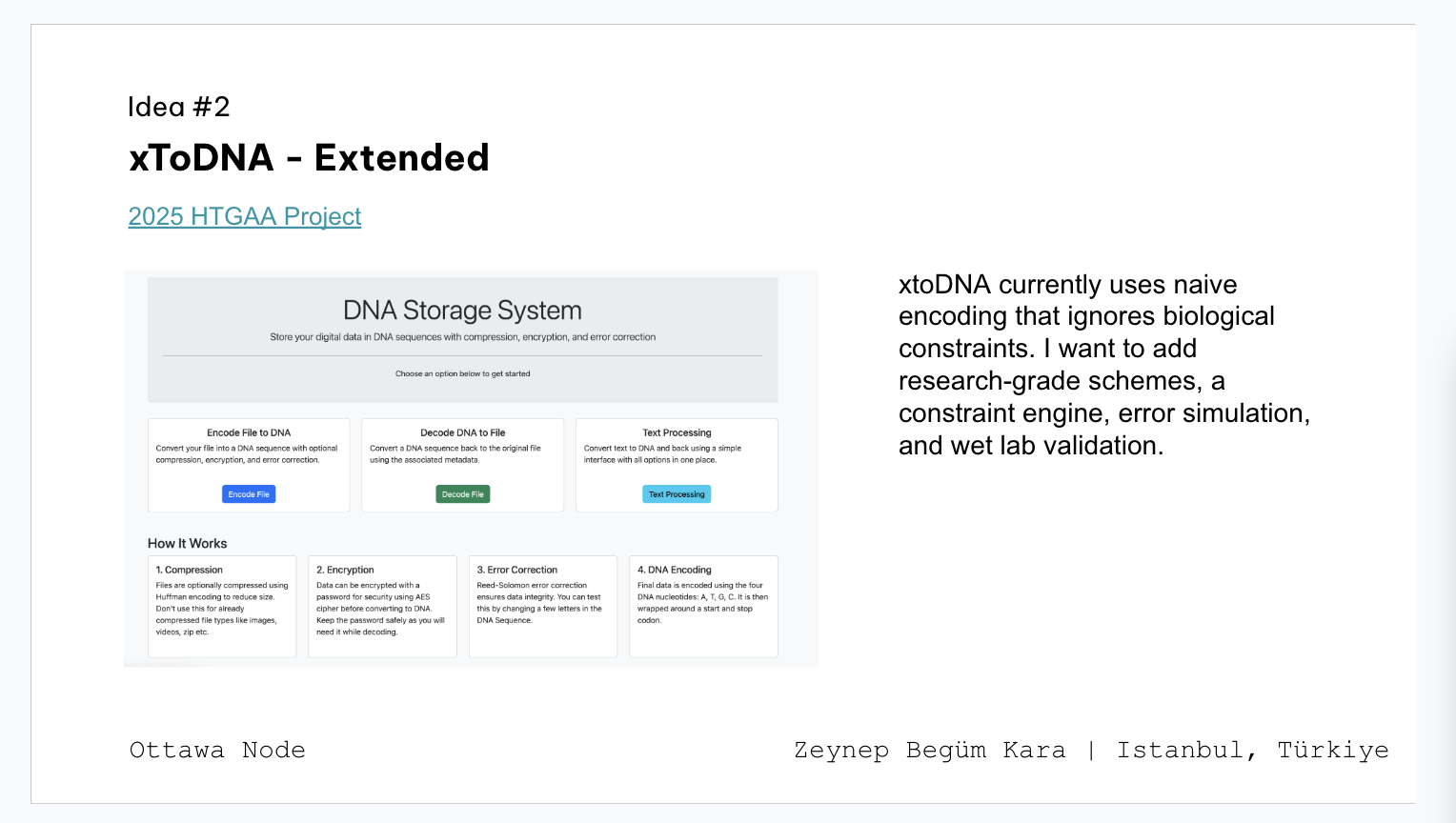
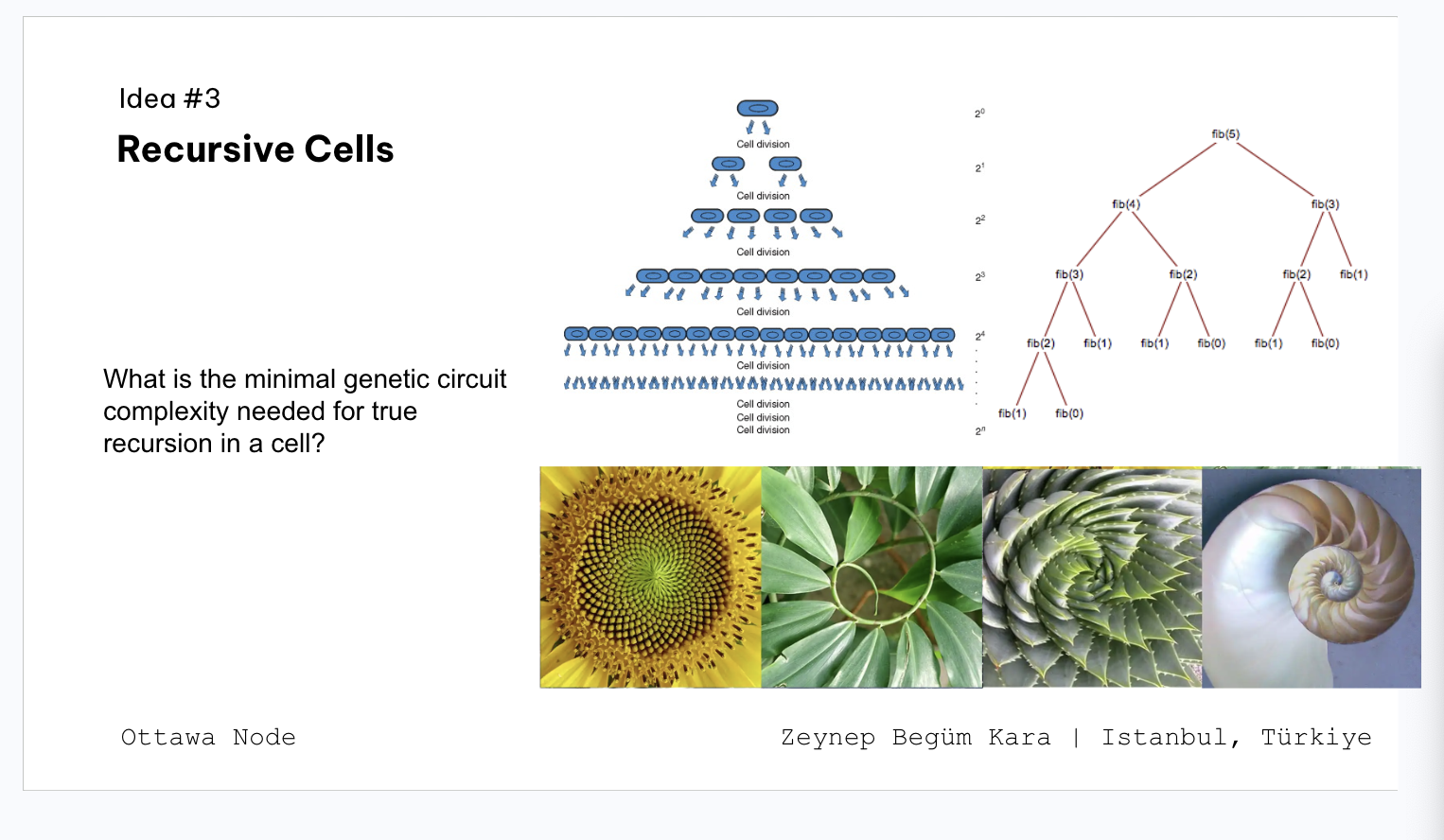
Class assignment 1. First, describe a biological engineering application or tool you want to develop and why. As a CS/AI master’s student, I find it exciting that I can use AI protein design tools like AlphaFold to work on something that actually matters.
Part 1: Benchling & In-silico Gel Art For this exercise, the full genome of Bacteriophage Lambda (GenBank accession J02459.1, 48,502 bp) was imported into Benchling from NCBI. A virtual restriction enzyme digestion was performed using seven enzymes: EcoRI, HindIII, BamHI, KpnI, EcoRV, SacI, and SalI. Each enzyme was applied individually to identify its recognition sites across the Lambda genome. The digest results were visualized using Benchling’s simulated gel electrophoresis tool. To get a nice visual, I tried different ladders and different lane orderings. The final output consisting of for each enzyme’s fragment pattern is below.
Part 1: Python Script Opentron Artwork Opentrons Colab for Source Code
My Jelly Smiley Design
Part 2: Post-Lab Questions Question 1: Find and describe a published paper that utilizes the Opentrons or an automation tool to achieve novel biological applications.
Part A. Conceptual Questions How many molecules of amino acids do you take with a piece of 500 grams of meat? (on average an amino acid is ~100 Daltons)
Meat is roughly 20% protein by mass, so 500 g of meat contains about 100 g of protein. Since one Dalton equals 1 g/mol, an average amino acid at 100 Da means 100 g/mol. That gives 100 g ÷ 100 g/mol = 1 mole of amino acid residues, or about 6 × 10²³ amino acid molecules. So eating a steak hands you roughly Avogadro’s number of amino acids, a staggering count that just means “about a mole.”
Part 1: Generate Binders with PepMLM I retrieved the human SOD1 sequence from UniProt (P00441) in FASTA format.
Original canonical sequence:
>sp|P00441|SODC_HUMAN Superoxide dismutase [Cu-Zn] OS=Homo sapiens OX=9606 GN=SOD1 PE=1 SV=2 MATKAVCVLKGDGPVQGIINFEQKESNGPVKVWGSIKGLTEGLHGFHVHEFGDNTAGCTS AGPHFNPLSRKHGGPKDEERHVGDLGNVTADKDGVADVSIEDSVISLSGDHCIIGRTLVV HEKADDLGKGGNEESTKTGNAGSRLACGVIGIAQ I introduced the A4V mutation by changing Alanine (A) to Valine (V) at position 4 of the mature protein sequence (index 4, 0-based).
What are some components in the Phusion High-Fidelity PCR Master Mix and what is their purpose? Phusion HF PCR Master Mix is a ready-to-use 2X solution that contains everything needed for PCR except the template, primers, and water.
The most important component is Phusion DNA Polymerase, which is the enzyme that copies DNA during the reaction. The standard alternative is Taq polymerase, a widely used enzyme isolated from a heat-resistant bacterium called Thermus aquaticus. Taq survives the high temperatures of PCR but has no proofreading ability, so it cannot correct mistakes it makes while copying, resulting in a relatively high error rate. Phusion addresses this by including a proofreading domain that catches and fixes errors as they occur, making it about 50 times more accurate than Taq. This accuracy is important in a mutagenesis experiment where only the specific, intended mutations should be introduced and any unintended errors would compromise the result.
Assignment Part 1: Intracellular Artificial Neural Networks (IANNs) 1. What advantages do IANNs have over traditional genetic circuits, whose input/output behaviors are Boolean functions? Boolean genetic circuits treat signals like 0 or 1, meaning every input must pass a hard on/off threshold. However, this can lose useful information. This can be limiting because real biological signals are usually not binary. Inside cells, signals often exist as gradual changes such as in concentration level. IANNs are useful because they keep more of that analog information since they are not only asking “is this signal present or absent?”.
