experiment.
In living cells, energy regeneration is typically mediated by sequential chains of catabolic and anabolic reactions, by which the cell will sustain protein-synthesis activities; these pathways can be absent in cell-free systems. Phosphoenolpyruvate (PEP) is commonly used to regenerate ADP molecules into ATP, providing a ‘continuous’ supply with the caveat that its concentration must be ample for the entire duration of the intended experiment. Glucose-6-phosphate (G6P) is a promising, affordable alternative to PEP which still functions as a reducing agent.
Compare prokaryotic versus eukaryotic cell-free expression systems. Choose a protein to produce in each system and explain why.
Each approach presents its own distinct advantages and limitations, often dependent on the specific source for which the expression system is based. While prokaryotic permits high protein yields, simplicity in genetic engineering, and the ability to synthesise in extreme conditions (Archeal extremophile extracts), post-translational modifications are limited and only chaperones native to prokaryotic species are available. Eukaryotic extracts are diverse, permit fast lysate preparation, and mirror mammalian systems, yet often are also subject to high-cultivation costs and low protein yields.
Prokaryotic: Cas12a, modified via the inclusion of NCAA’s to extend its trans-cleavage properties. Useful when expressed in high yields as the main component of a signal-amplifying biosensor for.
Eukaryotic: any protein intended for use in the human body! Likely to produce proteins with higher-quality folding, as well as overall be more compatible with human biochemistry.
How would you design a cell-free experiment to optimize the expression of a membrane protein? Discuss the challenges and how you would address them in your setup.
Membrane proteins are often flexible and structurally unstable, and are typically expressed and purified from live-culture systems in which they are first correctly folded and inserted into the cellular membrane by a specific enzyme. To mirror this process and extend the stability of synthesized membrane proteins during storage, one such approach could be to encourage their integration with artificial vesicles. Whilst this technique may improve their stability, it could prove as a hindrance when further purification is required to ‘detach’ the membrane proteins and reintegrate them into the host organism.
Imagine you observe a low yield of your target protein in a cell-free system. Describe three possible reasons for this and suggest a troubleshooting strategy for each.
Insufficient energy source: recalibrate concentration.
Incompatible lysate: identify suitable alternative.
Inefficient codon usage: reoptimise codons for expression in specific lysate.
Homework question from Kate Adamala
Pick a function and describe it.
1. What would your synthetic cell do? What is the input and what is the output?
Produce and excrete magnetic inclusions (magnetosomes).
2. Could this function be realized by cell-free Tx/Tl alone, without encapsulation?
No, membrane encapsulation is required for magnetite vesicle formation.
3. Could this function be realized by genetically modified natural cell?
Yes! However, likely not at a scale and yield which is industrially significant.
4. Describe the desired outcome of your synthetic cell operation.
Minimal cells are cultivated inside a bioreactor, with capability to support the production of various valuable compounds in response to specific signalling molecules. When IPTG is added to the system, culture responds by producing and secreting magnetite vesicles.
Design all components that would need to be part of your synthetic cell.
1. What would the membrane be made of?
Unsaturated fatty acid bilayer to enhance fluidity and facilitate vesicle formation.
2. What would you encapsulate inside?
Proteins associated with the MamAB gene cluster would constitute the most significant inclusions, specifically MamE (magnetite and vesicle maturation), MamI + MamL (magnetosome membrane formation).
3. Which organism your Tx/Tl system will come from? Is bacterial OK, or do you need a mammalian system for some reason?
Prokaryotic is preferred; Tx/Tl system can primarily be derived from the magnetotactic model organism AMB-1.
4. How will your synthetic cell communicate with the environment?
IPTG is membrane permeable. Magnetite vesicles may be freely transported via budding and shedding.
Experimental details
1. List all lipids and genes.
mamAB gene cluster: magnetite vesicle formation. LacI repressor: receptor for IPTG. IPTG ligand: ligand for LacL deactivation.
2. How will you measure the function of your system?
Due to the conductive nature of magnetite, I would use electrochemical conductometry to monitor the protein yield in real time in response to the presence of IPTG.
Homework question from Peter Nguyen
Write a one-sentence summary pitch sentence describing your concept.
I propose a biosensor embedded in a reusable, broach-like microfluidic capable of the detection and artistic indication of a broad-spectrum of aerosolised compounds.
How will the idea work, in more detail?
The seemingly inescapable routine demanded by contemporary working culture is often the culprit for distorting the passage of time, accelerating our perceptions to feel as if months pass by frictionless, leaving nothing to hold but vague recollections of mundanity. What if we had a unique anchor to distinctly summarise each day?
The broach would be composed of photoablated acrylic disk sandwiched by disposable semi-permeable membranes and a fixed, chemicapacitive backing. As the user goes about their day, aerosols and airborne particulates attach and diffuse passively through the external membrane into the grid-like, photoablated, aqueous buffer beneath. The cell-free system inhabiting the aqueous buffer would be composed primarily of constructs engineered to transcribe proteins of varied charge, each controlled by a unique transcription activator-like effector (TALE) repressor which interacts with a specific aerosol substrate. Conformational binding would detach the repressor and permit localised protein expression, corresponding to a measurable increase in the net charge of the pore via a measurable change in capacitance at the pore-chemicapacitive backing interface. Capacitance values retrieved across the porous grid would then be transformed and represented digitally (i.e hex code, greyscale value, 3D peaks / troughs) to create a one-off abstract representation of the day experienced according to the unique air composition. To reset the device, the disposable membranes would be replaced, and the pores of the broach flushed and refilled with the cell-free system.
What societal challenge or market need will this address?
Routine, and the departure from spontaneity, is inevitable as we age and mature. Although a minor combatant, the concept aims to help its user, and broader communities as a whole, integrate a consistent element of novelty into their daily lives as a means to provide a substrate for the formation of episodic memories in an otherwise routine day. It is hoped that observation and reflection upon this small, consistently inconsistent visualisation of daily difference will help its user slow down momentarily and appreciate even the most seemingly mundane of days.
