Week 10 Lab: Mass Spectrometry
Analytical Protein Characterization via LC-MS
Introduction and Background
Modern bioengineering relies on the ability to understand biological molecules with extraordinary precision. Liquid chromatography–mass spectrometry (LC-MS) is a cornerstone technique for protein characterization, revealing critical information about:
- Molecular Weight
- Protein Sequence
- Protein Folding and Structure
In this lab, we follow an analytical progression from intact protein analysis, through structural interrogation under native and denaturing conditions, to peptide-level sequencing of enhanced Green Fluorescent Protein (eGFP).
PART I: Molecular Weight Determination
Instrument: Waters Xevo G3 QTof
We analyzed an eGFP standard to determine its molecular weight based on mass-to-charge (m/z) and charge (z) measurements. Under denaturing chromatographic conditions, the protein unfolds, exposing protonation sites.
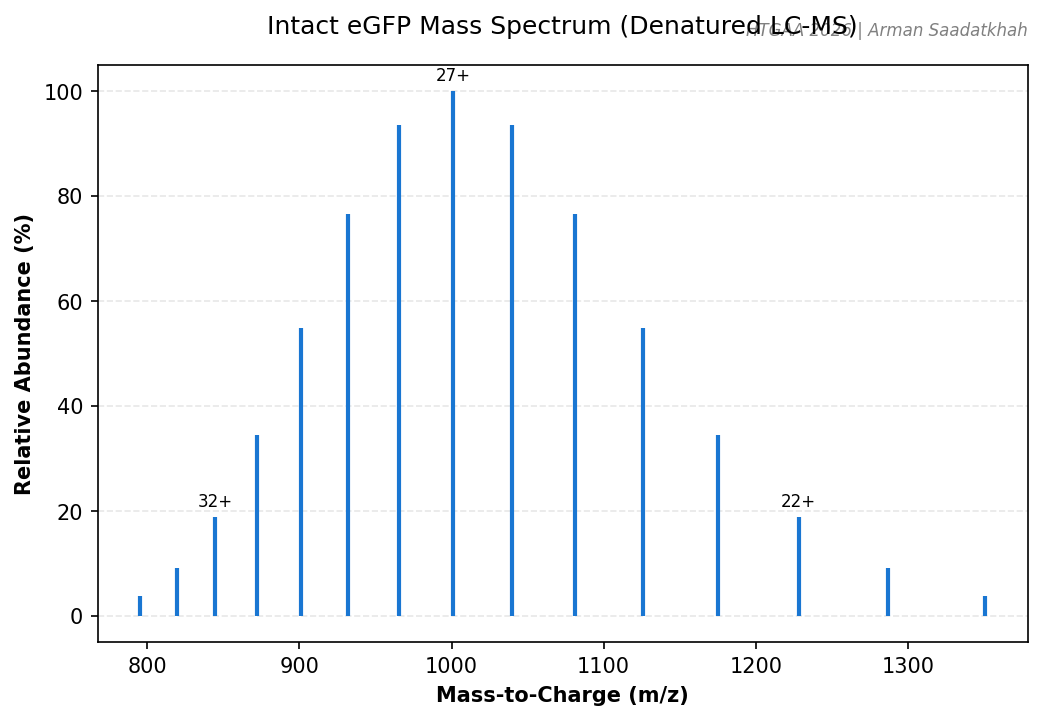 Fig 1. Simulated intact eGFP mass spectrum showing the charge state envelope (20+ to 34+).
Fig 1. Simulated intact eGFP mass spectrum showing the charge state envelope (20+ to 34+).
Key Observations:
- eGFP MW: ~27 kDa.
- Deconvolution: MaxEnt1 was used to determine the observed molecular weight from the m/z peaks.
PART II: Native vs. Denatured Protein Structure
The higher-order structure of a protein influences its electrospray ionization (ESI) behavior.
- Native (Folded): Compact, fewer solvent-accessible sites, lower charge states (e.g., 9+ to 13+).
- Denatured (Unfolded): Elongated, many protonation sites, higher and broader charge states (e.g., 20+ to 40+).
