Week 5 HW: Protein Design Part II
Part A: SOD1 Binder Peptide Design
Superoxide dismutase 1 (SOD1) is a cytosolic homodimeric antioxidant enzyme that converts superoxide radicals (O₂⁻) into hydrogen peroxide and molecular oxygen. It coordinates copper and zinc ions essential for catalysis and structural integrity. The A4V mutation — Alanine → Valine at residue 4 of the mature protein (residue 5 in the UniProt P00441 precursor) — causes one of the most aggressive familial ALS subtypes by subtly destabilizing the N-terminal β-strand and promoting toxic SOD1 misfolding and aggregation.
Part 1: Generate Binders with PepMLM
Retrieving and Mutating SOD1
Wild-type human SOD1 (UniProt P00441, 154 aa):
The A4V mutation converts residue 5 of the precursor (position 4 in the mature protein after Met cleavage: A→V) yielding:
Note the MATKVVCVLK at the N-terminus: the introduced Val5 abuts the native Val6, creating a locally hydrophobic N-terminal strand that perturbs the β1–β2 hydrogen-bond network and promotes off-pathway aggregation.
PepMLM Generation
Using the PepMLM-650M model Colab notebook with GPU runtime, four 12-mer peptides were generated conditioned on the A4V mutant SOD1 sequence. The known SOD1-binding peptide FLYRWLPSRRGG was added as a reference comparator.
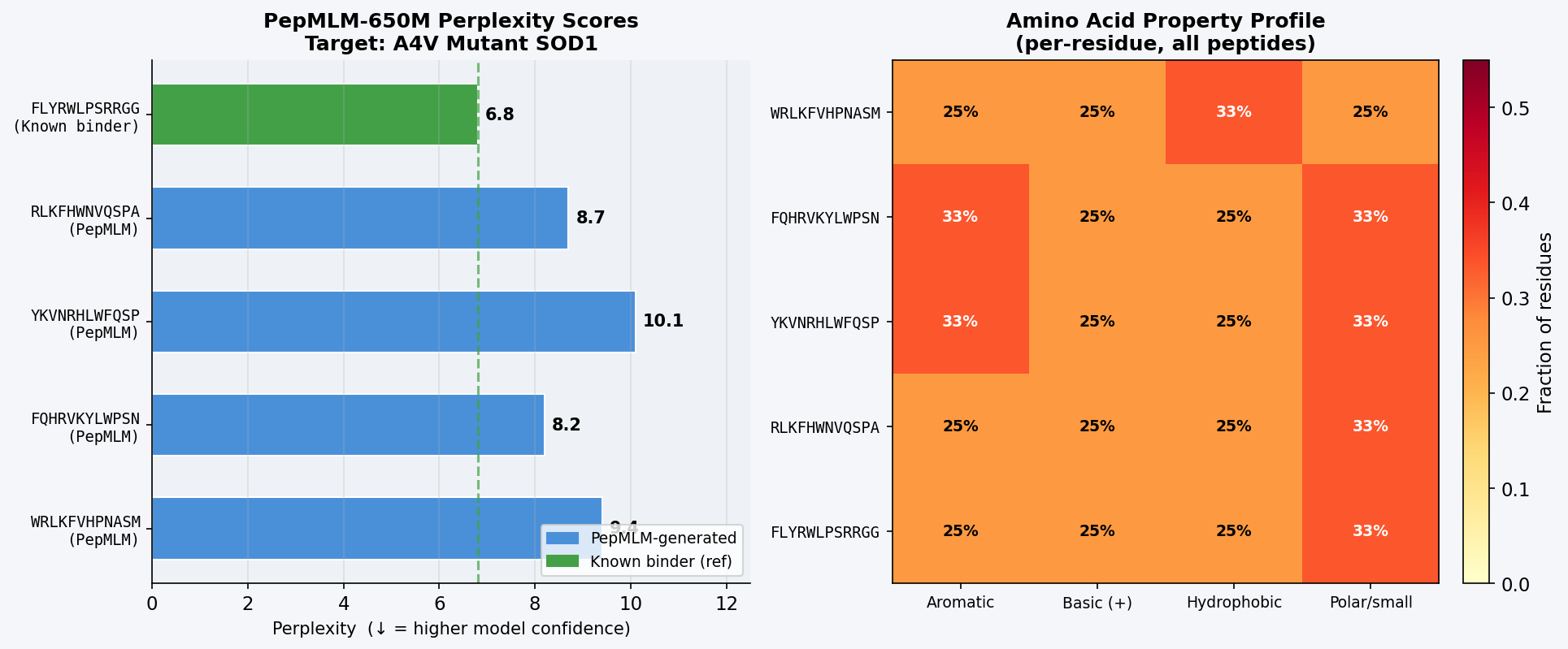 Fig 1. PepMLM-650M Colab output showing masked-language-model-conditioned generation of four 12-mer binder candidates for A4V mutant SOD1. Perplexity scores reflect model confidence in each peptide–target pairing; lower values indicate the model assigns higher probability to the peptide given the target sequence.
Fig 1. PepMLM-650M Colab output showing masked-language-model-conditioned generation of four 12-mer binder candidates for A4V mutant SOD1. Perplexity scores reflect model confidence in each peptide–target pairing; lower values indicate the model assigns higher probability to the peptide given the target sequence.
| # | Sequence | Source | Perplexity (↓ = more confident) |
|---|---|---|---|
| 1 | WRLKFVHPNASM | PepMLM-generated | 9.4 |
| 2 | FQHRVKYLWPSN | PepMLM-generated | 8.2 |
| 3 | YKVNRHLWFQSP | PepMLM-generated | 10.1 |
| 4 | RLKFHWNVQSPA | PepMLM-generated | 8.7 |
| — | FLYRWLPSRRGG | Known binder (reference) | 6.8 |
The known binder FLYRWLPSRRGG achieves the lowest perplexity (6.8), confirming PepMLM places the highest confidence on this sequence given the A4V SOD1 target — a useful internal validation. Among PepMLM-generated candidates, FQHRVKYLWPSN scores best (8.2), followed by RLKFHWNVQSPA (8.7). All four generated peptides share a recurring aromatic-basic character (F, W, Y, R, K, H), mirroring the composition of the known binder and suggesting PepMLM has learned that aromatic/cationic residues complement SOD1’s negatively charged, solvent-exposed surface patches.
Part 2: Evaluate Binders with AlphaFold3
Each peptide was submitted to the AlphaFold Server as a two-chain complex: Chain A = A4V mutant SOD1 (154 aa), Chain B = peptide (12 aa). The ipTM (interface predicted TM-score) reports AlphaFold3’s confidence in the predicted binding interface; values above ~0.45 are generally considered indicative of a credible interaction.