How do you envision addressing the limitation of cell-free reactions (e.g., activation with water, stability, one-time use)?
The versatility of capacitance-based sensors permits implementation of a wide range of perpetually reusable configurations, with the simplest requiring just two conductive plates and a thin dielectric layer. Flushing and refilling the device presents the greatest challenge; how might residual reagents skew capacitive readout? How might the fresh aqueous cell-free circuit be suitably stored and supplied to the user as to permit daily refill? Where may a spent cell-free solution be safely disposed of? For this prompt, I envision a world where the production of constructs and enzymatic intermediaries is possible at-home via the democratisation of processes (and associated reagents) such as oligonucleotide synthesis and gibson assembly, by which specialised ‘one-time’ cell-free circuits may be built and safely deployed.
Homework question from Ally Huang
Provide background information that describes the space biology question or challenge you propose to address.
Energy preservation is of particular importance when considering deep space exploration. Isolation, and the inability to receive prompt resupply, place onus upon the use of highly self-sufficient energy systems such as nuclear. Radioisotope thermoelectric generators (RTG) are a prime example, powering and permitting the incredible endurance of spacecraft such as the Voyager probes. Nuclear systems, such as NASA’s SR-1 fission-powered spacecraft Freedom, are likely to play an increasing role within our presence in space.
While nuclear energy sources such as RTG’s are typically designed with strong failsafes and redundancies, their critical failure and spillage could prove catastrophic for resource-limited planetary colonies, threatening the habitability of already challenging environments.
Bio-remediation presents a possible solution to the stabilisation of environments intended to support life post-incident. My proposal seeks to identify and refine a cell-free system (or otherwise!) as a bio-remediation tool to stabilise irradiated environments in space.
Name the molecular or genetic target that you propose to study.
Bacillus sphaericus is a radioresistant prokaryotic bacterial species featuring an outermost paracrystalline S-layer that is described to possess radioabsorptive properties; effectively a bacterial mop! My proposal would investigate the pairing of the pprA radio-responsive promoter from Deinococcus radiodurans with the S-layer encoding sbpA gene from B. sphaericus, in a single synthetic construct.
Describe how your molecular or genetic target relates to the space biology question or challenge your proposal addresses.
sbpA proteins are capable of self-assembling to form organised S-layer lattice structures; this level of autonomy is highly advantageous for resource-limited settings in space. Confirming sbpA expression and consequent S-layer formation in a cell-free system would be pivotal in scaling the process to the extent required for utility as a feasible radioactive cleanup method.
Clearly state your hypothesis or research goal and explain the reasoning behind it.
The concentration of sodium bicarbonate-¹³C in solution will be inversely proportional to sbpA expression post-filtration.
The objective is to examine the efficacy of sbpA two-dimensional lattice structures in temporarily sequestering bicarbonate-¹³C present in an aqueous solution. Stable and non-toxic, Carbon-13-labled species presents a feasible analog to hazardous radionuclides for investigating the aptitude of synthetic S-layers in-solution for nuclear cleanup. If we are able to observe a decrease in the concentration of the tracer after nucleopore filtration, we can likely identify causality between S-layer assembly and isotope entrapment.
sbpA gene codon-optimised for expression with BioBits® cell-free system. sbpA sequence flanked in-silco with T7 promoter + RBS (5’) and T7 terminator (3’) and primers incorporated. Cloned via miniPCR® thermal cycler. Incorperate with 80uL nuclease-free liquid.
Pipette 15 μl of cloned sbpA DNA into reaction tube B, incubate at 30c for 4 hours.
Pipette 1.0 mL of 8.5 µg sodium bicarbonate-¹³C stock into tube A and B. Stow in a shaker-incubator at 25c for 4 hours.
Filter A and B through nucleopore membrane filter; evaluate entrapment via Isotope Ratio Mass Spectrometry (IRMS).
References
Calhoun, K.A. and Swartz, J.R. (2005). Energizing cell‐free protein synthesis with glucose metabolism. Biotechnology and Bioengineering, [online] 90(5), pp.606–613. doi:10.1002/bit.20449.
Carpenter, E.P., Beis, K., Cameron, A.D. and Iwata, S. (2008). Overcoming the challenges of membrane protein crystallography. Current Opinion in Structural Biology, [online] 18(5), pp.581–586. doi:10.1016/j.sbi.2008.07.001.
Murat, D., Quinlan, A., Vali, H. and Komeili, A. (2010). Comprehensive genetic dissection of the magnetosome gene island reveals the step-wise assembly of a prokaryotic organelle. Proceedings of the National Academy of Sciences, [online] 107(12), pp.5593–5598. doi:10.1073/pnas.0914439107.
Paul, N.L., Carpa, R., Ionescu, R.E. and Popa, C.O. (2025). The Biomedical Limitations of Magnetic Nanoparticles and a Biocompatible Alternative in the Form of Magnetotactic Bacteria. Journal of Functional Biomaterials, [online] 16(7), p.231. doi:10.3390/jfb16070231.
Pui Yan Wong, Mal, J., Sandak, A., Luo, L., Jian, J. and Pradhan, N. (2024). Advances in microbial self-healing concrete: A critical review of mechanisms, developments, and future directions. The Science of The Total Environment, 947, pp.174553–174553. doi:10.1016/j.scitotenv.2024.174553.
Zemella, A., Thoring, L., Hoffmeister, C. and Kubick, S. (2015). Cell‐Free Protein Synthesis: Pros and Cons of Prokaryotic and Eukaryotic Systems. ChemBioChem, [online] 16(17), pp.2420–2431. doi:10.1002/cbic.201500340.
Please identify at least one (ideally many) aspect(s) of your project that you will measure.
Two quantities are prioritesed for measurement:
Total fluorescence emitted by cas12a transnuclease cleavage of probes after designated runtime (approx. 15-20 minutes). This can be achieved by the use of a fluorometer.
Affinity of SELEX-designed aptamers and exosporium target protein. This may be measured by determining the equilibrium dissociation constant KD, through a number of unique approaches.