General Homework Questions 1. Explain the main advantages of cell-free protein synthesis over traditional in vivo methods, specifically in terms of flexibility and control over experimental variables. Name at least two cases where cell-free expression is more beneficial than cell production. Cell-free protein synthesis (CFPS) skips the cell membrane barrier, so the user has direct access to the reaction mixture. This means parameters like pH, redox state, magnesium and potassium concentrations, temperature, and amino acid pools can all be tuned freely without worrying about cell viability. There is also no need for cloning, transformation, or growing cultures, so the time from DNA template to protein is hours instead of days.
Homework: Waters Part I — Molecular Weight An eGFP standard was analyzed on a Waters Xevo G3 QTof MS system to determine the molecular weight of intact eGFP and observe its charge state distribution in the native and denatured (unfolded) states.
What is the calculated molecular weight? eGFP Sequence:
Part A: The 1,536 Pixel Artwork Canvas | Collective Artwork Unfortunately, I missed the deadline, I guess I have to become a TA next year (hurray)! ¯(ツ)/¯
I thought the sense of community behind the project was really well designed, and I appericated the idea of many people contributing small pieces to one shared bioartwork.
This also made me think about my own project on protein steganography. It could be interesting to create a message-encoding project where people enter their names, messages, or short memories, and we encode them into DNA or protein sequences. Although this would not be as immediately visual as the pixel canvas, it could still be very aligned with HTGAA. One possible version could even involve producing the encoded DNA in bacteria and sharing it with committed listeners. Adrian from the Ottawa node also suggested that it might be beautiful to encode the information into a plant seed or something similar—a living memorial for HTGAA. :')
Subsections of Homework
Week 1 HW: Principles and Practices
Class assignment
1. First, describe a biological engineering application or tool you want to develop and why.
As a CS/AI master’s student, I find it exciting that I can use AI protein design tools like AlphaFold to work on something that actually matters.
A recent study by Trudler et al. (2024) at Scripps Research used patient-derived brain organoids (“mini-brains”) to show that mutations in the MEF2C gene — responsible for a severe form of ASD — disrupt the expression of specific microRNAs (miR-9, miR-124, miR-128). These miRNAs normally guide developing brain cells to become the right type of neuron; when they are dysregulated, the balance between excitatory and inhibitory neurons is lost, leading to hyperexcitability associated with autism.
Reference: Trudler, D. et al. “Dysregulation of miRNA expression and excitation in MEF2C autism patient hiPSC-neurons and cerebral organoids.” Molecular Psychiatry 30, 1479–1496 (2025). DOI: 10.1038/s41380-024-02761-9
I want to use AI protein design tools (AlphaFold, ESMFold, Rosetta — covered in HTGAA Weeks 4-5) to design a small protein or peptide that can bind to and modulate these autism-associated miRNAs. The designed protein would be expressed using cell-free synthesis (Week 9) and characterized with mass spectrometry (Week 10). The gene would be ordered from Twist Bioscience. I want to develop this because it sits right at the intersection of my CS/AI background and the wet lab techniques taught in HTGAA, and because AI-designed proteins targeting neurodevelopment could open up new research directions for autism.
2. Next, describe one or more governance/policy goals related to ensuring that this application or tool contributes to an “ethical” future.
My primary goal is to make sure AI-designed proteins targeting neurodevelopment are safe, responsibly used, and beneficial to the autism community. The sub-goals are:
Prevent AI-designed molecules from causing unintended biological harm.
Ensure traceability of AI-generated designs so problems can be traced back.
Keep AI protein design tools accessible and not locked behind excessive regulation.
Ensure the autism community benefits and is not exploited by research done in their name.
AI-designed proteins have the possibility to be used for great benefit in understanding and eventually treating neurodevelopmental conditions, but could also be misused or cause harm if not properly governed.
3. Next, describe at least three different potential governance “actions” by considering the four aspects below (Purpose, Design, Assumptions, Risks of Failure & “Success”).
Action 1: Mandatory biosafety screening for AI-designed proteins at DNA synthesis providers.
Purpose: Currently, synthesis companies like Twist screen orders against databases of known pathogens. But AI-designed novel proteins are not in these databases. We would extend screening to include computational toxicity and function prediction for novel sequences before synthesis.
Design: Synthesis providers (Twist, IDT, GenScript) would add a screening layer using protein function prediction tools to flag designs resembling known toxins or immune modulators. The IGSC would coordinate standards. Funding could come from industry fees and government biosecurity grants.
Assumptions: This assumes computational tools can reliably predict function from sequence alone, which is improving but still imperfect. It also assumes providers would voluntarily adopt this or that regulation would compel them.
Risks: If screening is too aggressive, it could block legitimate research orders (like my HTGAA project). If successful, it could create a false sense of security — “it passed screening” does not mean “it is safe.” Bad actors could also bypass providers using benchtop synthesis.
Action 2: Institutional ethics review for AI-designed therapeutics targeting neurodevelopment.
Purpose: Currently, IBCs review recombinant DNA work and IRBs review human subjects research. But there is no specific review for AI-designed molecules targeting brain development. We would add a lightweight ethics checklist when projects involve AI-designed proteins and neurodevelopmental targets.
Design: Universities would add a checklist to existing IBC review covering scientific justification, community benefit assessment, and transparent reporting. This could be piloted at MIT and Harvard first. The cost would be minimal since it piggybacks on existing infrastructure.
Assumptions: This assumes institutions will adopt the extra step without external mandate, and that the review can be lightweight enough not to discourage student projects.
Risks: If the review is too burdensome, students may avoid neurodevelopmental projects altogether. If too light, it becomes a rubber stamp. There is also a risk of paternalism — who decides what “benefits” the autism community?
Action 3: Open sharing of AI protein design methods and results through conferences and preprints.
Purpose: To share beneficial use cases of AI-designed proteins, foster collaboration between CS/AI and biology researchers, and disseminate safety learnings so the community can self-correct.
Design: This requires coordinating the AI protein design and synthetic biology communities. Organizations like iGEM or the Protein Society could host dedicated sessions. Researchers would be encouraged to publish designs, validation results, and failure cases openly.
Assumptions: I assume that researchers would be interested in attending and discussing, and that open sharing does more good than harm.
Risks: Open sharing of protein designs could be misused. Ethics could be overlooked in favor of scientific progress. However, keeping designs secret is arguably more dangerous because it prevents community oversight.
4. Next, score (from 1-3 with, 1 as the best, or n/a) each of your governance actions against your rubric of policy goals.
Does the action:
Action 1: Synthesis Screening
Action 2: Ethics Review
Action 3: Open Sharing
Enhance Biosecurity
• By preventing incidents
1
2
2
• By helping respond
1
3
1
Foster Lab Safety
• By preventing incident
1
2
2
• By helping respond
2
2
1
Protect the environment
• By preventing incidents
1
2
2
• By helping respond
2
2
1
Other considerations
• Minimizing costs and burdens to stakeholders
3
2
1
• Feasibility?
2
1
1
• Not impede research
3
2
1
• Promote constructive applications
2
2
1
5. Last, drawing upon this scoring, describe which governance option, or combination of options, you would prioritize, and why.
I think a combination of Action 2 (ethics review) and Action 3 (open sharing) would work best as immediate steps, with Action 1 (synthesis screening) as a longer-term goal.
Action 1 scores best on biosecurity and safety, but it is the most burdensome and could slow down student research. I would not want my own Twist order for this course to get delayed or rejected by an overly aggressive screening algorithm. This is better pursued as a long-term industry-wide initiative rather than something individual institutions can do on their own.
Action 2 is the most feasible to implement right now. MIT already has IBC infrastructure, and adding a lightweight neurodevelopment checklist requires no new funding or legislation. Action 3 is also easy to start and promotes the kind of interdisciplinary collaboration that makes AI protein design safer through community oversight.
Here many assumptions are made — mainly that AI-designed proteins pose meaningfully different risks from traditionally designed ones, which may not be true yet but will become more relevant as the tools improve. There is also a tension between accessibility and oversight. As a CS student entering biology for the first time through HTGAA, I benefit a lot from open tools and low barriers. Over-regulation could discourage exactly the kind of interdisciplinary work this course promotes. I also understand why the autism community can be wary of researchers who study autism without engaging with autistic people. These uncertainties can be mitigated by keeping review processes lightweight and making sure they include input from the autism community itself.
Week 2 lecture prep
Homework Questions from Joe Jacobson
Nature’s machinery for copying DNA is called polymerase. What is the error rate of polymerase? How does this compare to the length of the human genome? How does biology deal with that discrepancy?
DNA polymerase has a raw error rate of about 10⁻⁴ to 10⁻⁶ per base pair during nucleotide insertion. With the built-in 3’→5’ proofreading exonuclease, accuracy improves to around 10⁻⁷ to 10⁻⁸. The human genome is roughly 3.2 × 10⁹ base pairs (about 6.3 billion for the diploid genome), so at that rate there would still be many errors per copy. Biology solves this through mismatch repair mechanisms (such as MutS) that catch errors proofreading missed, bringing the final error rate down to about 1 per 10⁹ to 10¹⁰ nucleotides — low enough to reliably copy a genome this large.
How many different ways are there to code (DNA nucleotide code) for an average human protein? In practice what are some of the reasons that all of these different codes don’t work to code for the protein of interest?
The genetic code is degenerate — 64 codons encode 20 amino acids plus 3 stop signals. The slides mention the average human protein is about 1036 base pairs, which is roughly 345 codons / 345 amino acids. With about 3 possible codons per amino acid on average, that gives roughly 3³⁴⁵ ≈ 10¹⁷⁹ possible DNA sequences for one protein — a huge number.