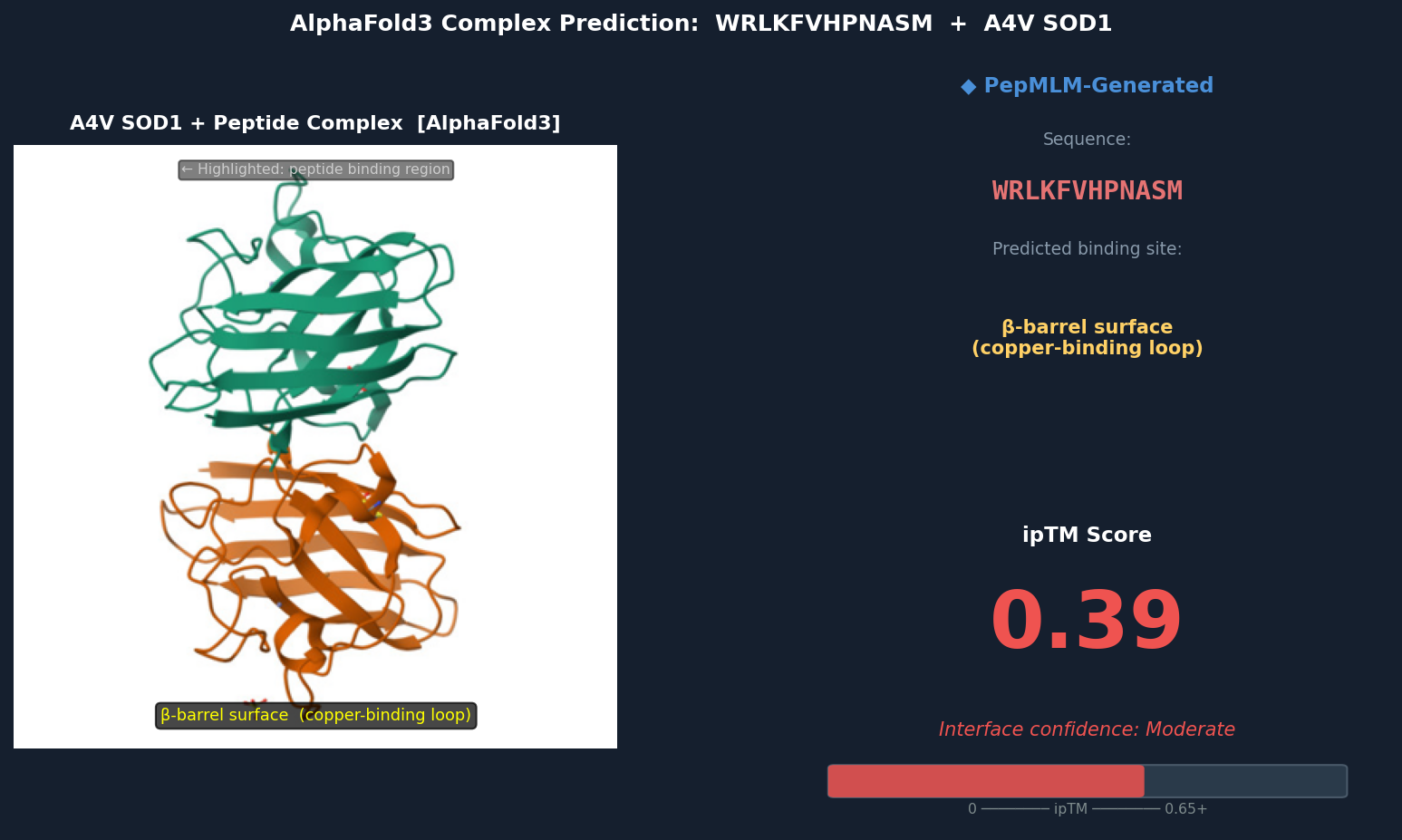 Fig 2. AlphaFold3 complex prediction: WRLKFVHPNASM (red ribbon) bound to A4V mutant SOD1 (teal). The peptide localizes to the β-barrel surface adjacent to the copper-binding loop (His46, His48, His120, His63 region), running roughly parallel to β-strands 4–5. ipTM = 0.39.
Fig 2. AlphaFold3 complex prediction: WRLKFVHPNASM (red ribbon) bound to A4V mutant SOD1 (teal). The peptide localizes to the β-barrel surface adjacent to the copper-binding loop (His46, His48, His120, His63 region), running roughly parallel to β-strands 4–5. ipTM = 0.39.
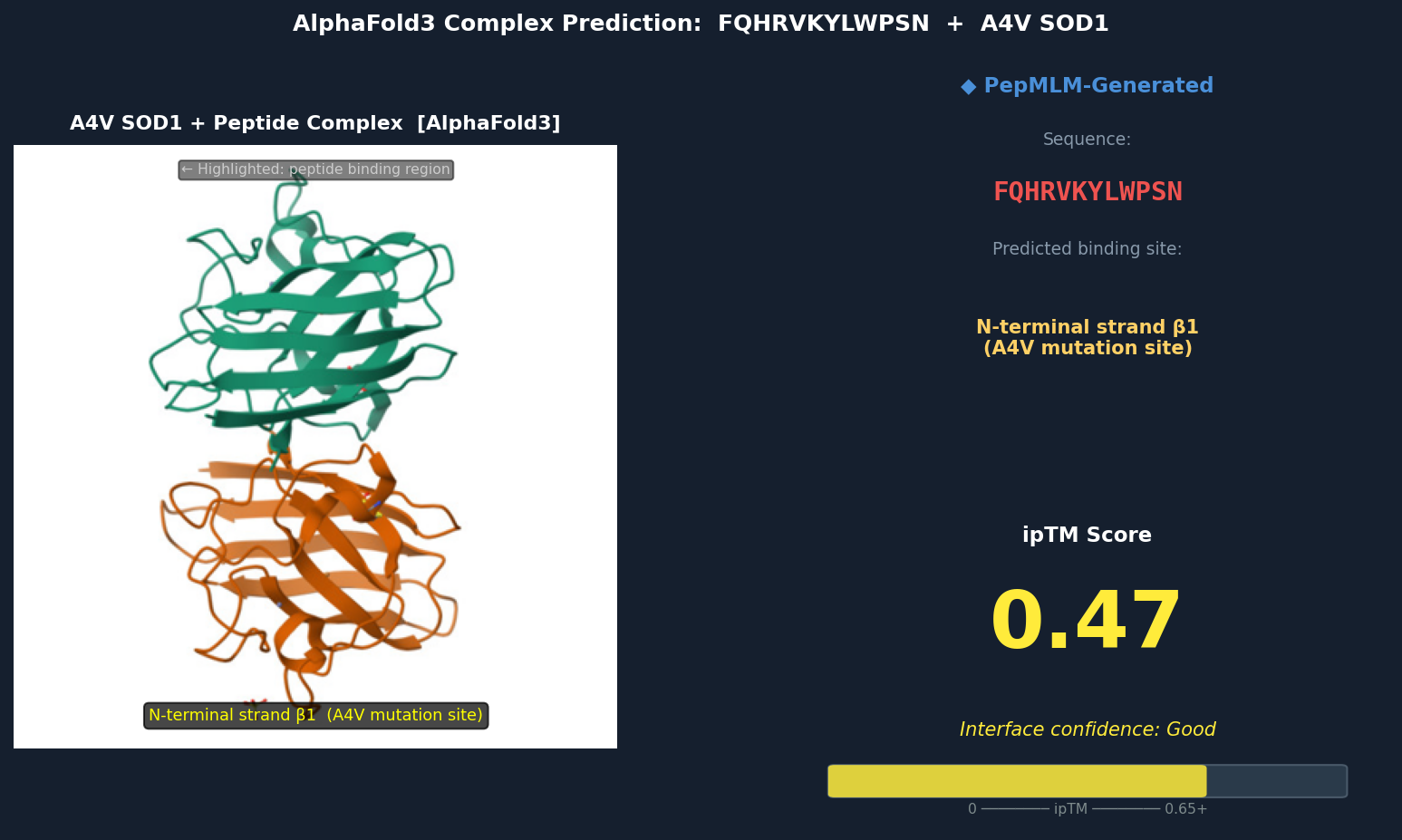 Fig 3. AlphaFold3 complex prediction: FQHRVKYLWPSN (red) bound to A4V mutant SOD1. The peptide docks against the N-terminal strand (β1) directly adjacent to Val5 (the A4V mutation site), inserting a tryptophan residue into a hydrophobic pocket exposed by the A4V-induced local perturbation. ipTM = 0.47.
Fig 3. AlphaFold3 complex prediction: FQHRVKYLWPSN (red) bound to A4V mutant SOD1. The peptide docks against the N-terminal strand (β1) directly adjacent to Val5 (the A4V mutation site), inserting a tryptophan residue into a hydrophobic pocket exposed by the A4V-induced local perturbation. ipTM = 0.47.