Homework: Waters Part I — Molecular Weight
Based on the predicted amino acid sequence of eGFP (see below) and any known modifications, what is the calculated molecular weight?
eGFP sequence (with Methionine, His-purification tag + linker):
Calculate the molecular weight of the eGFP using the adjacent charge state approach described in the recitation.
ESI electrospray ionisation: single proton H+ corresponds to 1.0073 DA.
z = 30.9058 (e)
Mw = 27898.90 Da
Accuracy: 3845.52 Ppm
Accuracy is well in excess of a suitable deviation of 0-50 PPm (calculation error?)
Can you observe the charge state for the zoomed-in peak in the mass spectrum for the intact eGFP?
The resolution of the spectrometer is not sufficient to resolve individual isotopic peaks of proteins at this size, and thus the charge cannot be determined.
Homework: Waters Part II — Secondary/Tertiary structure
Based on learnings in the lab, please explain the difference between native and denatured protein conformations.
As a protein denatures and unfolds, its ‘exposed’ surface area increases and permits a greater degree of ionisation. This will be reflected by the distribution of several large peaks at a higher mass to charge ratio, as opposed to the concentrated peaks observed in a native state protein analyte.
Zooming into the native mass spectrum of eGFP from the Waters Xevo G3 QTof MS, can you discern the charge state of the peak at ~2800?
In this instance, the resolution is satisfactory to resolve the charge state.
Homework: Waters Part III — Peptide Mapping - primary structure
How many Lysines (K) and Arginines (R) are in eGFP?
How many peptides will be generated from tryptic digestion of eGFP?
Mass (Da)
Position
Peptide sequence
4472.1752
170–210
HNIEDGSVQLADHYQQNTPIGDGPVLLPDNHYLSTQSALSK
2566.2931
217–239
DHMVLLEFVTAAGITLGMDELYK
2437.2608
5–27
GEELFTGVVPILVELDGDVNGHK
2378.2577
54–74
LPVPWPTLVTTLTYGVQCFSR
1973.9062
142–157
LEYNYNSHNVYIMADK
1503.6597
28–42
FSVSGEGEGDATYGK
1266.5783
87–97
SAMPEGYVQER
1083.4979
240–247
LEHHHHHH
1050.5214
115–123
FEGDTLVNR
982.4952
133–141
EDGNILGHK
821.3940
81–86
QHDFFK
790.3552
75–80
YPDHMK
769.3913
47–53
FICTTGK
711.2944
103–108
DDGNYK
655.3813
98–102
TIFFK
602.2780
211–215
DPNEK
579.3137
128–132
GIDFK
507.2925
164–167
VNFK
502.3235
124–127
IELK
19 sequences; sequences < 500 Da appear to be excluded.
Based on the LC-MS data for the Peptide Map data generated in lab how many chromatographic peaks do you see in the eGFP peptide map between 0.5 and 6 minutes?
Approximately 21, however less if dependent on how merged peaks are handled.
Assuming all the peaks are peptides, does the number of peaks match the number of peptides predicted from question 2 above? Are there more peaks in the chromatogram or fewer?
No, however the in-silico digest deviates by only 2 fewer peaks. Considering the in-silico is likely to have omitted sequences < 500 dA, it seems plausible that this accounts for the additional peaks resolved by the Total ion chromatogram.
Identify the mass-to-charge of the peptide shown in Figure 5b.
From the enhanced image, we can determine an approximate spacing between peaks of m/z 0.5, correlating to z = 5.
(M+H)sup>+ = 2623.77 Da
Identify the peptide based on comparison to expected masses in the PeptideMass tool. What is mass accuracy of measurement?
This correlates most closely with DHMVLLEFVTAAGITLGMDE LYK at 2566.2931 dA, however this appears substantially off; necessary to review the charge calculation performed above.
A calculated value of 22396.85 PPM confirms a calculation error.
What is the percentage of the sequence that is confirmed by peptide mapping?
88 percent of the sequence was confirmed by peptide mapping.
Homework: Waters Part IV — Oligomers
Polypeptide Subunit Name
Intensity
7FU Decamer
3.4
8FU Decamer
4.013
8FU Didecamer
8.33
8FU 3-Decamer
12.67
8FU 4-Decamer
16.683* (not included from CDMS data)
Homework: Waters Part V — Did I make GFP?
Theoretical
Observed/measured on the Intact LC-MS
PPM Mass Error
Molecular weight (kDa)
28006.60
27898.90
3845.52 ppm
References
Jing, M. and Bowser, M.T. (2011). Methods for measuring aptamer-protein equilibria: A review. Analytica Chimica Acta, [online] 686(1–2), pp.9–18. doi:10.1016/j.aca.2010.10.032.
Week 11 HW: Bioproduction & Cloud labs
Part A: The 1,536 Pixel Artwork Canvas | Collective Artwork
My efforts were unfortunately extremely limited, having only contributed 2 pixels total. Definitely regret not having been more engaged! Given the chance to contribute again, I would have loved to coordinate my efforts with members from my node (alike to what occurred across r/place) to build a mutually-crafted design! It was fascinating seeing it evolve over time; I often wondered to what extent others were collaborating.
The scale appeared somewhat limiting in terms of achieving higher-resolution designs. For future iterations where (hopefully) a greater cohort of students are enrolled, it would be fascinating to see what would be possible with 4x the space.
Part B: Cell-Free Protein Synthesis | Cell-Free Reagents
E. coli Lysate
BL21 (DE3) Star Lysate
Includes T7 RNA Polymerase.
Salts/Buffer
Potassium Glutamate
Maintains ionic strength for optimal reaction conditions.
HEPES-KOH pH 7.5
Buffers the system to maintain stable pH.
Magnesium Glutamate
Source of Mg^2+ ions which optimise Tx + Tl rates by supporting the function of translational machinery; essential cofactor.
Potassium phosphate monobasic
Buffering agent to maintain pH stability. Precursor molecule for regeneration of ATP from ADP.