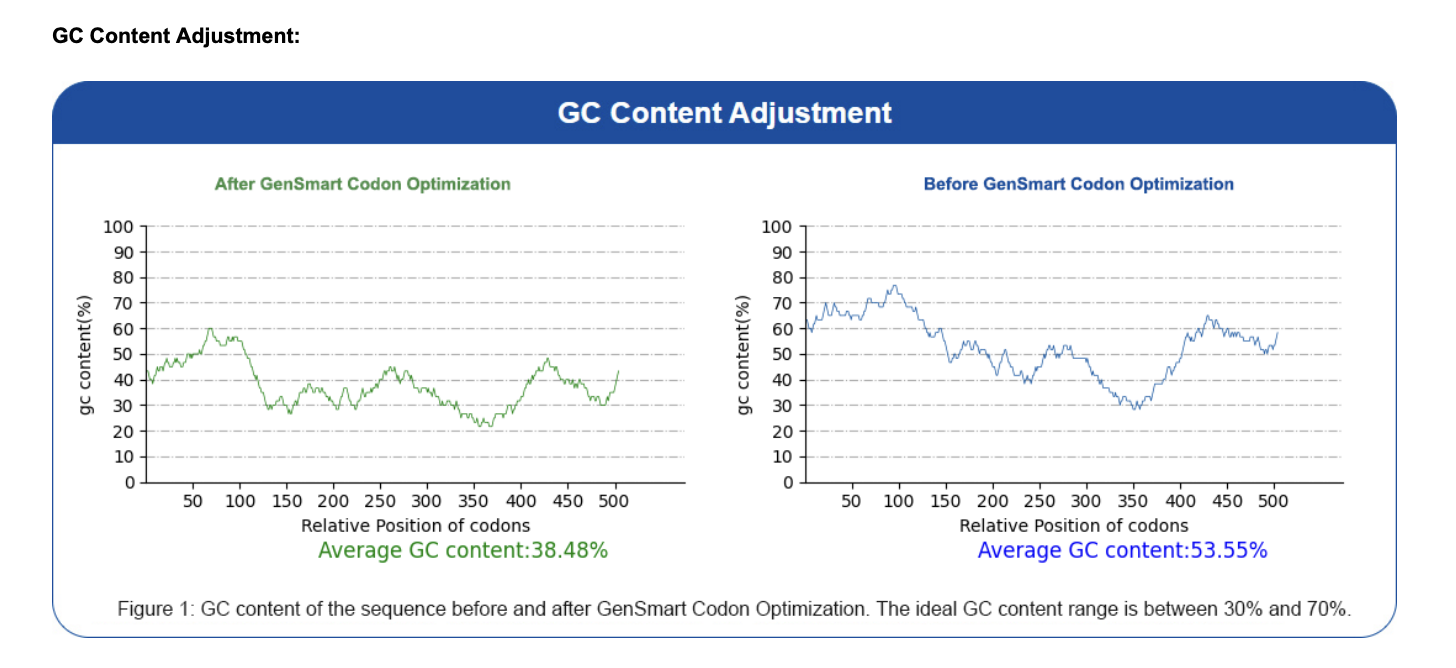
In practice most don’t work because organisms prefer certain codons that match their tRNA pools (codon usage bias), and using rare codons slows the ribosome and drops protein yield. Other issues include mRNA secondary structure and GC content blocking transcription or translation, accidental creation of regulatory signals like splice sites, and changes in translation speed that alter co-translational protein folding — affecting structure, solubility, or stability even when the amino acid sequence is identical. This is why codon optimization is standard practice in protein engineering.
Homework Questions from Emily Leproust
What’s the most commonly used method for oligo synthesis currently?
The phosphoramidite method — solid-phase chemical synthesis originally developed by Caruthers. Oligos are built stepwise on a solid support like controlled pore glass (CPG), adding one nucleotide at a time through cycles of detritylation, coupling, oxidation, and capping. It is highly automatable and forms the basis of all modern oligo synthesis platforms.
Why is it difficult to make oligos longer than 200 nt via direct synthesis?
Each coupling step has an efficiency of about 99%, but these small errors compound exponentially. A 200-mer at 99% efficiency gives only ~13% theoretical full-length yield, and in practice it’s much lower. Longer sequences accumulate more deletions, truncations, and depurination from the repeated harsh chemical cycles. Longer strands also form secondary structures that hinder reagent diffusion and coupling on porous supports like CPG. The result is that failure sequences (n-1 mers, mutations) dominate the output, and purifying the correct full-length product becomes impractical.
Why can’t you make a 2000 bp gene via direct oligo synthesis?
The above answer explains why longer oligos become dramatically harder to make. At 2000 bp, cumulative coupling inefficiency and side reactions cause full-length product yield to approach zero. Unlike enzymatic replication, chemical synthesis has no proofreading, so deletions, insertions, and substitutions build up rapidly. Long sequences also form stable hairpins that block reagent access. Instead, modern gene synthesis assembles shorter oligos (~60–200 nt) into longer fragments using enzymatic methods like Gibson Assembly, followed by sequence verification.
Homework Question from George Church
What are the 10 essential amino acids in all animals and how does this affect your view of the “Lysine Contingency”?
The 10 essential amino acids are: lysine, methionine, tryptophan, threonine, valine, isoleucine, leucine, arginine, histidine, and phenylalanine. Animals cannot synthesize these and must obtain them from dietary sources.
The “Lysine Contingency” is from Jurassic Park — a genetic modification to make the dinosaurs unable to produce lysine so they would die without human-provided supplements. However, lysine is already one of the 10 essential amino acids, so animals cannot produce it anyway. The dinosaurs could simply get lysine by eating plants, meat, or bacteria, making this a scientifically dubious plot point.
That said, the concept highlights something real about food security. Lysine is a limiting amino acid in cereal-based diets — staple crops like maize, rice, and wheat are all lysine-deficient relative to animal nutritional needs (Galili G, 2002). Growth and health can be constrained by lysine availability even when total protein intake is sufficient. This is why biotechnological interventions like microbial lysine production or high-lysine crops can have outsized impacts on food security and animal productivity. The lysine contingency, while fictional, illustrates how molecular-level biochemical constraints shape global food systems and ecological dependencies.
Applied AI support for early-stage project ideation through informal, conversational brainstorming (including: “My background is [X]. I’m interested in autism in the HTGAA course—what can I do?”), as well as subsequent formatting, structural organization, and language refinement of written outputs.
Week 2 HW: DNA Read, Write and Edit
Part 1: Benchling & In-silico Gel Art
For this exercise, the full genome of Bacteriophage Lambda (GenBank accession J02459.1, 48,502 bp) was imported into Benchling from NCBI. A virtual restriction enzyme digestion was performed using seven enzymes: EcoRI, HindIII, BamHI, KpnI, EcoRV, SacI, and SalI. Each enzyme was applied individually to identify its recognition sites across the Lambda genome. The digest results were visualized using Benchling’s simulated gel electrophoresis tool. To get a nice visual, I tried different ladders and different lane orderings. The final output consisting of for each enzyme’s fragment pattern is below.
Note: As a possible extension, it could be cool to build an interactive version of Ronan’s website where you can pick genome, enzymes, drag lanes around, swap ladders, add timing, etc. Could be a nice tool for future HTGAA classes.
Note: As a possible extension, it could be cool to build an interactive version of Ronan’s website where you can pick genome, enzymes, drag lanes around, swap ladders, add timing, etc. Could be a nice tool for future HTGAA classes.
Part 3: DNA Design Challenge
3.1. Choose your protein
I chose PIGH (Phosphatidylinositol N-acetylglucosaminyltransferase subunit H), a human protein encoded by the PIGH gene on chromosome 14q24.1. PIGH is a 188-amino-acid subunit of the GPI-GnT complex, which catalyzes the first step of GPI-anchor biosynthesis — transferring N-acetylglucosamine to phosphatidylinositol on the cytoplasmic side of the endoplasmic reticulum. GPI anchors tether many important proteins to the cell surface.
I chose this protein because mutations in PIGH cause GPIBD17 (Glycosylphosphatidylinositol Biosynthesis Defect 17), a rare autosomal recessive disorder characterized by developmental delay, seizures, and autistic features. The gene was only linked to disease in 2018, so it is likely underdiagnosed. Its small size (188 aa) also makes it practical for the synthesis and expression exercises in this homework.
Using the Sequence Manipulation Suite reverse translation tool from bioinformatics.org, I converted the 188-amino-acid protein sequence into a 564 bp DNA sequence using the most likely codons:
>reverse translation of sp|Q14442|PIGH_HUMAN to a 564 base sequence of most likely codons.
atggaagatgaacgcagctttagcgatatttgcggcggccgcctggcgctgcagcgccgc
tattatagcccgagctgccgcgaattttgcctgagctgcccgcgcctgagcctgcgcagc
ctgaccgcggtgacctgcaccgtgtggctggcggcgtatggcctgtttaccctgtgcgaa
aacagcatgattctgagcgcggcgatttttattaccctgctgggcctgctgggctatctg
cattttgtgaaaattgatcaggaaaccctgctgattattgatagcctgggcattcagatg
accagcagctatgcgagcggcaaagaaagcaccacctttattgaaatgggcaaagtgaaa
gatattgtgattaacgaagcgatttatatgcagaaagtgatttattatctgtgcattctg
ctgaaagatccggtggaaccgcatggcattagccaggtggtgccggtgtttcagagcgcg
aaaccgcgcctggattgcctgattgaagtgtatcgcagctgccaggaaattctggcgcat
cagaaagcgaccagcaccagcccg
3.3. Codon Optimization
I used the GenSmart Codon Optimization Tool to optimize the PIGH coding sequence for expression in E. coli and Staphylococcus aureus. Codon optimization is necessary because different organisms have different preferences for which codons they use most efficiently related to tRNA availability. Each organism has a different pool of tRNA molecules, and some tRNAs are abundant while others are rare. When the ribosome hits a codon whose matching tRNA is scarce in that organism, translation slows down or stalls. A human gene expressed directly in E. coli may use rare codons that slow down or stall translation. Optimization replaces these with codons preferred by the host organism, improving expression levels without changing the protein sequence. Optimization report available here.
To produce the PIGH protein, the codon-optimized DNA sequence is incorporated into an expression construct containing regulatory elements — a promoter (BBa_J23106), ribosome binding site (BBa_B0034), start codon, coding sequence, 7×His tag for purification, stop codon, and terminator (BBa_B0015). This can be ordered as a synthetic plasmid from Twist Bioscience in a backbone like pTwist Amp High Copy.
Protein production follows the central dogma in two stages. During transcription, RNA polymerase binds the promoter and synthesizes a complementary mRNA strand. During translation, ribosomes assemble at the RBS and read the mRNA in three-nucleotide codons, with tRNA molecules delivering amino acids from the start codon (AUG/methionine) until the UAA stop codon is reached, producing the folded 188-amino-acid PIGH polypeptide.
Cell-Dependent Production: The plasmid could be transformed into E. coli BL21 via heat shock or electroporation, with ampicillin selection to identify successful transformants. The host cell’s machinery and metabolic resources would then drive protein expression. Since PIGH is relatively small and does not appear to require complex eukaryotic modifications, E. coli may be a suitable host. The His-tagged protein could then be purified via Ni-NTA chromatography. This approach tends to be scalable but requires time for cell growth and purification.
Cell-Free Production: Alternatively, a TX-TL cell-free system (e.g., PURExpress) could be used, where the linear expression cassette is mixed directly with ribosomes, polymerase, tRNAs, and amino acids from lysed cells. This would likely produce protein within hours without living cultures, making it potentially ideal for rapid prototyping. PIGH’s small size (~564 bp) should be well within the efficient range for cell-free systems, though yields are generally lower and cost per reaction tends to be higher.
Part 4: Prepare a Twist DNA Synthesis Order
Part 5: DNA Read/Write/Edit
5.1 DNA Read
I would want to sequence the PIGH gene in patients presenting with developmental delay, seizures, and autistic features who lack a molecular diagnosis. Sequencing PIGH in such individuals could potentially help identify pathogenic variants and improve diagnostic rates for GPI-anchor biosynthesis disorders.
Since the PIGH coding region is relatively short (~564 bp), Sanger sequencing would likely be a suitable approach. Sanger is generally considered a first-generation method, reading one sequence at a time rather than in parallel like next-gen methods. I would extract genomic DNA from a patient sample, PCR-amplify the PIGH region using forward and reverse primers, and use the purified product as input for the Sanger reaction. A forward and reverse read should be sufficient to cover the full sequence with overlap.
The Sanger reaction mix contains normal dNTPs along with fluorescently labeled ddNTPs. During extension, the polymerase occasionally incorporates a ddNTP, terminating the chain at that position. This produces fragments of various lengths, each capped with a color for A, T, G, or C. Capillary electrophoresis separates them by size, and a detector reads the color at each position. The output is a chromatogram with color-coded peaks and a derived nucleotide sequence that could be compared against the PIGH reference to look for mutations.