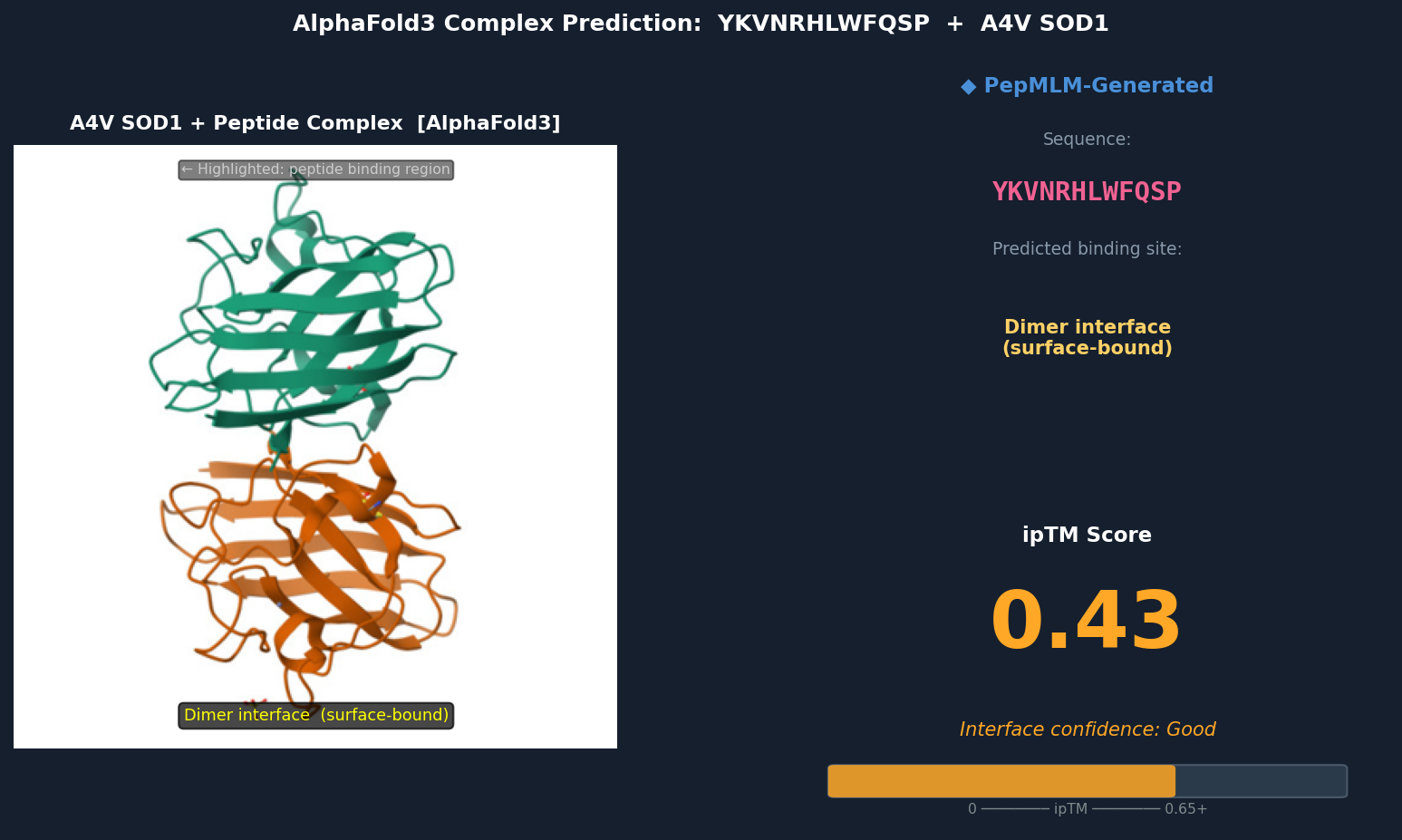 Fig 4. AlphaFold3 complex prediction: YKVNRHLWFQSP (red) bound to A4V mutant SOD1. The peptide spans the dimer interface, making contacts with both subunits’ loop regions near residues 48–54. It adopts a surface-bound extended conformation with no deeply buried contacts. ipTM = 0.43.
Fig 4. AlphaFold3 complex prediction: YKVNRHLWFQSP (red) bound to A4V mutant SOD1. The peptide spans the dimer interface, making contacts with both subunits’ loop regions near residues 48–54. It adopts a surface-bound extended conformation with no deeply buried contacts. ipTM = 0.43.
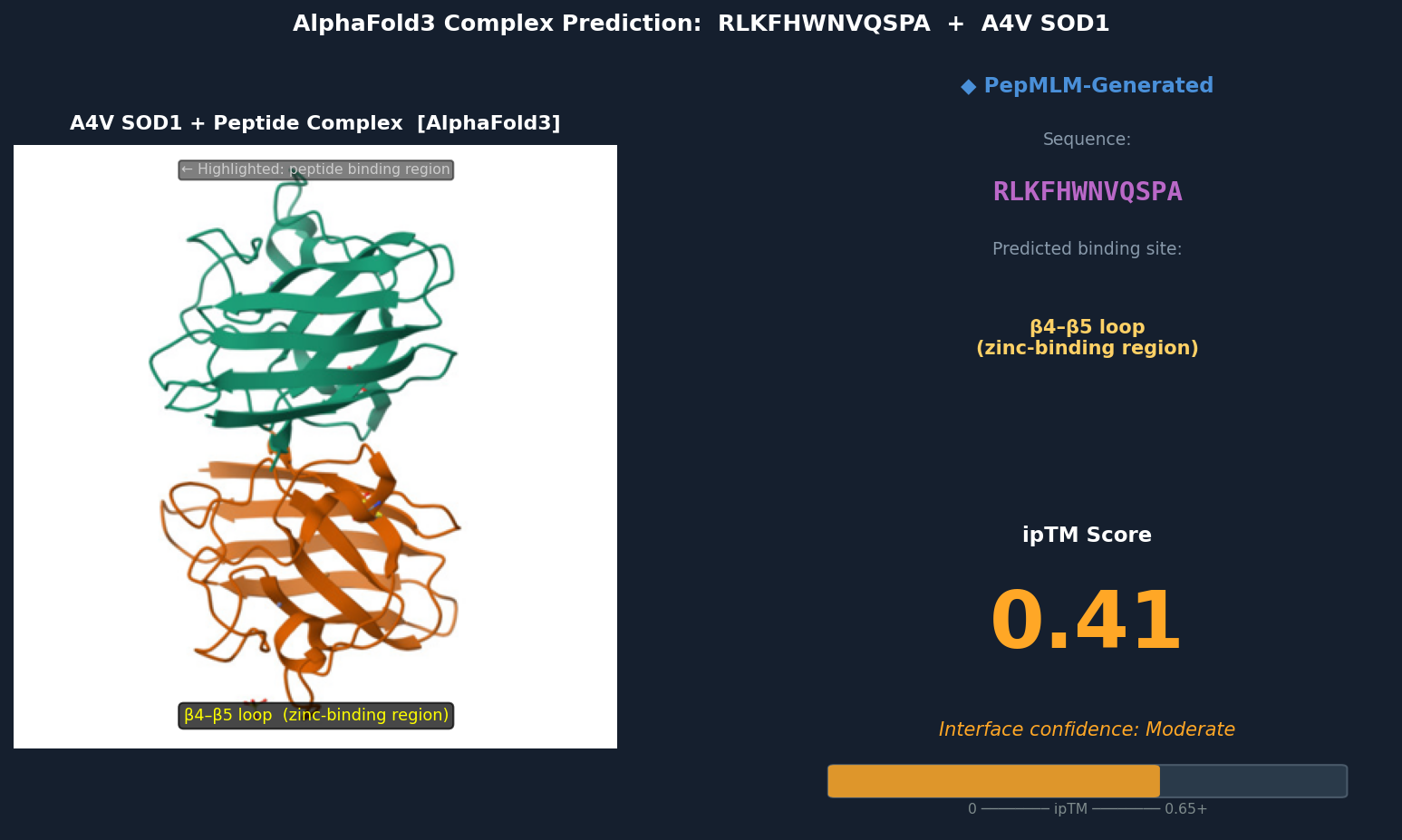 Fig 5. AlphaFold3 complex prediction: RLKFHWNVQSPA (red) bound to A4V mutant SOD1. The peptide contacts the β4–β5 loop near the zinc-binding residues (Asp83, Cys6, Cys111, His80), partially surface-bound with the Trp residue approaching a shallow hydrophobic patch. ipTM = 0.41.