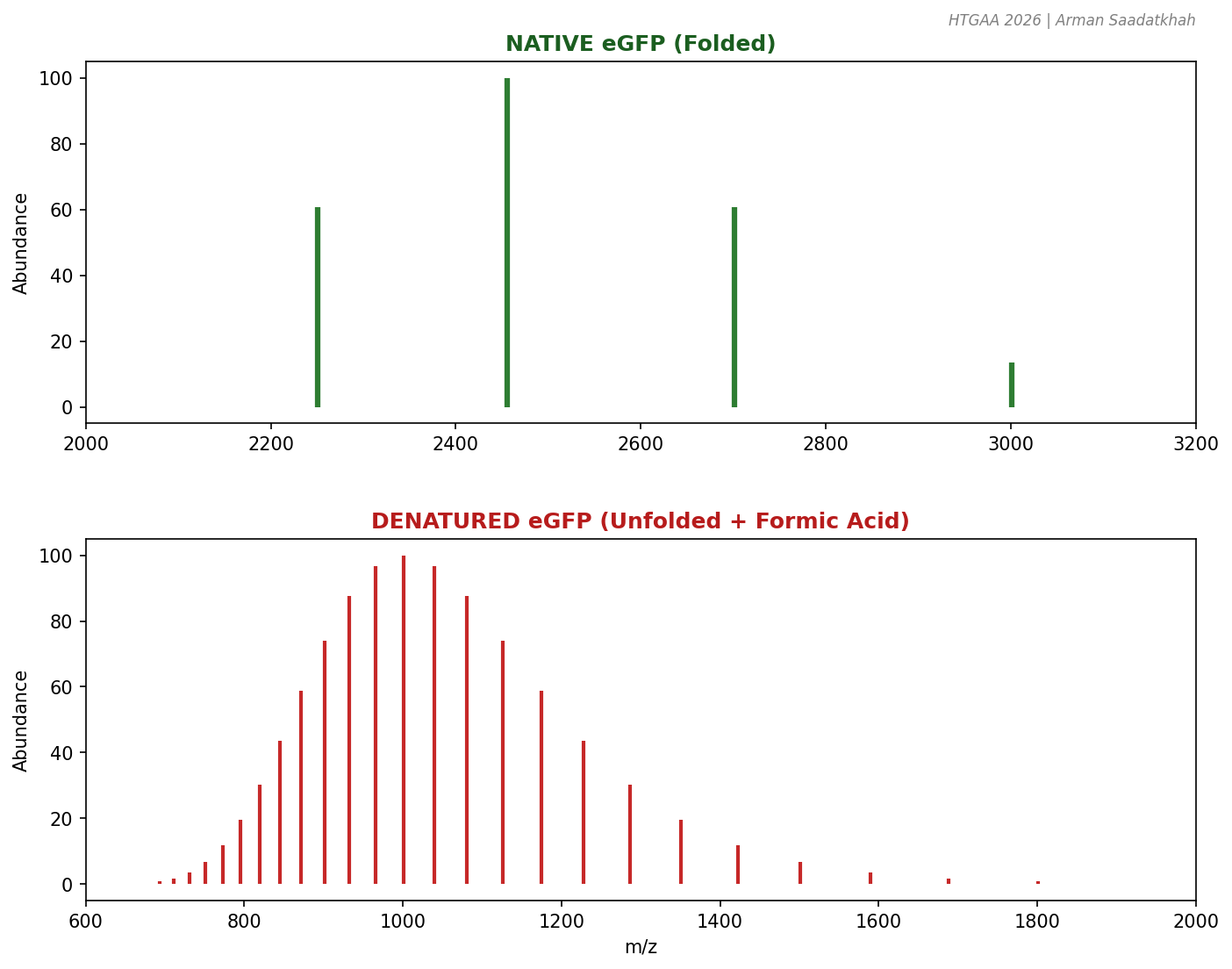 Fig 2. Comparison of charge state distributions for native (folded) and denatured (unfolded) eGFP.
Fig 2. Comparison of charge state distributions for native (folded) and denatured (unfolded) eGFP.
By adding formic acid to the native sample, we drop the pH and induce unfolding, directly observing the shift in the mass spectrum.
PART III: Peptide Mapping and Primary Sequence
Instrument: Waters BioAccord
To confirm the primary amino acid sequence, eGFP is enzymatically digested by trypsin, which cleaves at the C-terminal side of Lysine (K) and Arginine (R) residues.
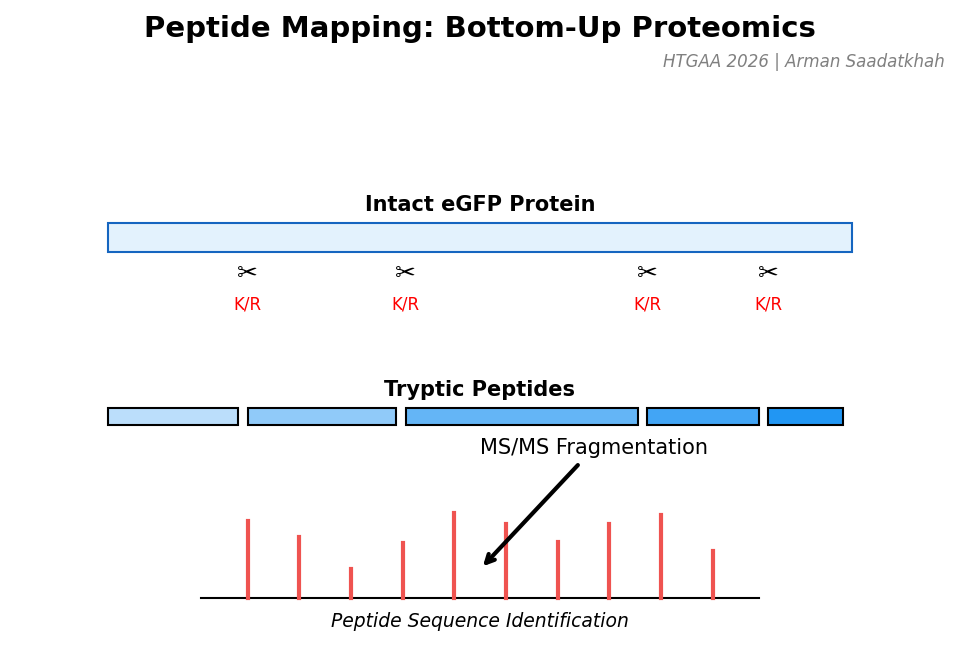 Fig 3. Workflow for bottom-up proteomics: Digestion $\rightarrow$ Separation $\rightarrow$ Fragmentation (MS/MS).