5.2 DNA Write
I would want to synthesize the codon-optimized PIGH expression cassette from Parts 3–4. It could also be useful to order variant versions carrying known disease-associated mutations alongside wild-type to compare their effects on protein function. I would likely use phosphoramidite oligonucleotide synthesis as offered by companies like Twist Bioscience. Short oligos are built base-by-base on a solid support through repeated cycles of de-protection, coupling, capping, and oxidation, then assembled into the full-length gene and sequence-verified. Error rate tends to increase with length, so longer constructs may need to be built from smaller fragments. For PIGH (~564 bp), this should be well within a comfortable range.
5.3 DNA Edit
I would want to correct the M1L mutation (c.1A>T) in the PIGH gene. This change appears to disrupt the start codon and may prevent normal protein production. An adenine base editor (ABE) could be a reasonable choice for this kind of single-base correction. ABE generally uses a nickase Cas9 fused to an adenosine deaminase. A guide RNA directs the complex to the target site, the deaminase converts the target base without creating a double-strand break, and the cell’s repair machinery completes the correction. Inputs would include the ABE protein or mRNA, a guide RNA targeting the mutation region, and cells carrying the variant. Some limitations include the narrow editing window of the deaminase, dependence on a nearby PAM sequence, possible bystander edits on other adenines in the window, and variable delivery efficiency across cells.
Question 1: Find and describe a published paper that utilizes the Opentrons or an automation tool to achieve novel biological applications.
Fushimi, K., Nakai, Y., Nishi, A., Suzuki, R., Ikegami, M., Nimura, R., Tomono, T., Hidese, R., Yasueda, H., Tagawa, Y., & Hasunuma, T. (2025). Development of the autonomous lab system to support biotechnology research. Scientific Reports, 15, 6648. https://doi.org/10.1038/s41598-025-89069-y
This paper by Fushimi, Nakai et al. (2025) tackles the problem of optimizing how engineered bacteria produce glutamic acid, a commercially valuable amino acid used in MSG, medicine, and cosmetics. The researchers engineered E. coli by knocking out four genes and overexpressing ten others to redirect its metabolism toward glutamic acid production. However, finding the right growth medium recipe is extremely difficult because the bacteria tightly regulate their own glutamic acid levels to protect themselves from stress like pH changes and osmotic pressure. With dozens of possible nutrient combinations to test, manual experimentation is too slow and the biology is too complex for human intuition alone.
To solve this, they built the Autonomous Lab (ANL) using an Opentrons OT-2 liquid handler, an incubator, centrifuge, plate reader, mass spectrometer, and a robotic arm connecting them all. A Bayesian optimization AI suggests which combinations of calcium, magnesium, cobalt, and zinc concentrations to test, the robots autonomously prepare the media, culture the bacteria, and measure both growth and glutamic acid output, then feed the results back to the AI for the next round.
The system found a medium that nearly doubled E. coli growth using only 24 AI-picked experiments, outperforming a brute-force search of 256 conditions. This is the first fully autonomous closed-loop system for culture medium optimization, demonstrating that affordable tools like the Opentrons can power self-driving biology labs capable of navigating complex biological regulation faster than any human could, opening the door to optimizing production of many other valuable biomolecules the same way.
Part 3: Final Project Ideas
Week 4 HW: Protein Design I
Part A. Conceptual Questions
How many molecules of amino acids do you take with a piece of 500 grams of meat? (on average an amino acid is ~100 Daltons)
Meat is roughly 20% protein by mass, so 500 g of meat contains about 100 g of protein. Since one Dalton equals 1 g/mol, an average amino acid at 100 Da means 100 g/mol. That gives 100 g ÷ 100 g/mol = 1 mole of amino acid residues, or about 6 × 10²³ amino acid molecules. So eating a steak hands you roughly Avogadro’s number of amino acids, a staggering count that just means “about a mole.”
Why do humans eat beef but do not become a cow, eat fish but do not become fish?
Digestion breaks dietary protein down to individual amino acids (or short peptides) in our gut, stripping away all the cow or fish specific information that lived in the sequence of those proteins. Once the amino acids are absorbed, they are just interchangeable building blocks, the same 20 letters every organism uses. Our ribosomes then assemble new proteins using our own mRNA templates, which come from our own DNA. So the food provides raw material, but your genome determines what gets built. Therefore, information is in the sequence, not the bricks. Thanks to that we are not turning to a fish!
Why are there only 20 natural amino acids?
The genetic code uses three nucleotide codons drawn from a 4 letter DNA alphabet, giving 4³ = 64 possible codons, comfortably enough to encode 20 amino acids with redundancy. Twenty also turns out to be enough chemical diversity to fold proteins and run all of biochemistry: you get charged residues, polar ones, hydrophobic ones, aromatics, and structurally special cases like proline, glycine, and cysteine. Beyond that, it is largely a frozen accident. Early life locked in this set, and the entire translation machinery (tRNAs, synthetases, ribosomes) co-evolved around it, so deviating became evolutionarily costly. A few exceptions exist (selenocysteine and pyrrolysine), showing the system is not completely rigid.
Can you make other non-natural amino acids? Design some new amino acids.
Yes, non-natural amino acids (ncAAs) are a major area of synthetic biology, pioneered by Peter Schultz and Jason Chin, who have expanded the genetic code to incorporate hundreds of new residues using engineered tRNA/synthetase pairs and reassigned stop codons. Existing examples include p-azido-phenylalanine (a click chemistry handle), photo-crosslinking benzophenone amino acids, fluorinated residues for ¹⁹F NMR, and phosphoserine for tunable phosphorylation. Some design ideas: a metal chelating residue (terpyridine side chain) for built-in catalysis; a photoswitchable azobenzene amino acid that lets light flip a protein between two conformations; a bioorthogonal alkyne residue for labeling; or a heavy tryptophan analogue with extra fused rings to deepen hydrophobic cores. The principle is that as long as the side chain does not break the backbone geometry the ribosome expects, you can design almost any chemistry into a protein.
Where did amino acids come from before enzymes that make them, and before life started?
Prebiotic chemistry. Amino acids form spontaneously under reasonable early Earth conditions. Stanley Miller and Harold Urey demonstrated this in 1953 by sparking a flask of methane, ammonia, hydrogen, and water; within days the flask contained glycine, alanine, and several other amino acids. Amino acids also rain down from space: the Murchison meteorite, which fell in Australia in 1969, carries over 70 amino acids including most of the 20 biological ones. Hydrothermal vents and mineral surfaces likely also drove abiotic synthesis. So before life, a primordial soup already had a pool of amino acids ready for early replicators (probably RNA based) to put to use.
If you make an α-helix using D-amino acids, what handedness (right or left) would you expect?
Left handed. Natural α-helices are right handed because they are built from L-amino acids, and the steric and torsional constraints of L chirality force the backbone into a right handed twist. D-amino acids are the exact mirror image, so a helix made entirely of them would be the mirror image of a natural one, left handed, with otherwise identical pitch, rise, and hydrogen bond geometry. This is well established and confirmed in synthetic D-protein chemistry.
Can you discover additional helices in proteins?
There are already several beyond the canonical α-helix: the 3₁₀-helix (tighter, 3 residues per turn, common at helix ends), the π-helix (wider, rarer), the left handed polyproline II helix (PPII, abundant in collagen and disordered regions), and the collagen triple helix itself. Coiled coil oligomers, dimers up through 7 helix bundles like in GPCRs (Thras showed this progression in lecture: laminin trimer, 4 helix bundles, the coronavirus spike’s 6 helix, 7 helix GPCRs), are higher order assemblies built from α-helices but with their own structural identities. With non-natural amino acids or β-amino acids you can build helices with completely different geometries (foldamers), so the catalogue is genuinely open ended.
Why are most molecular helices right-handed?
Because their building blocks are chiral and biology picked one chirality. L-amino acids have a specific spatial arrangement around their alpha carbon, and the allowed backbone dihedral angles (the φ/ψ region of the Ramachandran plot) heavily favor a right handed twist. A left handed α-helix made of L-amino acids is sterically strained and almost never seen. The deeper question of why life uses L-amino acids and not D ones is unresolved: it might be a frozen accident amplified from a tiny initial bias, possibly seeded by parity violation in the weak nuclear force or by chiral selection on mineral surfaces. Either way, once L was locked in, right handed helices followed automatically.
Why do β-sheets tend to aggregate?
A β-sheet has two edges that expose unsatisfied backbone hydrogen bond donors (N–H) and acceptors (C=O) sticking out into solvent. These edges are chemically sticky; the cheapest way to satisfy them is to recruit another β-strand to hydrogen bond against. Hydrophobic side chains on the sheet faces add a second driving force: they want to be buried, so two sheets stack face to face to exclude water. The result is that β-sheets greedily extend and stack, growing into multi-stranded assemblies, while α-helices can satisfy their hydrogen bonds internally and do not have this same edge problem.
What is the driving force for β-sheet aggregation?
Two cooperating forces. First, backbone hydrogen bonding: every additional strand zips a row of N–H···O=C bonds along the sheet edge, and these bonds are highly directional and energetically favorable. Second, the hydrophobic effect: water is entropically penalized when forced to organize around exposed hydrophobic side chains, so burying those side chains between stacked sheets releases water and increases entropy. Together these make β-sheet aggregates extraordinarily thermodynamically stable, which is exactly why amyloids resist heat, detergents, and proteases.
Why do many amyloid diseases form β-sheets?
Because the cross-β fold is a generic, sequence tolerant low energy sink that almost any polypeptide can fall into under the right conditions. The hydrogen bonds doing the heavy lifting are between backbone atoms, which every amino acid has, so the propensity is not unique to any one protein, it is a property of the polypeptide backbone itself. Once a small amyloid seed forms, it templates further misfolding (a prion like mechanism), so the process is self amplifying. That is why structurally unrelated proteins, Aβ in Alzheimer’s, α-synuclein in Parkinson’s, tau in tauopathies, prion protein in CJD, all converge on cross-β fibrils despite having totally different native folds.
Can you use amyloid β-sheets as materials?
Absolutely. Thras hit this in lecture when he compared spider silk (a β-sheet protein) to Kevlar, noting that humans engineered Kevlar inspired by the same backbone hydrogen bonding geometry that makes silk one of nature’s strongest materials. Functional amyloids exist naturally too: bacterial curli fibers form biofilm scaffolds, and mammalian Pmel17 organizes melanin in melanosomes. Engineered uses include self assembling peptide hydrogels for tissue scaffolding (Shuguang Zhang’s RADA16 is the canonical example), conductive amyloid nanowires, drug delivery fibrils, and adhesive coatings. The same property that makes amyloids pathological, extreme stability, makes them excellent biomaterials when you control the assembly.
Design a β-sheet motif that forms a well-ordered structure.
The trick is an amphipathic alternating pattern: hydrophobic and hydrophilic residues on alternating positions, so each β-strand has one hydrophobic face and one hydrophilic face. Shuguang Zhang’s RADA16, Ac-(RADA)₄-NH₂, the sequence Arg-Ala-Asp-Ala repeated four times, is the textbook example: alanines pack into a hydrophobic core, while alternating arginines (+) and aspartates (−) form complementary salt bridges on the polar face, locking the sheets into ordered nanofibers in water. A simple variant I would design: Ac-(VEVK)₄-NH₂, where valine gives the hydrophobic face and alternating glutamate/lysine give a charge zipper polar face; this should self assemble into stable antiparallel β-sheets at neutral pH. To enforce well ordered (rather than messy aggregated) structure, cap the ends with proline or glycine to prevent runaway elongation, and keep the strand length around 8 to 16 residues so a single sheet can register cleanly without frame shifts.