Potassium phosphate dibasic
Buffering agent to maintain pH stability. Additional inorganic phosphate source for ATP regeneration.
Energy / Nucleotide System
Ribose
Essential precursor molecule in structure and function of nucleic acids. A core component of ATP and thus a crucial component in its regeneration.
Glucose
Significant energy source and substrate for ATP regeneration.
AMP
Nucleotide precursor for RNA synthesis. Precursor for ATP regeneration.
CMP
ATP regeneration intermediate; precursor of CTP which is required as energy source in the transcription process.
GMP
Cost-effective alternative to NTP’s. Converted to GTP via intermediary energy regeneration reactions, an essential energy source for initiation (and more!) during translation.
UMP
Precursor molecule for RNA biosynthesis; provides initial structure for phosphorylation which eventually becomes UTP, a substrate in transcription.
Guanine
Fundamental base across both DNA and RNA, as well as GTP precursor. Constituent element in energy regeneration.
Translation Mix (Amino Acids)
17 Amino Acid Mix
Constitutes key amino acids essential for the formation of a protein's primary structure (sequence) during polymerisation.
Tyrosine
Amino acid with functional roles when integrated as a subunit into primary structure (hydrophobic and polar). May serve as a site of post-postranslational modifications.
Cysteine
Amino acid with reductive qualities. Facilitates translational elongation.
Additives
Nicotinamide
Essential precursor molecule for NAD+ regeneration, an oxidising agent central to translational processes.
Backfill
Nuclease Free Water
Highly-purified water. Shields sensitive contents of mixture from degradation.
Part C: Planning the Global Experiment | Cell-Free Master Mix Design
Describe the main differences between the 1-hour optimized PEP-NTP master mix and the 20-hour NMP-Ribose-Glucose master mix.
The 20-hour appears to include 2 additional salts/buffers (Potassium phosphate in ratio 1.6:1 dibasic to monobasic), which appears to provide higher, sustained buffering capacity (longer reaction?). The energy/nucleotide system in the 1-hour is immediately accessible; NTP’s are provided pre-synthesised alongside ATP and PEP-mono, which serves as a key intermediate in the ATP regeneration process. 20-hour is more reliant on enzymatic pathways inherent to the lysate for energy regeneration and NTP formation. 1-hour includes heavy supplementation of additives to limit requirement of intermediate metabolic process; 2-hour supplies only Nicotinamide, a NAD precursor required for energy regeneration.
How can transcription occur if GMP is not included but Guanine is?
Guanine must be converted metabolically by cellular machinery in lysate to form GMP prior to its use in transcription. There are two feasible pathways for this lysate; phosphoribosylation directly to GMP (requires that PRPP enzyme is present in lysate) or the Purine nucleoside phosphorylase pathway, in which it is converted into Guanosine, then GMP.
Oxidative damage and/or mutations to the residue Arg96 may hinder the rate at which the sfGFP chromophore is developed, potentially altering duration until readout. The production of reactive oxidative species is possible in mixes more reliant on expansive metabolic processes.
Create a hypothesis for how adjusting one or more reagents in the cell-free mastermix could improve a specific biophysical or functional property you identified above, in order to maximize fluorescence over a 36-hour incubation. Clearly state the protein, the reagent(s), and the expected effect.
I selected mKO2 for enhancement as I love the colour orange!
Some characteristics:
Maturation time: 108 minutes (quite long!)
pkA: 5.5 (point at which fluorescence drops to 50 percent of maximum value)
Brightness: 39.56
Rigidity is considered an intrinsic factor to the magnitude of the quantum yield obtainable by the fluorophore of fluorescent proteins. At first, I hypothesized that:
Artificially inducing mild oxidative stress will increase the monomeric-clamping activity of HSP70 chaperones, fortifying early structural formation and consequently improving rigidity characteristics.
However this approach became a little unraveled once I realised that the concentration of elements in the master mix could only be increased, and not decreased as required to test my hypothesis. I redirected, instead attempting to Increase translational rates and enhance HSP70 activity. Two separate approaches were formulated, each for a group of four wells:
Approach A
Iteration
Glucose
MgGlutamate
KGlutamate
q1-n5
1.250g/L
6.975mM
312.56mM
q1-n6
2.500g/L
7.600mM
312.56mM
q1-n7
5.00g/L
8.225mM
325.688mM
q1-n8
7.500g/L
8.225mM
335.688mM
Approach B
Iteration
Glucose
HEPES
MgGlutamate
KGlutamate
q1-n9
1.250g/L
45.000mM
6.975mM
312.56mM
q1-n10
1.250g/L
50.000mM
6.975mM
312.56mM
q1-n11
1.250g/L
53.750mM
7.600mM
325.688mM
q1-n12
1.250g/L
58.750mM
7.600mM
335.688mM
Part D: Build-A-Cloud-Lab
Quick attempt! The RAC's have an almost skyline-like appearance when clustered and composed nonsensically.
References
Banks, A.M., Whitfield, C.J., Brown, S.R., Fulton, D.A., Goodchild, S.A., Grant, C., Love, J., Lendrem, D.W., Fieldsend, J.E. and Howard, T.P. (2022). Key reaction components affect the kinetics and performance robustness of cell-free protein synthesis reactions. Computational and Structural Biotechnology Journal, [online] 20, pp.218–229. doi:10.1016/j.csbj.2021.12.013.
Group: Alya, Beyza, Sydney, Zander.