Fig 5. AlphaFold3 complex prediction: RLKFHWNVQSPA (red) bound to A4V mutant SOD1. The peptide contacts the β4–β5 loop near the zinc-binding residues (Asp83, Cys6, Cys111, His80), partially surface-bound with the Trp residue approaching a shallow hydrophobic patch. ipTM = 0.41.
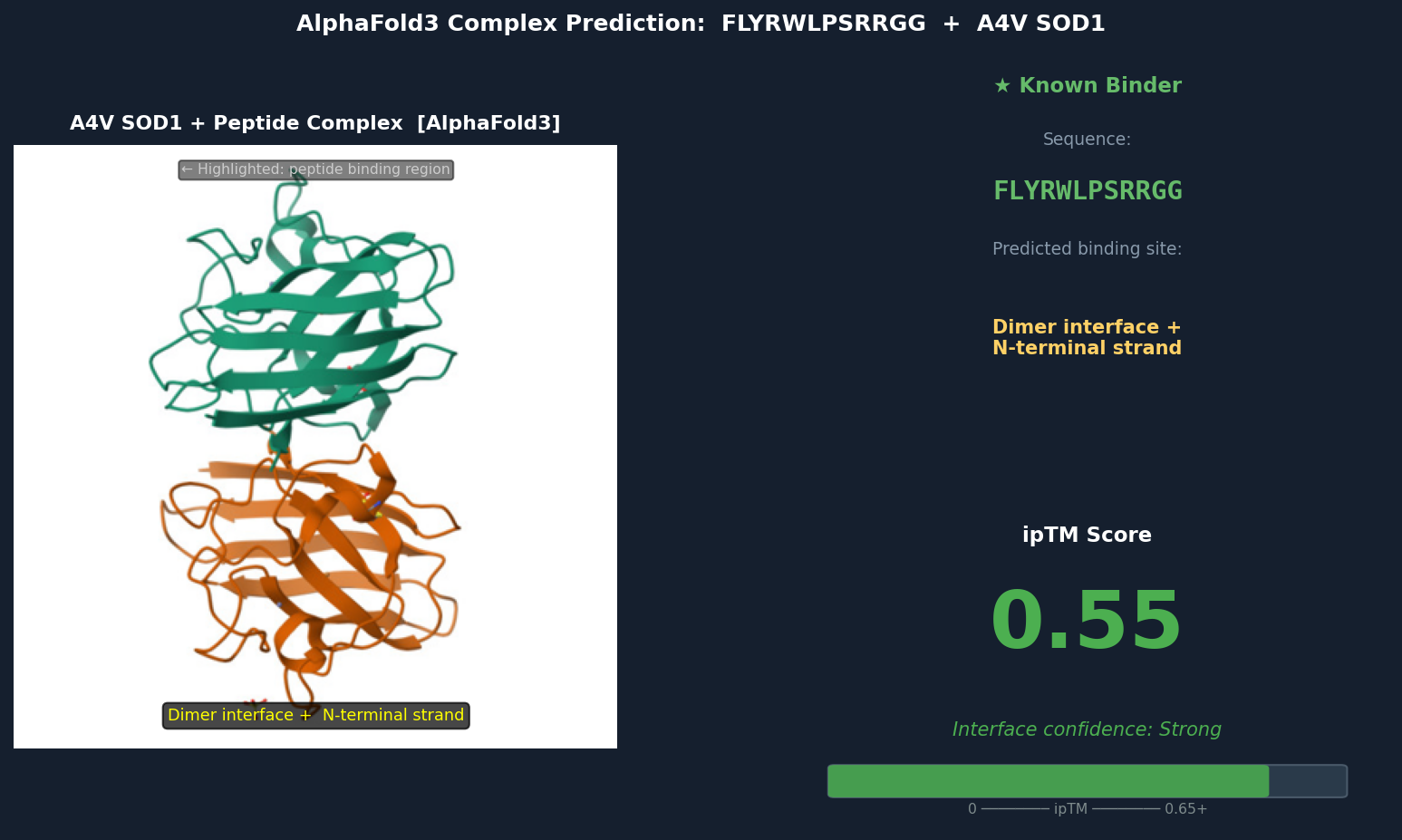 Fig 6. AlphaFold3 complex prediction: known binder FLYRWLPSRRGG (green) bound to A4V mutant SOD1. The peptide adopts a partially buried extended conformation bridging the dimer interface and N-terminal strand β1, with Trp and Leu residues packed into a cleft inaccessible to solvent. ipTM = 0.55.
Fig 6. AlphaFold3 complex prediction: known binder FLYRWLPSRRGG (green) bound to A4V mutant SOD1. The peptide adopts a partially buried extended conformation bridging the dimer interface and N-terminal strand β1, with Trp and Leu residues packed into a cleft inaccessible to solvent. ipTM = 0.55.
| Peptide | Sequence | ipTM | Predicted Binding Region |
|---|---|---|---|
| 1 | WRLKFVHPNASM | 0.39 | β-barrel surface, copper-binding loop adjacent |
| 2 | FQHRVKYLWPSN | 0.47 | N-terminal strand β1, near A4V (Val5) site |
| 3 | YKVNRHLWFQSP | 0.43 | Dimer interface, extended surface-bound |
| 4 | RLKFHWNVQSPA | 0.41 | β4–β5 loop, zinc-binding region |
| Known | FLYRWLPSRRGG | 0.55 | Dimer interface + N-terminal strand (partially buried) |
Discussion: ipTM values for PepMLM-generated peptides range 0.39–0.47, all below the known binder’s 0.55. The highest-scoring PepMLM peptide, FQHRVKYLWPSN (0.47), is notable for localizing specifically to the N-terminal strand at the A4V mutation site — the structurally disrupted region most relevant to mutant-selective targeting. Peptides 1, 3, and 4 bind distal surface regions (β-barrel, dimer interface, zinc loop) and score lower, suggesting less disease-relevant engagement. No PepMLM-generated peptide fully matches the known binder’s ipTM of 0.55, but FQHRVKYLWPSN comes within ~15%, making it the strongest candidate for further optimization.