Part B. Protein Analysis and Visualization
For this assignment, I selected Neuroligin-4X (NLGN4X) from human (Homo sapiens). I chose this protein because it has both sequence information and an experimentally solved 3D structure, which makes it suitable for practicing a protein analysis workflow. NLGN4X is also related to synaptic biology, so it is a useful example for connecting protein structure to a biological context. The main sequence resource I used is UniProt Q8N0W4, and the main structure resource is RCSB PDB 3BE8, titled “Crystal structure of the synaptic protein neuroligin 4.” The RCSB entry identifies the protein as human Neuroligin-4, X-linked, and links it to the gene name NLGN4X and the UniProt accession Q8N0W4. NCBI also describes NLGN4X as a neuronal cell-surface protein. This background gives the protein some biological relevance, but the main goal of this report is to analyze its sequence and structure using online databases and PyMOL.
For the sequence analysis, I used the full-length human NLGN4X sequence from UniProt. The full protein is 816 amino acids long. For the structure analysis, I used PDB 3BE8. This structure does not contain the entire full-length protein; instead, it contains the extracellular cholinesterase-like domain of NLGN4X. This means that the full UniProt sequence is used for the amino acid sequence and frequency analysis, while the PDB structure is used for 3D visualization. The sequence of the crystallized construct can also be viewed through the RCSB FASTA page for 3BE8.
The full-length amino acid sequence of human NLGN4X is shown below in FASTA format:
After counting the amino acids in the full-length sequence, the most frequent amino acid is leucine (L), which appears 75 times. The next most common amino acids are threonine (T), which appears 72 times, and proline (P), which appears 61 times. The full amino acid count is: L = 75, T = 72, P = 61, G = 50, D = 50, V = 47, A = 46, I = 46, N = 45, S = 44, E = 37, K = 36, Y = 36, Q = 35, R = 32, F = 32, H = 27, M = 22, W = 13, and C = 10. This result shows which amino acids are most common in the sequence, but amino acid frequency alone should not be over-interpreted because it does not fully explain the protein’s structure or function.
To search for sequence homologs, the NLGN4X sequence can be submitted to the UniProt BLAST tool. A reasonable approach is to paste the full FASTA sequence into UniProt BLAST, search against UniProtKB, and record significant matches using a cutoff such as E-value < 1e-5. The results are expected to include NLGN4X orthologs from other species and related human neuroligin proteins such as NLGN1, NLGN2, NLGN3, and NLGN4Y. The exact number of homologs may vary depending on the BLAST settings and database version, so this number should be filled in after running the search:
Using UniProt BLAST with human NLGN4X as the query, I found ___ significant homologs using an E-value cutoff of ___.
NLGN4X belongs to the neuroligin protein family. Neuroligins are cell-surface proteins involved in synaptic adhesion. NLGN4X also contains a cholinesterase-like extracellular domain and is related to the type-B carboxylesterase/lipase protein family. Although the fold resembles some enzyme families, NLGN4X is generally discussed as a synaptic adhesion protein rather than as a classical enzyme.
The RCSB structure used for this report is PDB ID 3BE8. This structure was solved by X-ray diffraction and has a resolution of 2.20 Å. In protein crystallography, smaller resolution values usually indicate better structural detail. Since the assignment describes 2.70 Å as a good-resolution cutoff, the 2.20 Å resolution of 3BE8 is good for this project. The structure was deposited on 2007-11-16 and released on 2008-01-29.
The solved structure contains the Neuroligin-4X extracellular domain as a homodimer, meaning that two copies of the protein domain are present together. The structure also contains some non-protein molecules, including N-acetylglucosamine (NAG) sugar residues and small crystallographic molecules or ions such as citrate and phosphate. Therefore, the structure is mainly a protein dimer, but it also includes additional molecules that are not part of the protein chain.
In terms of structural classification, NLGN4X can be described as a cell-adhesion protein with an alpha/beta hydrolase-like or cholinesterase-like fold. This type of fold contains both alpha helices and beta sheets. For this report, I describe NLGN4X as a neuroligin family protein with a mixed alpha/beta structural fold.
I visualized the structure using PyMOL. First, I loaded the structure with the following command:
fetch 3BE8, async=0
The cartoon view was generated using:
hide everything
show cartoon
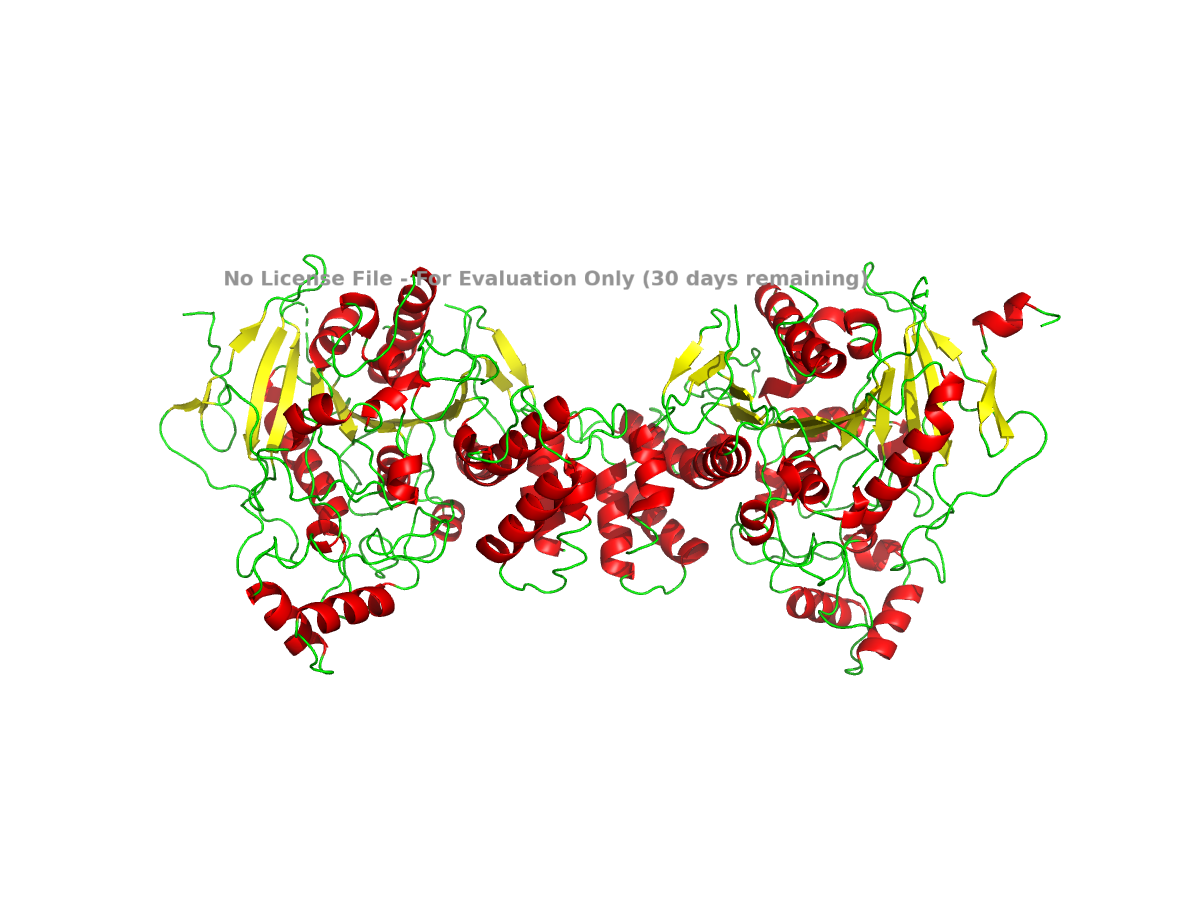
color gray
Figure 1. Cartoon view of human NLGN4X from PDB 3BE8.
The ribbon view was generated using:
hide everything
show ribbon
color gray
Figure 2. Ribbon view of human NLGN4X from PDB 3BE8.
The ball-and-stick view was generated using:
hide everything
show sticks
show spheres
Because showing the entire protein in ball-and-stick style can look crowded, a close-up region is usually easier to interpret.
Figure 3. Ball-and-stick close-up view of part of human NLGN4X.
I also colored the protein by secondary structure in PyMOL. In this view, alpha helices are colored red, beta sheets are colored yellow, and loops are colored green:
hide everything
show cartoon
dss
color red, ss h
color yellow, ss s
color green, ss l+''
Figure 4. NLGN4X colored by secondary structure. Helices, sheets, and loops are shown in different colors.
Based on this view, NLGN4X appears to be a mixed alpha/beta protein. It is not purely helical or purely beta-sheet. The structure contains a beta-sheet-containing core along with several alpha-helical regions around it.
To examine residue type distribution, I colored hydrophobic residues orange and hydrophilic or polar/charged residues cyan:
hide everything
show cartoon
select hydrophobic, resn ALA+VAL+LEU+ILE+MET+PHE+TRP+PRO
select hydrophilic, resn ARG+LYS+ASP+GLU+ASN+GLN+SER+THR+TYR+HIS+CYS
color orange, hydrophobic
color cyan, hydrophilic
In this view, hydrophobic residues are expected to appear more often in buried parts of the structure, while hydrophilic residues are more likely to appear on the surface where they can interact with water. This is a general pattern for many soluble or extracellular protein domains. For this protein, I would use the visualization to check whether this pattern is visible rather than treating it as a definitive conclusion from the sequence alone.