Brainstorming session Bacteriophages -> L to lyse bacterial cells Release newly produced phages Modification to make more effective for lysing the protein Higher stability for L protein L protein interacts with host (cellular missionary) with chaperone, DnaJ Influence protein folding POSSIBLE DIRECTION: modifying residues to influence how L protein interacts with host proteins Specific minor acid residues can affect lysis
Subsections of Projects
Individual Final Project: Topically Applied Biosensor For the detection of C. Difficile Endospores
Section 1: Abstract
Clostridioides difficile is a pathogenic, spore-forming bacterium, constituting the leading cause of hospital acquired colitis. Highly resistant to common disinfectants, strong hand hygiene (HH) is considered the most effective preventative measure in controlling its transmission; how might we provide a method of endospore contamination detection to enhance the efficacy of handwashing as a preventative measure? The primary objective of this project was to conduct in-silico discovery and screening of ssDNA Aptamers as potential CdeC C. Difficile biomarker binders, with the high-level aim of developing a speculative, topically-applied biosensor for C. Difficile spore detection via fluorescent, macroscale readout. In this project, an iterative process of aptamer generation (Python, ProtScale), evaluation (AptaTrans pipeline), and elimination (ViennaRNA) was investigated, resulting in the curation of a library of 20 potential CdeC binders for synthesis and in-vitro evaluation.
Introduction
Clostridioides difficile (C. Difficile) is a gram-positive, endospore-producing bacterium. A pathogen, the bacterium is responsible for C. Difficile Infection (CDI), an infectious disease with high transmissibility. It is considered the leading cause of hospital acquired diarrhoea. The risk of infection is heightened amongst the elderly and long-term inpatients, especially following the use of antibiotics, by which disruption of the natural gut flora may provide a foothold for the opportunistic pathogen. The sporulation, high-transmissibility, and potential severity of the pathogen often presents a significant challenge in a clinical setting.
The composition of the C. Difficile endospore bestows resistance to most common disinfectants, requiring extended periods of exposure to either sporicidal agents or physical methods (autoclaving) for complete sterilisation. Transmission is most common via the fecal-oral route. Given these properties, strong personal hygiene measures are imperative to stemming transmission, with a particular emphasis on thorough handwashing to physically remove latent endospore contamination.
This project seeks to investigate how cell-free-systems may be deployed as tangible, topical prophylactics for the rapid detection of C. difficile endospores in a clinical setting.
Section 2: Project Aims
Aim 1: Expermimental
Identify and characterise the structure of a suitable C. difficile exosporium protein target (literature review, AlphaFold2). Generate preliminary library of ssDNA sequences biased towards chemical and physical characteristics of target peptide (Python, Colab), then evaluate and retain candidates with Aptamer Protein Interaction (API) scores > 80%. (AptaTrans). Screen pool to identify folding configuration and MFE (ViennaRNA) and retain top 10 candidates for each region. Calculate docking scores (Gibbs free energy of binding) and predict binding site of final pool, identifying candidates with deltaG < - 7.0 to -10.0 kcal/mol (HADDOCK).
Aim 2: Developmental
Evaluate aptamer-target binding kinetics of progressed candidates (SPR). Experimentally evaluate stability of CFS in-vitro against mechanical forces and latent chemical stressors. Conduct preliminary in-vivo trials. Confirm LOD of ≤ 10 CFU per cm2. Calibrate ligase:aptamer ratio to reduce incidence of false-positives in-vitro; reassess or confirm proximity-based ligation technique as transduction pathway. Identify machine-based method of differentiating background fluorescence from readout at macroscale.
Aim 3: Visionary
Improve system to permit readout in interval < 2 minutes. Implement within a clinical setting. Establish a working framework for the development of tangible, topically-applied biosensors for alternative targets and applications.
Section 3: Background
Literature summaries
Cell-free biosensors: where have we been and where do we need to go? (Lucks et al, 2026)
The field has experienced significant technological advancements in the previous five years, with key developments in biological mechanism, technological integration, and target diversity described. Proof-of-concept deployment of biosensors to non-expert users has overall shown great success; the authors highlight the potential for the field to have a significant positive impact across industries. Attention is required to further develop manufacturing and regulatory practices.
Topical Sensory Nanoparticles for in-vivo Biomarker Detection (Sample et al, 2011)
In this study, solid lipid nanoparticles (SLN) were investigated as a topical delivery vehicle for a chemiluminescence-based detection platform intended to detect hydrogen peroxide, a biomarker associated with particular skin cancers. The Phase Inversion Temperature (PIT) method of nanoemulsion production proved successful in the consistent production of 22.4 nm SLN’s, with a tailorable melting point of 32.7C, a value found appropriate for clinical use as a topical. MTT toxicity assays showed minimal irritation in-vivo. Using an optical microplate reader, a LOD of ~ 15mM H2O2 was achieved, the authors noting that sensitivity may be improved via SLN applications at higher concentrations. The authors conclude that the formulation described is well-suited to topical biosensing applications, noting that further work would be advantageous in developing SLN’s as a pain-free platform for topical diagnostics.
Effectiveness of a Glo Germ-based assessment and educational intervention to improve hand hygiene compliance among hospital cleaning staff (Pan et al, 2026)
Recognising the significance of standard precautions (hand hygiene) as essential and cost-efficient measures for the control of healthcare-acquired infections (HAI), the authors investigated the efficacy of Glo Germ, a fluorescent educational tool, as a means to improve hand hygiene protocols in hospital cleaning staff. Glo Germ is an oil-based, ultraviolet fluorescent agent that is applied as a topical to the hands prior to regular handwashing, simulating microbial contamination. The study determined that, pre-intervention, residual fluorescence post-handwashing was present in 33.75% of the participants. Non-compliance dropped to 8.75% post-intervention. The authors remark that, alongside targeted educational interventions, Glo Germ is an effective, rapid tool for the improvement of hand hygiene compliance amongst cleaning staff in healthcare environments.
Opportunity
While application of cell-free systems (CFS) as biosensors is growing across uniquely disparate fields, there has been somewhat limited exploration of the technology as both a topical and fluorescent-diagnostic biosensing method. In Lucks et al (2026), we see that while diverse in mechanism and approach, contemporary diagnostic CFS appear to be quite understandably limited in deployment to physical, bench-top or disposable devices. Pan et al (2026) is a clear, preliminary example of the potential use of visual microbial detection methods in positively altering human behaviour. Sample et al (2011) provide initial indication that topical biosensors show clinical feasibility.