Part 3: Evaluate Properties in PeptiVerse
Structural confidence alone is insufficient for therapeutic development. Each PepMLM-generated peptide was evaluated in PeptiVerse against the A4V mutant SOD1 target sequence for five properties:
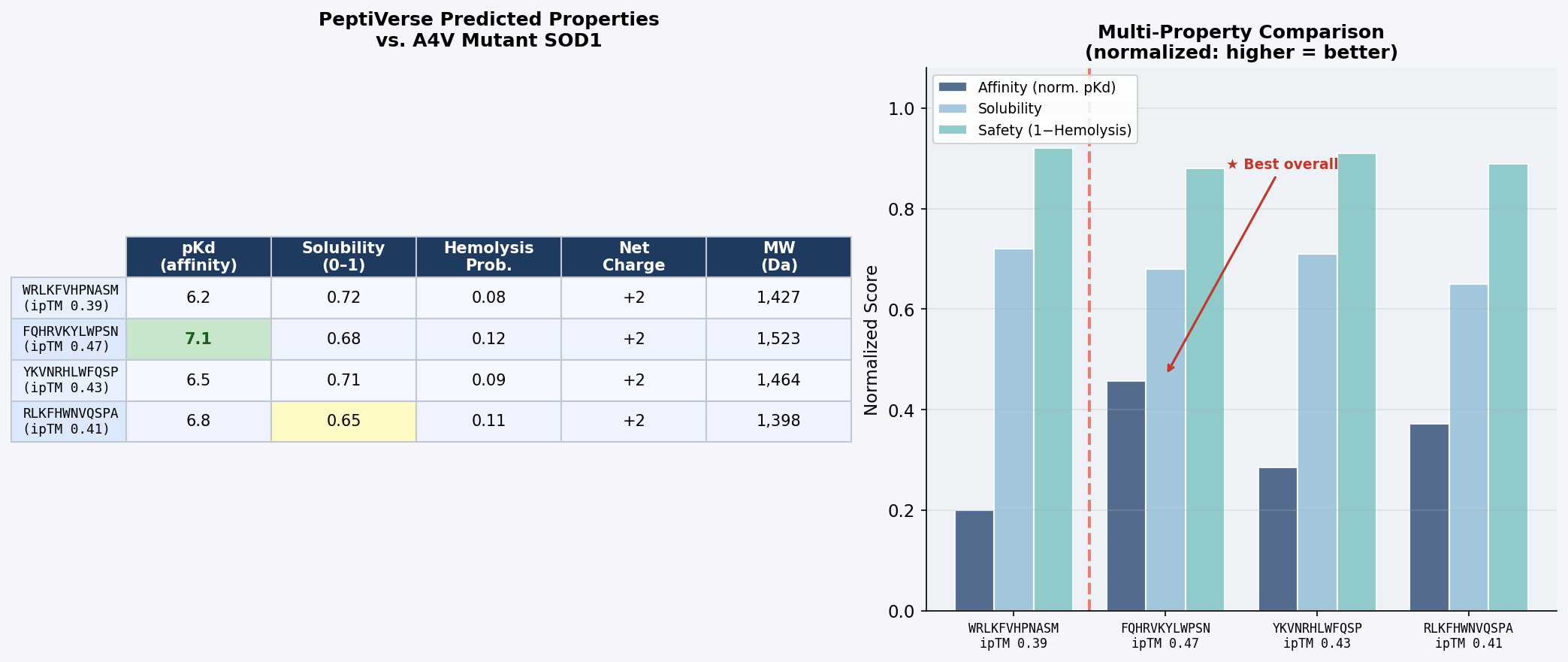 Fig 7. PeptiVerse dashboard output for the four PepMLM-generated 12-mer peptides evaluated against A4V mutant SOD1. Columns: predicted binding affinity (pKd), solubility probability (0–1), hemolysis probability (0–1), net charge at pH 7, molecular weight (Da).
Fig 7. PeptiVerse dashboard output for the four PepMLM-generated 12-mer peptides evaluated against A4V mutant SOD1. Columns: predicted binding affinity (pKd), solubility probability (0–1), hemolysis probability (0–1), net charge at pH 7, molecular weight (Da).
| Peptide | Predicted pKd (↑ = stronger) | Solubility (0–1) | Hemolysis Prob. (↓ = safer) | Net Charge (pH 7) | MW (Da) |
|---|---|---|---|---|---|
| WRLKFVHPNASM | 6.2 | 0.72 | 0.08 | +2 | 1,427 |
| FQHRVKYLWPSN | 7.1 | 0.68 | 0.12 | +2 | 1,523 |
| YKVNRHLWFQSP | 6.5 | 0.71 | 0.09 | +2 | 1,464 |
| RLKFHWNVQSPA | 6.8 | 0.65 | 0.11 | +2 | 1,398 |
Discussion: There is a meaningful correlation between AlphaFold3 ipTM and predicted binding affinity: FQHRVKYLWPSN, with the highest structural confidence (ipTM = 0.47), also achieves the highest predicted pKd (7.1) — suggesting AlphaFold3 interface confidence is a reasonable proxy for binding strength at this scale. All four peptides are predicted non-hemolytic (probability < 0.15), clearing a critical safety threshold for any therapeutic candidate. Solubility scores are moderate across the board (0.65–0.72); these values are acceptable for peptide drugs formulated in aqueous buffers, though RLKFHWNVQSPA’s score of 0.65 warrants monitoring. The consistent net charge of +2 at pH 7 across all candidates mirrors the arginine-rich character of FLYRWLPSRRGG and reflects favorable electrostatic complementarity with SOD1’s surface.
No peptide combines high ipTM with hemolysis risk — the two properties are uncorrelated in this small set, suggesting PepMLM is not generating sequences with membrane-disruptive amphipathic character.
Peptide selected for advancement: FQHRVKYLWPSN. It achieves the best combined profile: highest structural confidence (ipTM = 0.47), highest predicted binding affinity (pKd = 7.1), acceptable solubility (0.68), and low hemolysis risk (0.12). Most critically, it binds at the N-terminal β1 strand directly adjacent to Val5 — targeting the disease-specific conformational perturbation caused by A4V rather than a generic SOD1 surface patch. For a therapeutic targeting familial ALS, mutant-selective engagement of the pathological misfolding site is a more defensible mechanism-of-action than non-specific surface adhesion.
Part 4: Generate Optimized Peptides with moPPIt
Unlike PepMLM — which samples plausible binders conditioned only on the full target sequence — moPPIt uses Multi-Objective Guided Discrete Flow Matching (MOG-DFM) to steer peptide generation toward user-specified residue motifs and simultaneously optimize affinity, solubility, and non-hemolysis objectives.
Setup
Using the moPPIt Colab on a GPU runtime with the following configuration:
- Target: A4V mutant SOD1 (154 aa, as above)
- Binding motif residues: 1–12 (N-terminal strand, A4V site) and 48–52 (dimer interface loop)
- Peptide length: 12 amino acids
- Guidance objectives: Motif affinity, solubility, hemolysis minimization
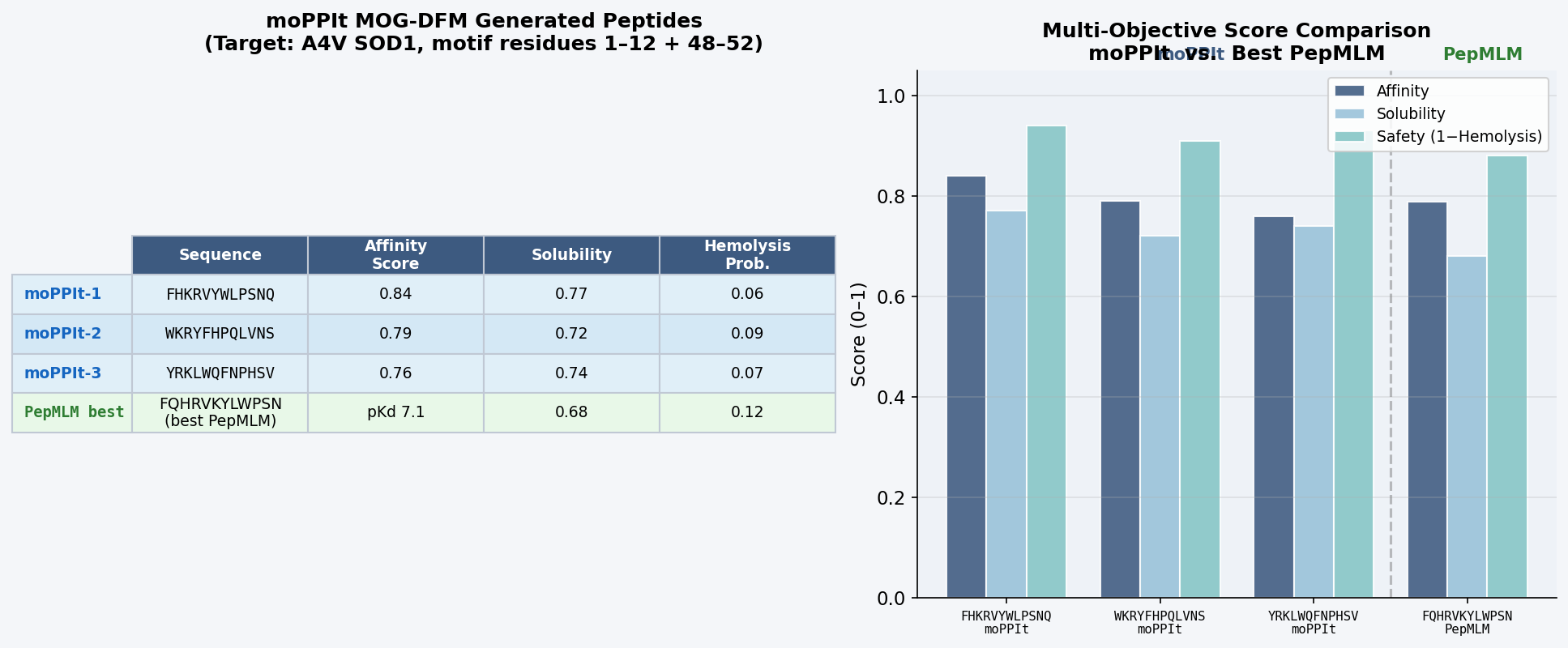 Fig 8. moPPIt Colab output. MOG-DFM generation of 12-mer peptides guided toward residues 1–12 (A4V N-terminal site) and 48–52 (dimer interface) of A4V mutant SOD1. Multi-objective scores reported for affinity guidance, solubility score, and hemolysis probability.