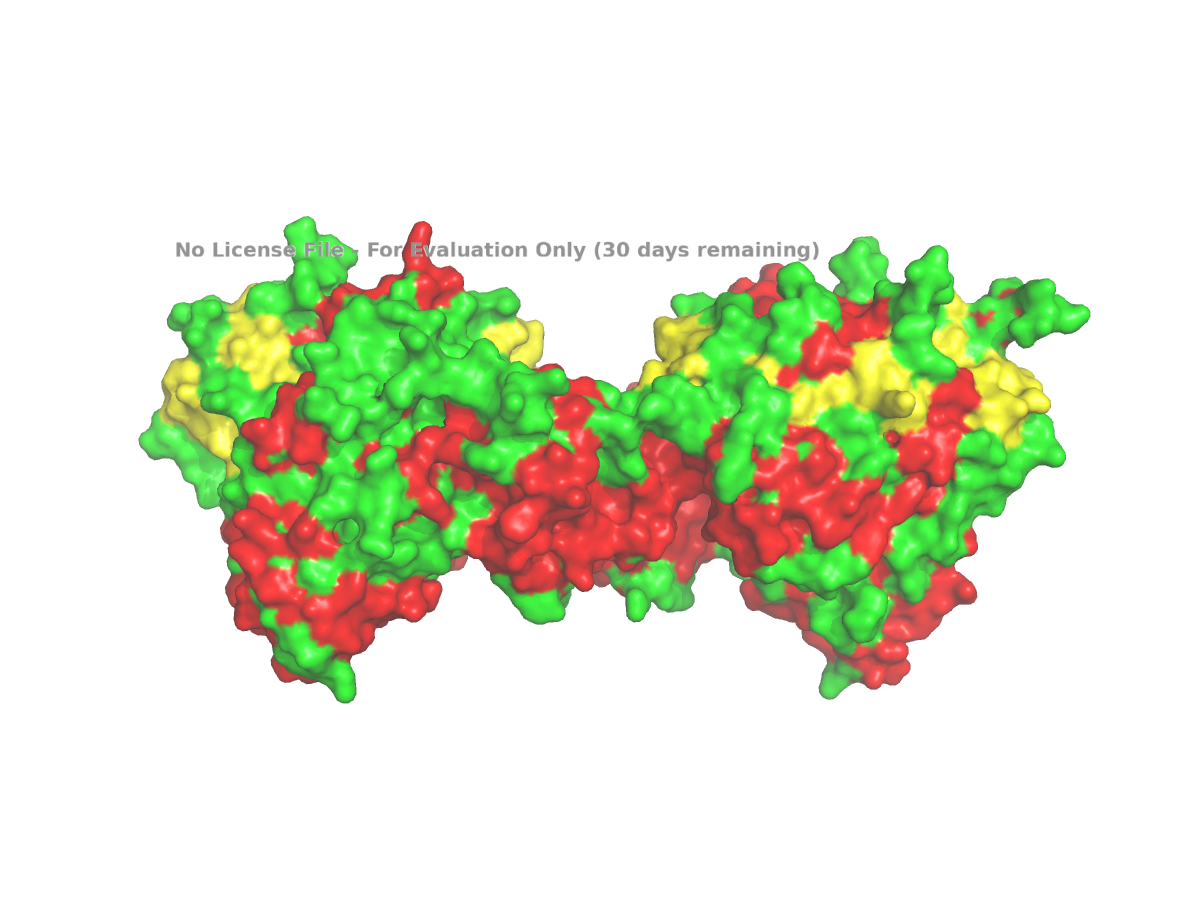
Finally, I used a surface representation to look for grooves, depressions, or possible pocket-like regions:
hide everything
show surface
set transparency, 0.25
For a clearer surface, I used:
set surface_quality, 1
set transparency, 0.35
Figure 5. Surface view of human NLGN4X from PDB 3BE8, used to inspect grooves or possible pocket-like regions.
The surface view shows that the protein is not completely smooth. There appear to be grooves and depressions on the surface. These can be described as possible pocket-like surface features, although I would avoid calling them enzyme active sites unless there is specific evidence for that. In this assignment, the surface view is mainly used to make a visual observation about the shape of the protein.
Overall, human NLGN4X is a suitable example for this assignment because its sequence and structure are available in standard databases. The full-length protein is 816 amino acids long, and leucine is the most frequent amino acid in the sequence. The selected structure, PDB 3BE8, represents the extracellular domain of NLGN4X and was solved by X-ray diffraction at 2.20 Å, which is better than the 2.70 Å cutoff given in the prompt. The structure shows a mixed alpha/beta fold, and PyMOL can be used to examine the protein in cartoon, ribbon, ball-and-stick, secondary-structure, residue-type, and surface views.
## Part C. Using ML-Based Protein Design Tools
I chose Thaumatin I from Thaumatococcus daniellii because it is a food-related sweet-tasting protein with a well-characterized 3D structure. Its sequence is available in UniProt P02883, and its structure is available as PDB 1RQW, with the corresponding RCSB FASTA sequence. UniProt lists Thaumatin I as 235 amino acids long, and RCSB reports the 1RQW structure as an X-ray structure with 1.05 Å resolution.
C1. Protein Language Modeling
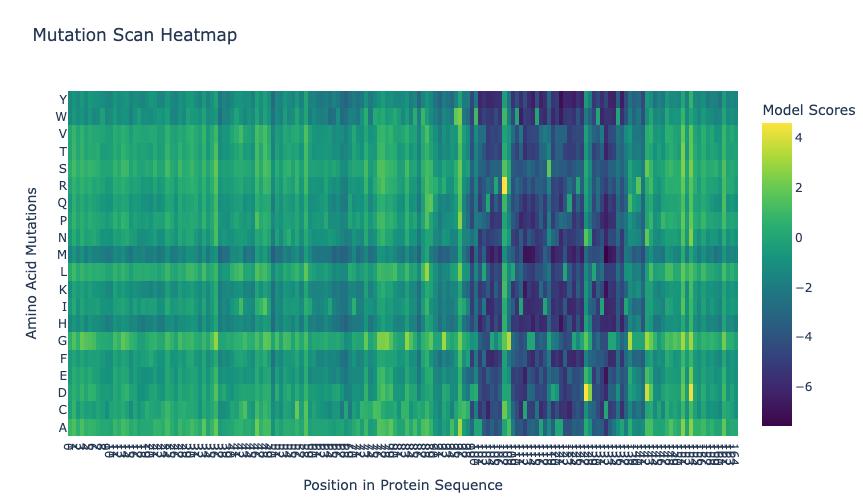
Deep Mutational Scans
The heatmap shows the ESM2 deep mutational scan for thaumatin. Each column represents a position in the protein sequence, and each row represents a possible amino acid substitution. Brighter colors correspond to higher LLR scores, meaning the model finds that mutation more compatible with the sequence context, while darker colors correspond to lower LLR scores. Overall, the heatmap shows that some regions are more tolerant to substitutions, while other regions contain many low-scoring mutations and may be more constrained.
In the ESM2 mutational scan, the most positive mutation score was for T2M, where threonine at position 2 is replaced by methionine. This mutation had an LLR score of 4.8903, meaning that the model scored methionine at this position as more likely than the original threonine. Since threonine is polar and methionine is larger and hydrophobic, this result may suggest that ESM2 finds a more hydrophobic residue compatible at this position. However, this should be interpreted cautiously because the scan is based on language-model likelihoods, not an experimental measurement of thaumatin stability or sweetness.
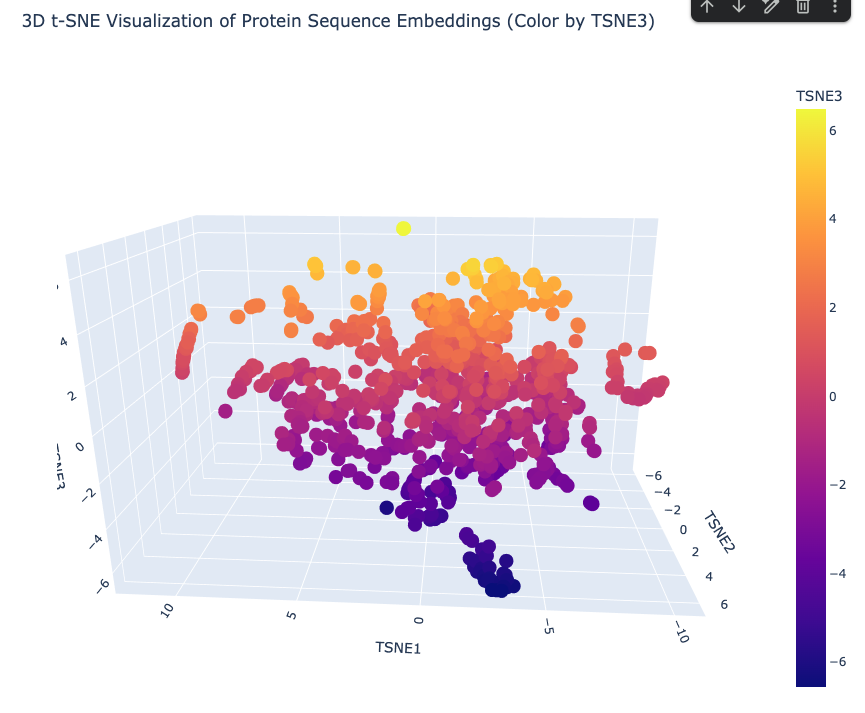
#### Latent Space Analysis
The latent-space analysis represents protein sequences as embeddings and then reduces them into three t-SNE dimensions for visualization. In the 3D t-SNE visualization of protein sequence embeddings, each point represents a protein sequence, and the axes represent reduced embedding dimensions. The plot shows several neighborhoods of proteins, suggesting that the embedding captures sequence-level similarities. Because the points are not distributed as one uniform cloud, the model appears to separate proteins into local groups based on features such as sequence similarity, domain composition, amino acid patterns, or protein family. I interpreted this plot as a qualitative map of protein similarity rather than an exact measurement of structural or functional relationships.
Week 5 HW: Protein Design II
Part 1: Generate Binders with PepMLM
I retrieved the human SOD1 sequence from
UniProt (P00441) in FASTA format.
The change is visible at positions 4-5: the original MATKA...
becomes MATKV...
Using the PepMLM-650M Colab,
I ran the model with the following parameters:
Parameter
Value
Target sequence
SOD1-A4V (mutant)
Peptide length
12
Top-K
3
Number of binders
4
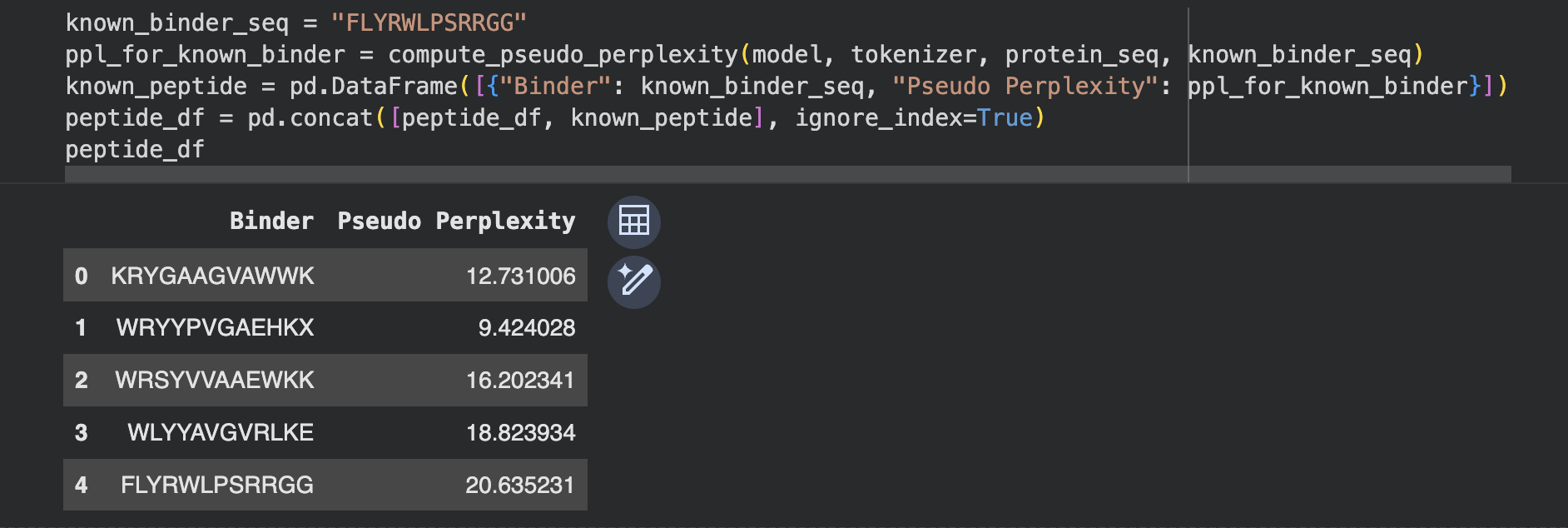
I generated 4 peptides conditioned on the SOD1-A4V sequence,
and added the known SOD1-binding peptide FLYRWLPSRRGG as a
positive control for comparison.