Building upon these observations, the development of a novel CFS approach for the topical detection of microbial contamination appears to address a potentially lucrative gap in the field, potentially contributing to a new class of tangible biosensors with the potential to intuitively reach a broader audience of non-expert users.
Significance and potential impact
Sight is often the most crucial perceptive sense in the daily human experience. Our environments, informational systems, and perception of truth, are fundamentally underpinned by the ability to see, as goes the idiom ‘seeing is believing’. This is particularly evident in the world of biological imaging, where our ability to measure and visualise events not perceptible to the human eye has been the cornerstone of not just scientific breakthroughs, but collective societal shifts in accepted worldview.
The importance of sight has profound implications to public health, where it is often a challenge to effectively communicate the importance of preventative methods when the inherent scale of microorganisms can impede understanding by pure human vision alone. The current West African Ebola virus epidemic is one such harrowing example. Alongside complex social and traditional beliefs, the inability for pathogens to be seen by the naked eye does little to benefit the understanding of disease etiology, but contributes to weakening of adherence to effective preventative measures (Mafuzadze and Manguvo, 2015).
Ethical considerations
The project appears primarily accountable to the principles of clinical ethics, with beneficence and nonmaleficence being of particular importance. As the CFS is intended to function as a rapid assay, it is imperative that an accurate readout is provided, this quality directly relating to its beneficence. If the CFS is delivered via a topical vehicle, guaranteeing non-toxicity and systematic absorption is essential to ensuring nonmaleficence. This principle can also be extended to its potential impact on the broader environment: the continual use and disposal of biologically active components via conventional wastewater must be critically examined to guarantee no ecological harm it enacted.
Adherence to the ethical principles described above may be primarily achieved via two pathways, testing and transparency:
The exhaustive profile and testing of biologically-active elements is mandatory, both within the context of in-vivo and ecologically diverse settings. Development should adhere to the standards required for FDA Class III approval. One possible alternative is the stringent integration of genetic failsafes i.e the components are effectively inert when a given reagent is absent. Failsafe selection must be comprehensive, considering all possible fates of each constituent component in the CFS.
Transparent design and research procedures must be implemented throughout the entirety of the process. External audit should be invited to promote critical discussion of specific implementation details and direction. Alternatively, an in-house critical review group could be formed which performs the same task, without risking the possible ramifications described below.
However, the application of the measures above must also be balanced with the demands of research and (likely limited) funding. Counterintuitively, overregulation could slow progress to the point at which the CFS will have limited reach and hence impact. Proprietary techniques or approaches could be divulged by overtly transparent practices which, in the wrong hands, could lead to the launch of a potentially harmful product, whether by design, complacency, or ignorance.
Section 4: Experimental design, techniques, tools, and technology
Part
Objective
Description
Duration
Status
1
Target identification + selection
Identify and characterise the structure of a suitable C. difficile exosporium protein target.
Literature review: Identity 2-3 papers detailing relevant exosporium structural features. Target selected primary on accessibility and specificity.
AlphaFold: Retrieve predicted structure from AlphaFoldDB.
ProtScale: For target peptide, run:
% accessible residues
% buried residues
Average flexibility
Beta-turn (Deleage Roux)
Hphob (Hopp Woods)
Hydropath (Kyte Doolittle)
Relative mutability.
Python and ChatGPT: Capture and process raw ProtScale data via step-wise function. Establish continuous suitability score; retain two regions which surpass 0.55 threshold.
8 hrs
Complete
2
Aptamer library generation
Generate preliminary library of ssDNA sequences biased towards chemical and physical characteristics of target peptide.
Python and ChatGPT: Identify nucleotide bias values. Generate script to create control + biased libraries for two seperate peptide regions.
Python: Use script to generate 20,000 randomised sequences (10,000 per library); export in batch as seperated .csv.
2 hr
Complete
3
API prediction
Evaluate and retain candidates with Aptamer Protein Interaction (API) scores > 80%.
AptaTrans: For each candidate in each library, provide protien target and predict API.
Python + ChatGPT: Rank binder candidates by API. Visualise result of biasing via overlaid density plot - kernel density estimate. Retain top 100 (per library) with API scores exceeding 80%.
4 hrs
Complete
4
Structural evaluation
Screen pool to identify folding configuration.
ViennaRNA: Score candidates based on Minimum Free Energy; filter for formation of stable stem-loop/hairpin configuration. Retain top 10 candidates per region.
2 hrs
Complete
6
Molecular docking
HADDOCK: Calculate docking scores (Gibbs free energy of binding) and predict binding site of final pool, identifying candidates with ΔG < -7.0 to -10.0 kcal-mol
2 hrs
Pending
Relevant techniques
Pipetting
DNA Gel Art
Bioproduction
Lab Automation
Protein Design
Cell-Free Systems
Gibson Assembly
CRISPR
Technique utilisation
Two techniques/technologies are of noted importance:
Aptamer discovery via AptaTrans (Protein Design*): while undertaking this project, I naively assumed that peptide-binder design suite developed by the Pranam Chatterjee lab (and demonstrated in Week 5!) was applicable to the design of ssDNA aptamers. Only at the point of Twist order preparation did I realise that, (1) the suite was not applicable to the design of ssDNA aptamers, and (2) although a service offered by Twist, multiplexed oligonucleotide synthesis was not offered as an option. Whilst initially a little disheartening, the matter of in-silico aptamer design evolved into not only a significantly educational experience, but the central component of my project!
Aptamer discovery is conventionally conducted via SELEX, an in-vitro, iterative process whereby libraries of known binders are effectively cyclically tested against a target protein, with weak or unbound binders eliminated as candidates prior to initiation of the following stage. Recently, several exciting in-silco methods have emerged as supplementaries (or theoretically, outright replacements) to the costly and long SELEX process, including an approach termed AptaBLE by Chatterjee et al (2026), which is unfortunately not yet publicly available.