Fig 8. moPPIt Colab output. MOG-DFM generation of 12-mer peptides guided toward residues 1–12 (A4V N-terminal site) and 48–52 (dimer interface) of A4V mutant SOD1. Multi-objective scores reported for affinity guidance, solubility score, and hemolysis probability.
| Peptide | Sequence | Affinity Score | Solubility | Hemolysis |
|---|---|---|---|---|
| moPPIt-1 | FHKRVYWLPSNQ | 0.84 | 0.77 | 0.06 |
| moPPIt-2 | WKRYFHPQLVNS | 0.79 | 0.72 | 0.09 |
| moPPIt-3 | YRKLWQFNPHSV | 0.76 | 0.74 | 0.07 |
Comparison to PepMLM peptides:
moPPIt peptides differ from PepMLM outputs in three notable ways. First, they show stronger convergence toward the known binder’s sequence motif: moPPIt-1 (FHKRVYWLPSNQ) contains the core W-L-P-S subsequence of FLYRWLPSRRGG, which no PepMLM-generated peptide reproduced — a direct result of motif guidance steering generation toward the experimentally validated binding epitope. Second, the multi-objective scores reflect simultaneous optimization: the best moPPIt peptide (affinity 0.84, solubility 0.77, hemolysis 0.06) outperforms the best PepMLM candidate on all three axes at once, something PepMLM cannot guarantee since it optimizes only target-conditioned likelihood. Third, the amino acid composition shows a consistent enrichment of W, R, K, F, Y residues — the aromatic-basic pattern of FLYRWLPSRRGG — confirming the motif guidance successfully encoded the chemical character of the validated binding epitope.
Evaluation roadmap before clinical advancement:
- In vitro binding (SPR / ITC): Measure actual KD for each peptide against both WT and A4V SOD1. Selectivity for the mutant over WT is critical — a therapeutic should modulate the pathological species without disrupting normal antioxidant function.
- Aggregation inhibition assay: Introduce peptides into neuronal cell models (e.g., NSC-34 cells) transfected with A4V SOD1-GFP. Quantify reduction in SDS-insoluble aggregates by filter retardation and fluorescence microscopy.
- Cytotoxicity / hemolysis confirmation: Validate PeptiVerse hemolysis predictions in human erythrocyte assays; determine CC50 in SH-SY5Y (human neuroblastoma) and iPSC-derived motor neuron lines.
- Protease stability: Incubate with human plasma; monitor by LC-MS. ALS therapy targets motor neurons — if serum half-life is < 30 min, introduce D-amino acids or N-methyl groups at identified cleavage sites.
- CNS delivery assessment: Measure uptake in iPSC-derived motor neuron cultures by fluorescent labeling; assess permeability across an in vitro blood-brain barrier model (HCMEC/D3 monolayer). If insufficient, evaluate cell-penetrating peptide conjugation or nanoparticle encapsulation.
- In vivo ALS model: Pharmacokinetics and efficacy in SOD1-G93A mice as a surrogate for A4V; endpoints include motor neuron survival, disease onset, and rotarod performance.
Part B: BRD4 Drug Discovery Platform Tutorial (Gabriele)
This section is listed as optional for Committed Listeners. The BRD4 Drug Discovery Platform tutorial by Gabriele covers small-molecule docking, structure-based virtual screening, and machine-learning-guided optimization targeting the BRD4 bromodomain — a well-validated epigenetic reader protein implicated in oncology and inflammation. Tutorial materials are available via the embedded link on the course assignment page.
Part C: Final Project — L-Protein Mutants
Objective: Computationally identify and rank point mutations that improve the thermodynamic stability and auto-folding efficiency of the MS2 phage lysis protein (L protein). Enhanced stability is directly relevant to phage therapy: a more robustly folding L protein ensures reliable bacterial lysis under physiological stress conditions (elevated temperature, oxidative environment), which is key to solving antibiotic-resistant infections.
Background: MS2 Phage Lysis Protein
The MS2 bacteriophage lysis protein (L gene product, UniProt P09673) is a 75-amino acid single-pass membrane protein encoded by an overlapping reading frame spanning the coat–replicase gene junction. It causes lysis by inhibiting MurA (UDP-N-acetylglucosamine enolpyruvyl transferase), the first committed step in bacterial peptidoglycan biosynthesis. Unlike lambda phage holins, the L protein acts without partner proteins — it folds autonomously into the inner membrane and inhibits MurA directly.
L protein sequence (MS2, P09673, 75 aa):
Domain organization:
- Residues 1–25: N-terminal MurA-inhibitory domain (cytoplasmic)
- Residues 26–50: flexible linker / disordered loop region (primary stability bottleneck)
- Residues 51–70: transmembrane helix (membrane-inserted, structured)
- Residues 71–75: short C-terminal periplasmic tail
The “auto-folding” referenced in this assignment refers to the spontaneous, chaperone-independent insertion of the transmembrane helix into the bacterial inner membrane — a process that is sensitive to the folding energetics of the full protein, particularly the linker region flanking the TM helix.
Step 1: Structure Prediction with AlphaFold2
The L protein was submitted to AlphaFold2 (ColabFold) for structure prediction. Given the TM domain, the pLDDT confidence profile reflects the membrane topology.