Perplexity indicates PepMLM’s confidence that a peptide
will bind the target — lower scores indicate higher confidence.
Week 6 HW: Genetic Circuits I
1. What are some components in the Phusion High-Fidelity PCR Master Mix and what is their purpose?
Phusion HF PCR Master Mix is a ready-to-use 2X solution that contains everything needed for PCR except the template, primers, and water.
The most important component is Phusion DNA Polymerase, which is the enzyme that copies DNA during the reaction. The standard alternative is Taq polymerase, a widely used enzyme isolated from a heat-resistant bacterium called Thermus aquaticus. Taq survives the high temperatures of PCR but has no proofreading ability, so it cannot correct mistakes it makes while copying, resulting in a relatively high error rate. Phusion addresses this by including a proofreading domain that catches and fixes errors as they occur, making it about 50 times more accurate than Taq. This accuracy is important in a mutagenesis experiment where only the specific, intended mutations should be introduced and any unintended errors would compromise the result.
The mix also contains dNTPs, which are the four DNA building blocks (dATP, dTTP, dGTP, dCTP) that the polymerase uses to construct new strands. MgCl₂ is included as well, providing magnesium ions that the polymerase requires to function and that also affect how tightly primers bind to the template. Finally, the HF Buffer maintains the right pH and salt levels so the enzyme works reliably across different templates.
2. What are some factors that determine primer annealing temperature during PCR?
The annealing temperature is typically set 2 to 5°C below the melting temperature (Tm) of the primer pair. The Tm is the temperature at which half of the primer-template bonds break apart, so setting the annealing temperature just below it ensures primers bind firmly without sticking to the wrong locations.
Several factors affect Tm. Primer length matters because longer primers form more bonds with the template and therefore require more heat to detach, resulting in a higher Tm. GC content also plays a role since G-C base pairs form three hydrogen bonds while A-T pairs form only two, meaning primers with more G and C bases bind more tightly and have a higher Tm.
Salt concentration in the buffer raises the effective Tm because magnesium and other ions stabilize the DNA duplex. Mismatches lower the Tm because they weaken binding. When a primer contains intentional mismatches, it grips the template less tightly, so a lower annealing temperature is needed to allow binding to occur. Primer concentration and secondary structures such as hairpins can also shift the effective annealing behavior.
3. Compare and contrast PCR and restriction enzyme digests as methods for producing linear DNA fragments.
Both PCR and restriction enzyme digestion produce linear DNA fragments, but they work through different mechanisms and are suited to different situations.
In terms of protocol, restriction digestion is the simpler method. The DNA is mixed with the enzyme in an appropriate buffer and incubated at 37°C for about an hour. PCR is more involved and requires primer design, a reaction setup with polymerase and dNTPs, and a thermocycling program that moves through denaturation, annealing, and extension steps, taking around 90 minutes in total.
The two methods also differ in how flexible they are. Restriction enzymes can only cut at their specific recognition sequences, so the location of the cut is fixed by whatever sites exist in the template. PCR can amplify any region defined by the primer binding sites, giving full control over fragment boundaries. PCR also amplifies the DNA exponentially from a very small starting amount, while restriction digestion simply cuts the DNA that is already present without increasing the quantity.
A key advantage of PCR is the ability to introduce mutations through mismatches built into the primers. The amplified product will carry those changes. Restriction enzymes cannot introduce new sequence and can only cut existing DNA.
PCR
Restriction Enzyme Digest
Requires primer design and a thermocycling program. More involved setup.
Simpler protocol. Mix with enzyme and buffer, incubate at 37°C.
Fragment boundaries are fully flexible, defined by primer placement.
Can only cut at specific recognition sequences already in the DNA.
Primers can introduce mutations into the product.
Cannot change sequence, only cuts existing DNA.
Amplifies DNA exponentially from very small amounts.
Does not amplify, only cuts what is already present.
Best when custom boundaries, mutations, or overhangs are needed.
Best when convenient cut sites already exist and no changes are needed.
In this lab PCR is the right choice because the chromophore mutations need to be introduced and overlapping ends need to be added for Gibson assembly, neither of which restriction digestion can do.
4. How can you ensure that the DNA sequences that you have digested and PCR-ed will be appropriate for Gibson cloning?
The most important requirement is that the fragments have overlapping ends of around 20 to 40 base pairs. These overlaps are what allow the fragments to recognize and stick to each other during Gibson assembly. When designing PCR primers, the overlapping regions need to be built into the primer sequences so that the amplified fragment carries them at its ends. The overlaps must also match the correct neighboring fragment so everything assembles in the right order.
Unlike restriction cloning, Gibson assembly does not require specific cut sites in the DNA. However, the ends must be carefully designed to be complementary to adjacent fragments. After PCR or digestion, the DNA should also be clean and free of contaminants so the assembly enzymes can work efficiently. Running a gel to confirm the fragments are the expected size is a useful check before proceeding to the assembly step.
5. How does plasmid DNA enter E. coli cells during transformation?
Plasmid DNA enters E. coli by making the cell membrane temporarily permeable so the DNA can pass through. There are two common methods for doing this.
The first is heat-shock transformation. The cells are treated with calcium chloride, which neutralizes the negative charges on both the DNA and the membrane. This reduces the natural repulsion between them and allows the DNA to sit against the cell surface. A sudden heat shock at 42°C then causes the membrane to expand rapidly, opening temporary pores. The DNA diffuses through those pores into the cell. The cells are immediately cooled so the membrane reseals, and then given nutrients and time to recover before being plated on selective media.
The second method is electroporation. Instead of heat, a short electrical pulse is applied to the cells. This creates tiny pores in the membrane through which the plasmid DNA can pass. Electroporation is generally more efficient than heat shock and is commonly used when transformation efficiency needs to be very high.
6. Describe another assembly method in detail: Golden Gate Assembly
Golden Gate Assembly is a one-pot cloning method that uses Type IIS restriction enzymes, most commonly BsaI, to join multiple DNA fragments in a single reaction without leaving any scar sequence at the junctions.
The key property of Type IIS enzymes is that they cut outside their recognition sequence rather than within it. This means the recognition site can be placed at the end of a fragment so that when the enzyme cuts, the recognition site itself is removed. What remains is a short custom 4-base-pair overhang at each junction, which the designer specifies in the primer sequence. Because every junction in the assembly has a different 4-base overhang, each fragment can only join its correct neighbor in the correct orientation. Order and direction are determined by the overhang sequences, not by chance.
The reaction contains all fragments, BsaI, and T4 ligase together in one tube. It cycles between 37°C, where the enzyme cuts, and 16°C, where the ligase seals compatible overhangs. Once two fragments are correctly joined, the BsaI recognition site is gone from that junction and the ligated product cannot be re-cut. This drives the reaction toward the fully assembled product over multiple cycles.
Compared to Gibson assembly, Golden Gate offers more precise control over junction sequences and scales well to 10 or more fragments in a single reaction. Gibson is simpler for small assemblies of 2 to 4 fragments because it only requires sequence overlap in the primers and runs isothermally at 50°C. Golden Gate requires that the BsaI recognition sequence does not appear anywhere inside the fragments being assembled, otherwise the enzyme will cut in the wrong place.
Golden Gate
Gibson
Overhang type
4-bp sticky ends, custom designed
~25 bp single-stranded overlap
Reaction temperature
Cycles between 37°C and 16°C
Isothermal at 50°C
Scarless junctions
Yes
Yes
Best for
10 or more fragments, ordered assembly
2 to 4 fragments, simple workflow
Requires RE site in primers
Yes
No
For this chromophore lab, Gibson is the more practical choice because only two fragments need to be joined. Golden Gate becomes the better option when assembling many parts in a defined order, such as in metabolic pathway engineering or combinatorial library construction.
Benchling
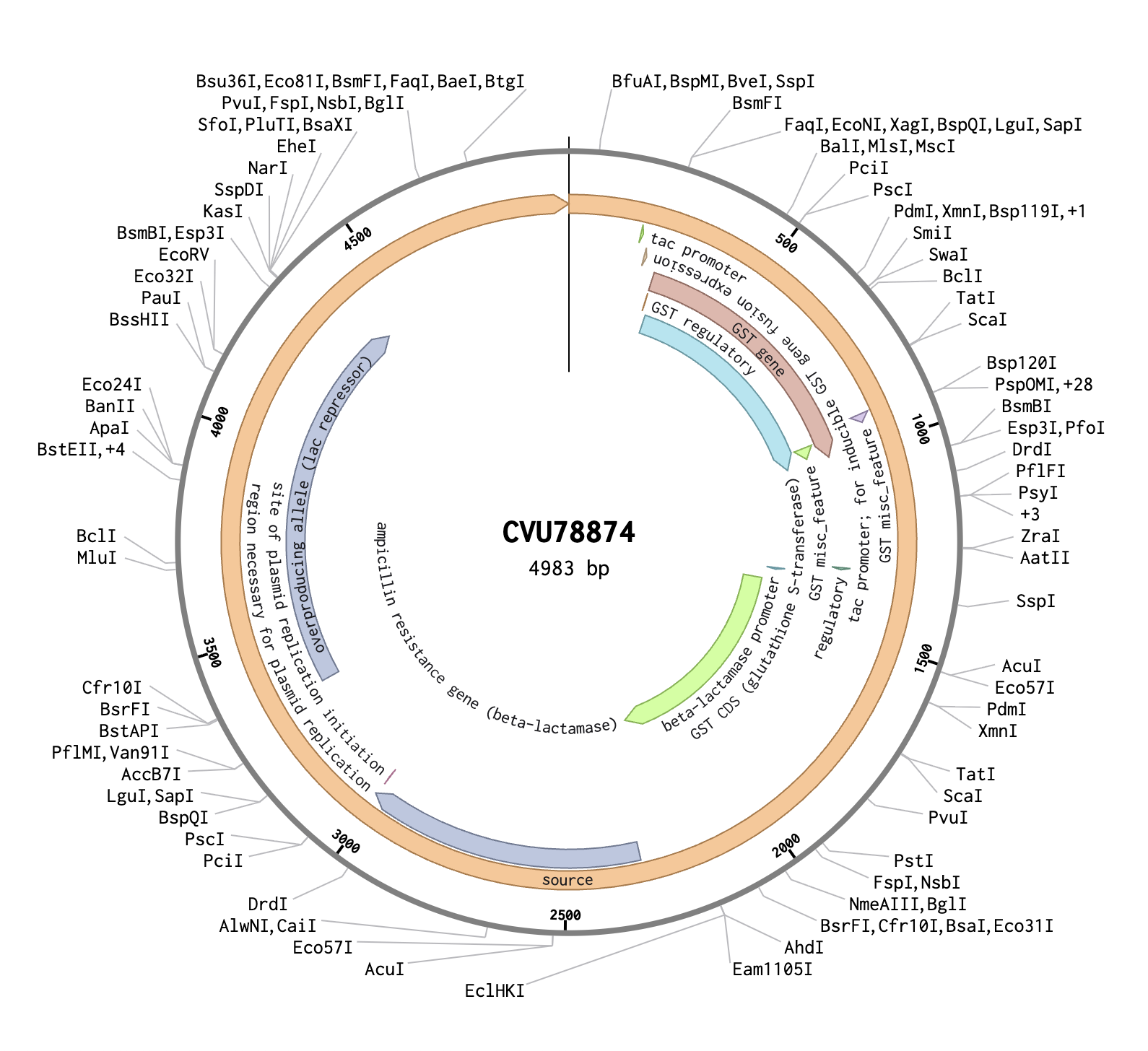
In Benchling, I created a new DNA sequence (type: DNA, topology: circular) to represent the pGEX plasmid. I imported it from the NCBI database using the accession number CVU78874, and confirmed it to be 4983 bp. The plasmid map showed several key annotated regions: the GST CDS (glutathione S-transferase), which is the orange region on the map and acts as a fusion tag so the inserted protein can be purified later using glutathione beads; the tac promoter, which drives expression of the insert and is switched on by adding IPTG to the growth media; and the ampicillin resistance gene, which allows only bacteria that successfully took up the plasmid to survive on selection plates.