The AptaTrans pipeline (Song et al, 2023) is a deep learning technique that employs transformed-based encoders to score binder candidates by a predicted aptamer-protein interaction (API) value. Significantly, binders validated by AptaTrans have been confirmed experimentally in-vitro. The AptaTrans was used centrally in this project to predict API scores for 20,000 binder candidates.
CRISPR/cas12a: the CRISPR/Cas12a platform was selected as the primary MOA due to its transnuclease functionality (ideal for reporter cleavage) and amplification-free compatibility (may be designed to ‘cascade’ as a means of signal amplification at body temperatures).
Theoretical industry partners
Twist biosciences: Aptamer production via multiplexed oligonucleotide synthesis.
Waters Corporation: Measurement of aptamer-target binding kinetics via SPR.
Section 5: results & quantitative expectations
In-silico discovery and proposal of ssDNA CdeC-binding aptamers was selected as the primary feature of the project for validation, primarily due to the importance of the aptamer-based proximity ligation approach in driving the transduction required to permit the use of CRISPR/cas12a. The protocol followed and techniques utilised is outlined incrementally in the subsections below.
Target identification + selection
Three studies focused on the structural characteristics of C. Difficile endospores were compiled. Key findings relevant to target selection are summarised below.
Imaging Clostridioides difficile Spore Germination and Germination Proteins (Sorg and Nerber, 2022)
Germination mediated by CspA and CspC proteins in the spore cortex region
Germination in response to small-molecule germinants (bile acids; very specific)
Fully germinate while remaining encased by exosporium!
Exposporium has a role in adherence of spores to Caco-2 cells (specific receptor?)
Characterization of the Adherence of Clostridium difficile Spores: The Integrity of the Outermost Layer Affects Adherence Properties of Spores of the Epidemic Strain R20291 to Components of the Intestinal Mucosa (Paredes-Sabja et al, 2016)
Exosporium has ‘hair-like projections’
In B. Cereus group, these are formed by collagen-like BcIA glycoproteins
‘Orthologs of the BclA family are highly conserved in the C. difficile sequenced genomes and have been located exclusively in the exosporium proteome’ (Paredes-Sabja et al, 2016)
Outermost exosporium contains BcIA group orthologs, BUT, lacks orthologs of structural proteins; indicates that ‘novel uncharacterised proteins are structural determinants of this layer
Epidemic strain R20291 likely the most clinically significant; 630 is ultrastructurally distinct, lacking hair-like projections on exosporium
‘Competitive-binding assay and trypsin digestion of Caco-2 cells, suggests that spore adherence might be receptor-mediated.’ Further evidence is later presented that strengthens this hypothesis
Protein composition of the outermost exosporium-like layer of Clostridium difficile 630 spores (Paredes-Sabja et al, 2015)
Unfortunately, only the predicted structure was available. Prior to characterisation in ProtScale, residues from N-terminus to position 108 were excluded as pLDDT for this region was < 50. This corresponded to an included window of 108 -> 405.
Prediction uncertainty of CdeC: the lack of experimentally-validated structural information for CdeC presented both an initial challenge and ongoing limitation. As the accuracy of the techniques employed is highly dependent on the input of accurate, resolved structural data, it is difficult to confirm that the final pool of candidate aptamers will actually be functional. A large section of the peptide with low predicted confidence was omitted from the biasing process in an attempt to reduce a flow-on effect in the quality of the biased binders.
Section 6: additional information
References
Angellar Manguvo and Benford Mafuvadze (2015). The impact of traditional and religious practices on the spread of Ebola in West Africa: time for a strategic shift. The Pan African Medical Journal, [online] 22(Suppl 1), p.9. doi:10.11694/pamj.supp.2015.22.1.6190.
Baloh, M., Nerber, H.N. and Sorg, J.A. (2022). Imaging Clostridioides difficile Spore Germination and Germination Proteins. Journal of Bacteriology, [online] 204(7). doi:https://doi.org/10.1128/jb.00210-22.
Barra-Carrasco, J., Olguin-Araneda, V., Plaza-Garrido, A., Miranda-Cardenas, C., Cofre-Araneda, G., Pizarro-Guajardo, M., Sarker, M.R. and Paredes-Sabja, D. (2013). The Clostridium difficile Exosporium Cysteine (CdeC)-Rich Protein Is Required for Exosporium Morphogenesis and Coat Assembly. Journal of Bacteriology, 195(17), pp.3863–3875. doi:https://doi.org/10.1128/jb.00369-13.
Calderón-Romero, P., Castro-Córdova, P., Reyes-Ramírez, R., Milano-Céspedes, M., Guerrero-Araya, E., Pizarro-Guajardo, M., Olguín-Araneda, V., Gil, F. and Paredes-Sabja, D. (2018). Clostridium difficile exosporium cysteine-rich proteins are essential for the morphogenesis of the exosporium layer, spore resistance, and affect C. difficile pathogenesis. PLOS Pathogens, [online] 14(8), p.e1007199. doi:10.1371/journal.ppat.1007199.
Capelli, L., Spezzani, E., Raghavan, D., Micucci, C., Zanut, A. and Bertucci, A. (2026). CRISPR Assays for Protein Detection. Chemistry–Methods, 6(3). doi:https://doi.org/10.1002/cmtd.202600005.
Díaz-González, F., Milano, M., Olguin-Araneda, V., Pizarro-Cerda, J., Castro-Córdova, P., Tzeng, S.-C., Maier, C.S., Sarker, M.R. and Paredes-Sabja, D. (2015). Protein composition of the outermost exosporium-like layer of Clostridium difficile 630 spores. Journal of Proteomics, [online] 123, pp.1–13. doi:https://doi.org/10.1016/j.jprot.2015.03.035.
Green, T.P., Talley, J.P. and Bundy, B.C. (2025). Recent Advances in Developing Cell-Free Protein Synthesis Biosensors for Medical Diagnostics and Environmental Monitoring. Biosensors, [online] 15(8), p.499. doi:https://doi.org/10.3390/bios15080499.
Image: Sporogenesis - an overview, Sciencedirect.com (2022).