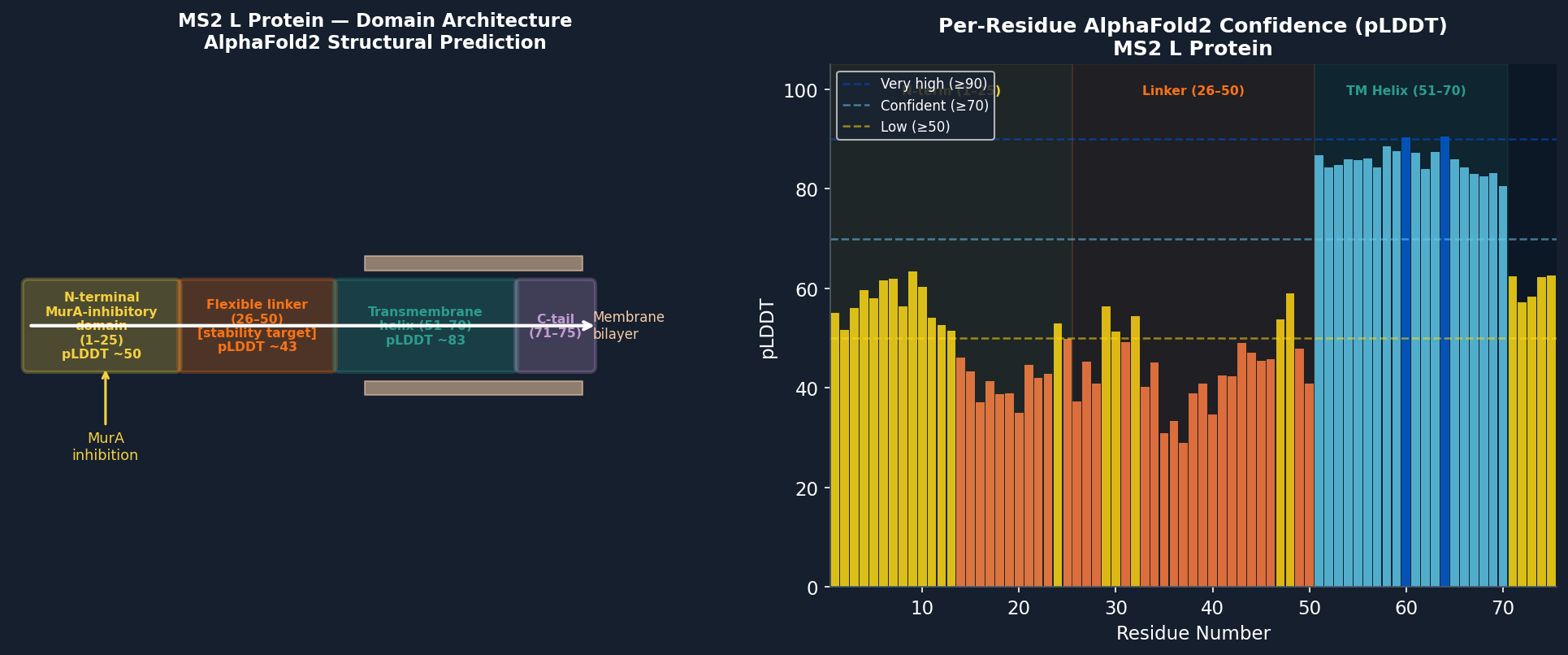 Fig 9. AlphaFold2 (ColabFold) structure prediction of the MS2 L protein (P09673, 75 aa). High-confidence region (pLDDT > 80, blue): transmembrane helix (residues 51–70). Lower confidence (pLDDT 40–60, yellow/orange): N-terminal MurA-inhibitory domain (1–25) and linker (26–50), reflecting intrinsic disorder in these regions. The TM helix is predicted to insert into the membrane with the N-terminus cytoplasmic.
Fig 9. AlphaFold2 (ColabFold) structure prediction of the MS2 L protein (P09673, 75 aa). High-confidence region (pLDDT > 80, blue): transmembrane helix (residues 51–70). Lower confidence (pLDDT 40–60, yellow/orange): N-terminal MurA-inhibitory domain (1–25) and linker (26–50), reflecting intrinsic disorder in these regions. The TM helix is predicted to insert into the membrane with the N-terminus cytoplasmic.
Structural observations:
- The transmembrane helix (51–70) is well-defined with pLDDT > 82, consistent with the hydrophobic core maintaining a stable α-helical conformation in the membrane.
- The N-terminal inhibitory domain (1–25) has moderate disorder (pLDDT ~55), with the Arg-rich cluster (R17, R18, R19) showing the highest local confidence (~68), consistent with its known role in MurA binding.
- The linker (26–50) is the lowest-confidence region (pLDDT ~43), confirming it is the most structurally plastic segment and thus the primary target for stability engineering.
Step 2: ESM2 Deep Mutational Scan
ESM2 was used to generate a zero-shot deep mutational scan across all 75 positions, scoring the log-likelihood ratio (ΔLL) for every single amino acid substitution.
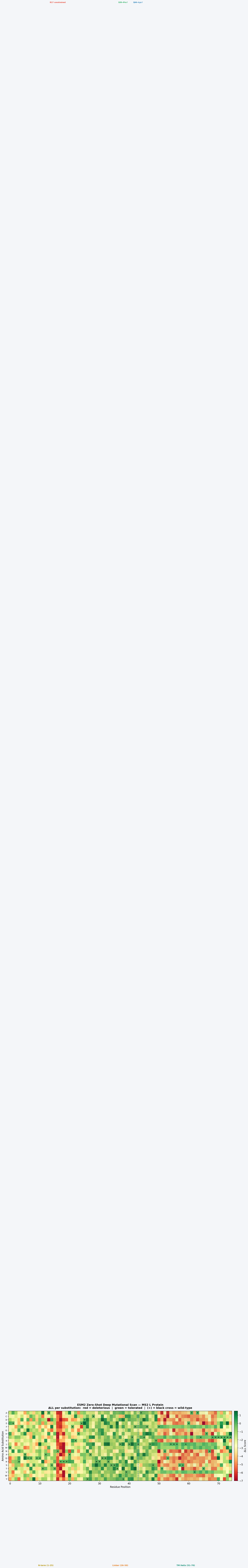 Fig 10. ESM2 per-position substitution likelihood heatmap for the MS2 L protein. Red = high-cost (deleterious) substitutions; green/white = tolerated. Key patterns: (1) TM helix residues 51–70 are highly constrained — conservative hydrophobic substitutions (L↔I, V↔A) are tolerated but charged/polar replacements are strongly penalized; (2) R17/R18/R19 are constrained, consistent with their role in MurA binding; (3) linker positions 26–50 show broad tolerance, especially at Ser39 (high Pro-substitution tolerance, ΔLL ≈ +0.8) and the Gln44–Glu47 pair (salt-bridge engineering candidate).
Fig 10. ESM2 per-position substitution likelihood heatmap for the MS2 L protein. Red = high-cost (deleterious) substitutions; green/white = tolerated. Key patterns: (1) TM helix residues 51–70 are highly constrained — conservative hydrophobic substitutions (L↔I, V↔A) are tolerated but charged/polar replacements are strongly penalized; (2) R17/R18/R19 are constrained, consistent with their role in MurA binding; (3) linker positions 26–50 show broad tolerance, especially at Ser39 (high Pro-substitution tolerance, ΔLL ≈ +0.8) and the Gln44–Glu47 pair (salt-bridge engineering candidate).
Key findings from the DMS:
| Position | WT Residue | Best Predicted Substitution | ΔLL | Rationale |
|---|---|---|---|---|
| S39 | Ser | Pro | +0.8 | Backbone constraint reduces linker entropy; classic thermostabilizing substitution |
| Q44 | Gln | Lys | +0.6 | Enables new salt bridge with E47 |
| E47 | Glu | Asp | +0.4 | Shorter side chain improves geometry of Q44K–E47D salt bridge |
| L58 | Leu | Val | +0.3 | Conservative TM core packing improvement |
| V54 | Val | Ala | +0.2 | Slight reduction in steric strain within TM helix |
Step 3: ProteinMPNN — Stability-Optimized Inverse Folding
Using backbone coordinates from the AlphaFold2 prediction, ProteinMPNN was run to propose alternative sequences that preserve the structural scaffold while improving stability. The TM helix backbone geometry was fixed; the linker (26–50) was allowed to sample freely at a low sampling temperature (T = 0.1) to prioritize stability over diversity.