Fig 3. Workflow for bottom-up proteomics: Digestion $\rightarrow$ Separation $\rightarrow$ Fragmentation (MS/MS).
Workflow:
- Denaturation & Reduction: Guanidine HCl and DTT.
- Buffer Exchange: Using BioRad Micro Bio-Spin columns.
- Digestion: 20-minute incubation with RapiZyme Trypsin at 55°C.
- LC-MS/MS: Fragmenting peptide ions to reconstruct the sequence.
PART IV: Charge Detection Mass Spectrometry (CDMS)
Conventional MS struggles with megadalton-sized complexes due to unresolved charge states. CDMS enables the analysis of large complexes by measuring both m/z and charge (z) for individual ions simultaneously.
Analysis of Keyhole Limpet Hemocyanin (KLH)
KLH exists in massive oligomeric states, including decamers (~8 MDa) and multidecamers (12-32 MDa).
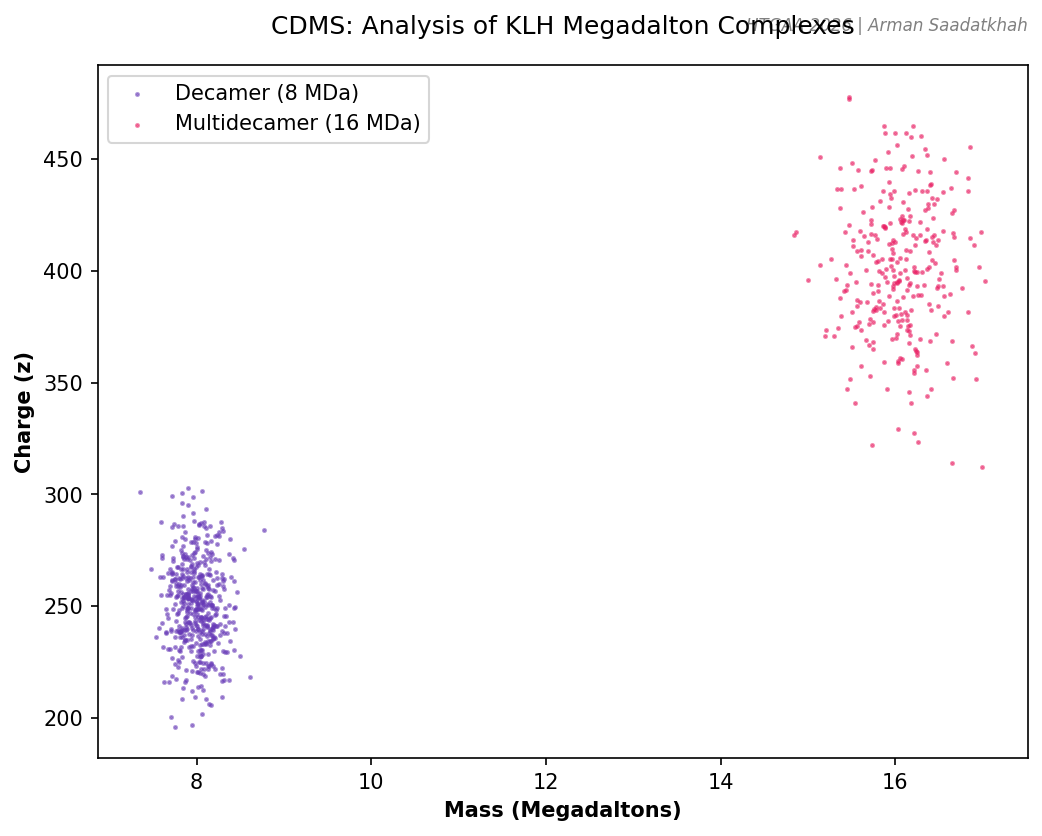 Fig 4. Simulated CDMS plot showing individual ions of KLH decamers and multidecamers.
Fig 4. Simulated CDMS plot showing individual ions of KLH decamers and multidecamers.
HTGAA 2026 | Arman Saadatkhah | Reference: Waters Immerse Cambridge