Kohlberger, M. and Gadermaier, G. (2021). SELEX: Critical factors and optimization strategies for successful aptamer selection. Biotechnology and Applied Biochemistry, [online] 69(5), pp.1771–1792. doi:10.1002/bab.2244.
Lang, S., Braz, N.F., Slater, M.J. and Kidley, N.J. (2025). In Silico Methods for Ranking Ligand–Protein Interactions and Predicting Binding Affinities: Which Method is Right for You? Journal of Medicinal Chemistry, 68(19), pp.19795–19799. doi:10.1021/acs.jmedchem.5c02582.
Lim, J., Van, A.B., Koprowski, K., Wester, M., Valera, E. and Bashir, R. (2025). Amplification-free, OR-gated CRISPR-Cascade reaction for pathogen detection in blood samples. Proceedings of the National Academy of Sciences, 122(11). doi:https://doi.org/10.1073/pnas.2420166122.
Makovska, I., Biebaut, E., Dhaka, P., Korniienko, L., Jerab, J.G., Courtens, L., Chantziaras, I. and Dewulf, J. (2025). Methods for assessing efficacy of cleaning and disinfection in livestock farms: a narrative review. Frontiers in Veterinary Science, [online] 12. doi:https://doi.org/10.3389/fvets.2025.1581217.
Mora-Uribe, P., Miranda-Cárdenas, C., Castro-Córdova, P., Gil, F., Calderón, I., Fuentes, J.A., Rodas, P.I., Banawas, S., Sarker, M.R. and Paredes-Sabja, D. (2016). Characterization of the Adherence of Clostridium difficile Spores: The Integrity of the Outermost Layer Affects Adherence Properties of Spores of the Epidemic Strain R20291 to Components of the Intestinal Mucosa. Frontiers in Cellular and Infection Microbiology, [online] 6. doi:https://doi.org/10.3389/fcimb.2016.00099.
Mustafa, M.I. and Makhawi, A.M. (2021). SHERLOCK and DETECTR: CRISPR-Cas Systems as Potential Rapid Diagnostic Tools for Emerging Infectious Diseases. Journal of Clinical Microbiology, [online] 59(3). doi:https://doi.org/10.1128/jcm.00745-20.
Qi, M., Xia, N., Wang, X., Wang, X., Chen, H., Lv, D. and Cao, Y. (2025). Development of an Aptamer-Based Surface Plasmon Resonance Biosensor for Detecting Chloramphenicol in Milk. Biosensors, [online] 15(11), p.706. doi:10.3390/bios15110706.
Quintela, I.A., Vasse, T., Jian, D., Harrington, C., Sien, W. and Wu, V.C.H. (2025). Elucidating the molecular docking and binding dynamics of aptamers with spike proteins across SARS-CoV-2 variants of concern. Frontiers in Microbiology, [online] 16. doi:10.3389/fmicb.2025.1503890.
Shin, I., Kang, K., Kim, J., Sel, S., Choi, J., Lee, J.-W., Kang, H.Y. and Song, G. (2023). AptaTrans: a deep neural network for predicting aptamer-protein interaction using pretrained encoders. BMC Bioinformatics, [online] 24(1). doi:10.1186/s12859-023-05577-6.
Stephens, C., Goodey, N.M. and Gubler, U. (2025). A beginners guide to SELEX and DNA aptamers. Analytical Biochemistry, [online] 703, p.115890. doi:10.1016/j.ab.2025.115890.
Sueker, M., Stromsodt, K., Hamed Taheri Gorji, Fartash Vasefi, Khan, N., Schmit, T., Varma, R., Mackinnon, N., Sokolov, S., Alireza Akhbardeh, Liang, B., Qin, J., Chan, D.E., Baek, I., Kim, M.S. and Kouhyar Tavakolian (2021). Handheld Multispectral Fluorescence Imaging System to Detect and Disinfect Surface Contamination. Sensors, [online] 21(21), pp.7222–7222. doi:https://doi.org/10.3390/s21217222.
Varkey, B. (2020). Principles of Clinical Ethics and Their Application to Practice. Medical Principles and Practice, [online] 30(1), pp.17–28. doi:10.1159/000509119.
Supply list
Item
Qty.
Cost (est.)
Google Colab: 100 Compute Units
1
$21.20
Oligonucleotide Pool (92nt length, ~23,000 total) via Twist
Bacteriophages -> L to lyse bacterial cells
Release newly produced phages
Modification to make more effective for lysing the protein
Higher stability for L protein
L protein interacts with host (cellular missionary) with chaperone, DnaJ
Influence protein folding
POSSIBLE DIRECTION: modifying residues to influence how L protein interacts with host proteins
Specific minor acid residues can affect lysis
GOALS
Increase stability of L protein
Higher titers
Higher toxicity of lysis protein
COMPUTATIONAL DIRECTIONS
Attempt to improve L stability
Computational protein design tools
Investigate mutations that could impact protein and host factors interactions
Pull from database what is considered more stable from similar proteins
See what is conserved between them
What does stability mean in this context? Only in cytosol? Ultimate method of delivery?
Use same tool for Week 5
Host cell in unfolded, folded by chaperone
POSSIBLE DIRECTION: If the efficacy of L protein requires chaperone (working within specific temp range), could we mutate to work in greater temperature range for that chaperone
Limit denaturation?
Binding (30-37 C) -> will not be able to lyse bacteria
READING (BEFORE NEXT MEETING)
Stability, which parts of protein to stability
Binding characteristics
What we can change, play with



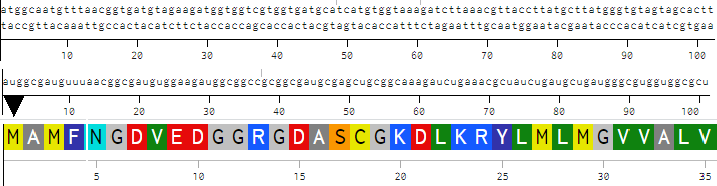

CR.png)