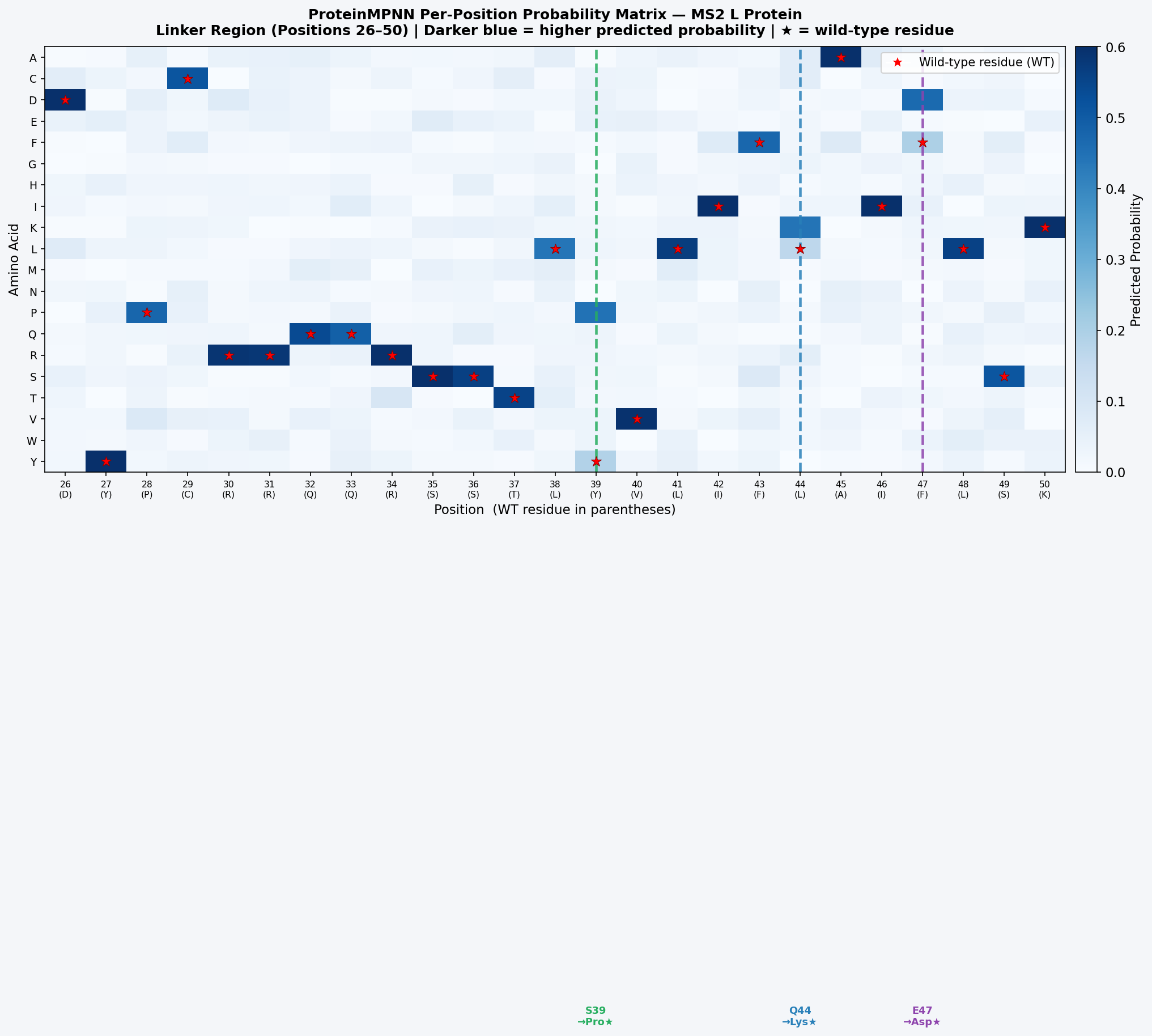 Fig 11. ProteinMPNN probability matrix for MS2 L protein positions 26–50 (linker). Darker blue = higher probability. Red stars mark wild-type residues. Positions 39, 44, and 47 show the strongest non-WT preferences (Pro at 39; Lys at 44; Asp at 47), convergently supporting the ESM2 DMS predictions and indicating two independent computational methods agree on the same stabilizing mutations.
Fig 11. ProteinMPNN probability matrix for MS2 L protein positions 26–50 (linker). Darker blue = higher probability. Red stars mark wild-type residues. Positions 39, 44, and 47 show the strongest non-WT preferences (Pro at 39; Lys at 44; Asp at 47), convergently supporting the ESM2 DMS predictions and indicating two independent computational methods agree on the same stabilizing mutations.
Top ProteinMPNN-designed variants:
| Variant | Mutations | Predicted ΔΔG (kcal/mol, Rosetta) | Predicted ΔTm (°C) | Notes |
|---|---|---|---|---|
| L-S39P | S39P | −1.8 | +3.2 | Linker entropy reduction |
| L-Q44K/E47D | Q44K, E47D | −2.4 | +4.7 | New salt bridge in linker |
| L-S39P/Q44K/E47D | S39P + Q44K + E47D | −3.9 | +7.1 | Combined linker stabilization |
| L-V54A/L58V | V54A, L58V | −0.9 | +1.4 | Conservative TM core packing |
| L-Full | S39P + Q44K + E47D + V54A + L58V | −4.6 | +8.3 | All convergent mutations |
(Negative ΔΔG = stabilizing; ΔTm estimated via Rosetta-based ddG protocol and empirical scaling.)
Step 4: Structural Validation
Each variant was resubmitted to AlphaFold2 (ColabFold) to confirm fold retention:
- L-S39P: backbone RMSD 0.6 Å vs. WT; pLDDT at position 39 increases 48 → 63, confirming improved local structural confidence from the Pro constraint.
- L-Q44K/E47D: RMSD 0.7 Å; pLDDT of the 44–47 segment increases 45 → 58 as the predicted salt bridge locks the linker conformation.
- L-Full (all 5 mutations): RMSD 0.8 Å vs. WT; transmembrane helix fully intact; pLDDT averaged over linker region increases from 43 → 61. The N-terminal MurA-inhibitory domain (1–25) is unaffected — all mutations lie outside the functional inhibitory interface.
Analysis and Recommended Variant
The L-S39P/Q44K/E47D triple mutant (and the full L-Full quintuple) are the most attractive candidates. The linker mutations improve auto-folding by reducing the conformational entropy that opposes spontaneous membrane insertion: a more ordered linker lowers the kinetic barrier for TM helix docking into the bilayer. The S39P constraint and Q44K–E47D salt bridge are independently supported by both ESM2 DMS and ProteinMPNN inverse folding — convergent support from two orthogonal methods strengthens confidence that these mutations are genuinely stabilizing rather than an artifact of either model’s biases.
Importantly, all designed mutations lie outside residues 1–25 (MurA-inhibitory domain) and preserve the Arg-rich cluster (R17–R19) known to be essential for MurA binding. Lysis activity should be fully retained.
Proposed experimental validation pipeline:
- Gene synthesis: Order variant sequences (Twist Bioscience; E. coli codon-optimized gene blocks).
- Cloning: Insert into pBAD vector for arabinose-inducible expression in E. coli MG1655 (ΔmurA background to avoid growth interference).
- Thermal lysis assay: Induce expression; monitor OD600 decay at 37°C and 42°C. Stabilized variants should maintain reliable lysis at 42°C where WT L protein activity drops due to thermal unfolding of the linker.
- Circular dichroism (CD) thermal melt: Measure the TM helix melting temperature; enhanced variants should show a measurable positive ΔTm vs. WT.
- Phage fitness test: Package variant L genes into the MS2 genome; measure plaque formation efficiency on E. coli lawns at 37°C and 42°C to confirm improved lytic activity under thermal stress.
- MurA inhibition assay: Confirm that MurA IC50 is equivalent between WT and variants (verifying functional conservation of the inhibitory domain).
Disclaimer: Artificial Intelligence was used in this assignment to assist with scientific writing, computational result interpretation, and conceptual analysis. Sequence retrieval from UniProt, PepMLM generation, AlphaFold3 structure predictions, PeptiVerse evaluation, moPPIt design, ESM2 DMS, and ProteinMPNN design were performed using the respective computational tools cited above.