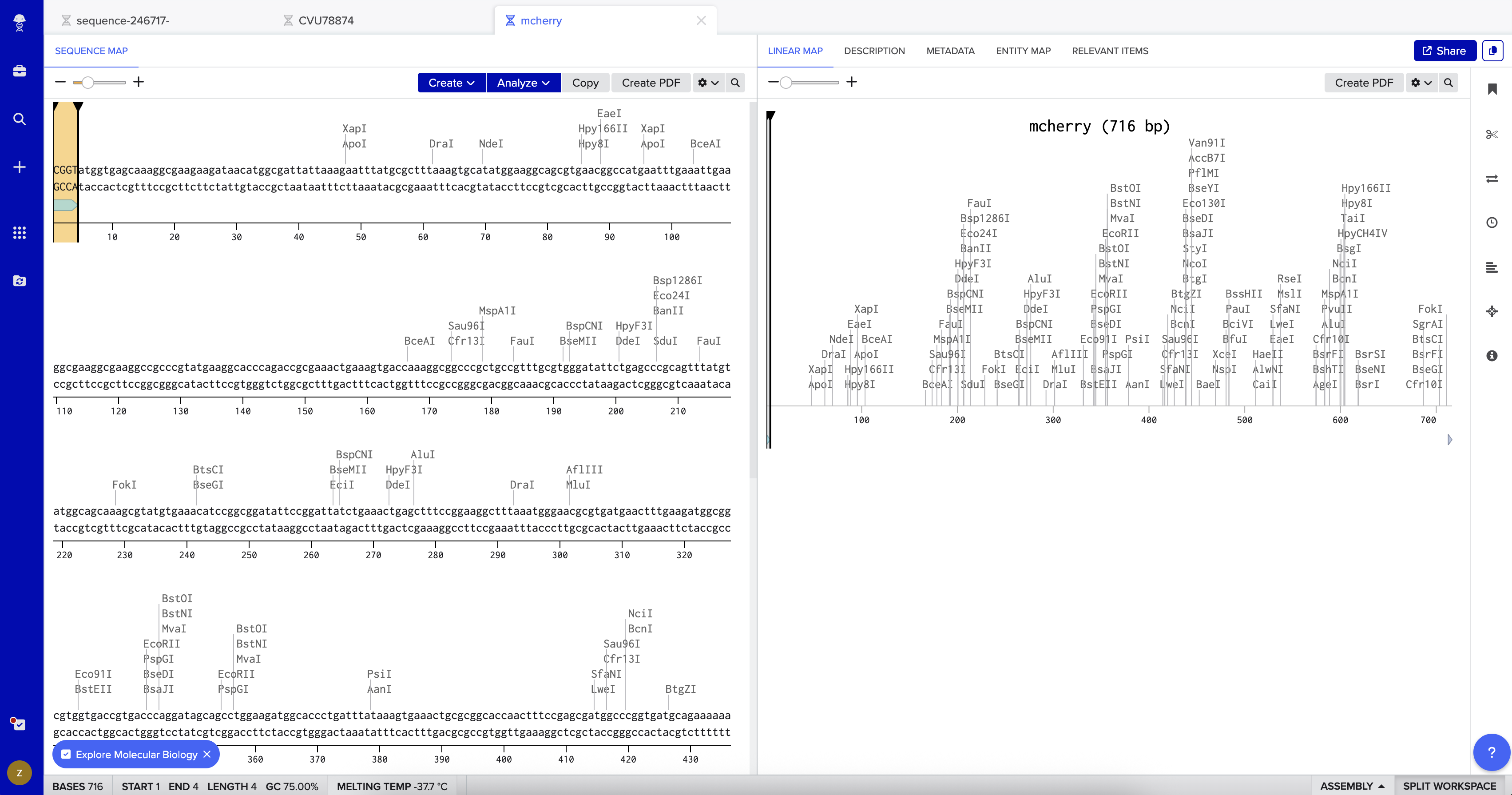
I then created a second DNA sequence (type: DNA, topology: linear) for the mCherry insert. From UniProt, I obtained the mCherry amino acid sequence of 236 amino acids and used the Reverse Translate tool at bioinformatics.org to convert it into a 708 bp DNA coding sequence optimized for E. coli. I added the BsaI-compatible overhangs to both ends of the sequence — CGGT at the 5’ end before the ATG start codon, and ACCG at the 3’ end after the stop codon — giving a final insert length of 716 bp.
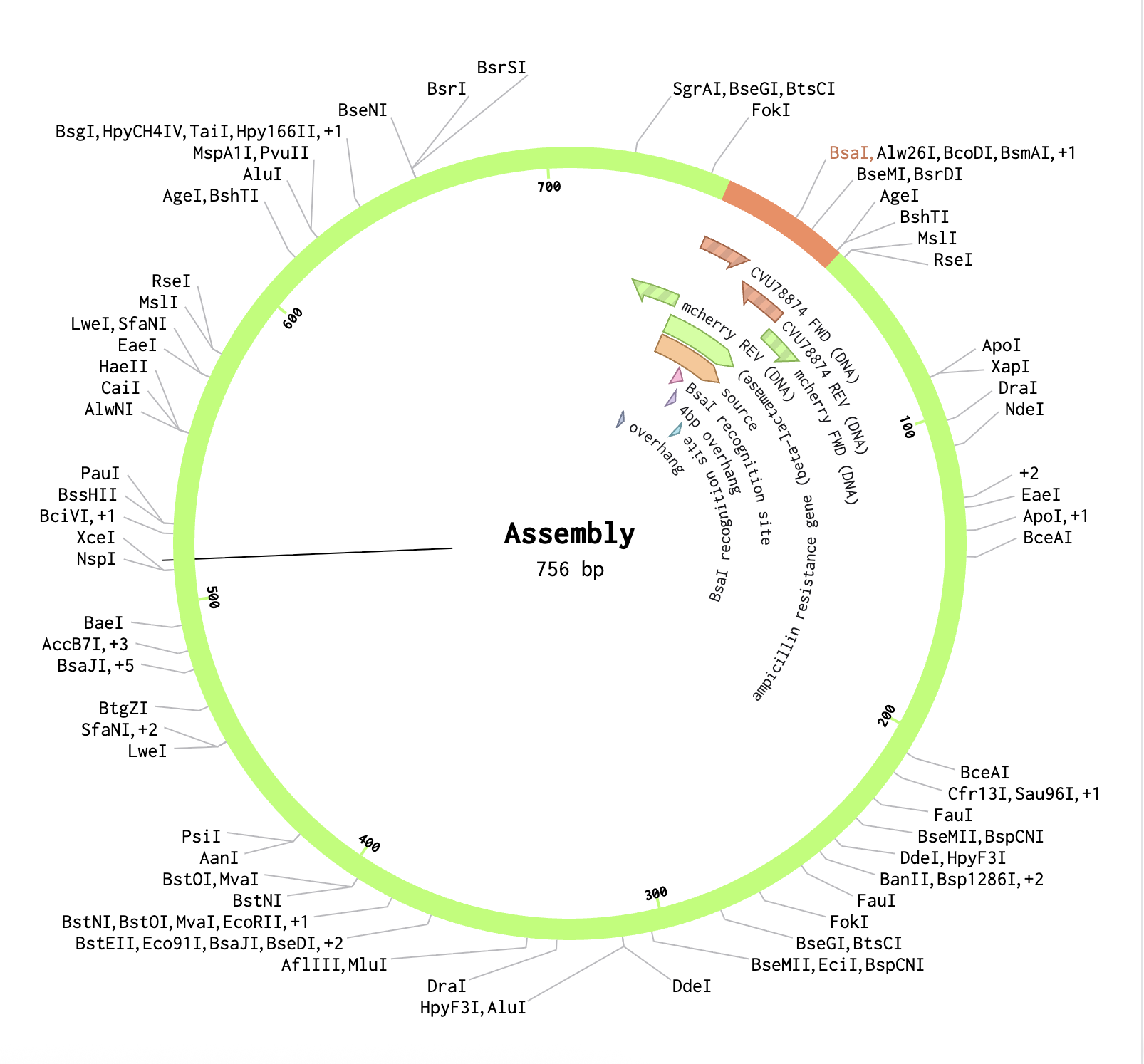
In the pGEX sequence, I ran a digest using the Type IIS enzyme BsaI and found that it cuts once at position 2113 with 100% efficiency at 37°C. I added annotations for the cut site: 2107–2112 for the BsaI recognition site (GGTCTC), and 2113–2116 for the 4 bp overhang (CGGT) that gets exposed after cutting. Because BsaI cuts 1 nt outside its own recognition sequence, the recognition site is removed after digestion, leaving only the clean 4-nt sticky end for ligation.
Finally, using the Assembly Wizard, I selected Golden Gate Assembly as the method, assigned pGEX as the backbone and mCherry as the insert, and set the enzyme to BsaI. I manually selected the region around positions 2090–2130 as the fragment to provide sufficient flanking sequence for primer binding. The assembled plasmid was generated successfully, with mCherry correctly inserted at the BsaI cut site in the pGEX backbone, producing a circular plasmid where the GST tag and mCherry are expressed together as a fusion protein.
Week 7 HW: Genetic Circuits II
Assignment Part 1: Intracellular Artificial Neural Networks (IANNs)
1. What advantages do IANNs have over traditional genetic circuits, whose input/output behaviors are Boolean functions?
Boolean genetic circuits treat signals like 0 or 1, meaning every input must pass a hard on/off threshold. However, this can lose useful information. This can be limiting because real biological signals are usually not binary. Inside cells, signals often exist as gradual changes such as in concentration level. IANNs are useful because they keep more of that analog information since they are not only asking “is this signal present or absent?”.
Main advantages of IANNs
They preserve more information. Boolean circuits lose information when they convert a signal into ON or OFF. IANNs keep the strength of the signal.
They are more robust to noise. A Boolean circuit can fail if a signal is close to its threshold. An IANN responds gradually, so small changes or noise are less likely to cause sudden failure.
They degrade smoothly. If an input changes slightly, the output changes slightly. In Boolean circuits, a small change can flip the whole output.
They can handle weighted inputs. Some signals can matter more than others. IANNs can assign different “weights” to different biological inputs.
They are more efficient for complex decisions. A single IANN neuron can combine several inputs in a way that might require many Boolean gates. Fewer gates can mean: less metabolic burden, less delay, or fewer parts that can fail.
2. Describe a useful application for an IANN; include a detailed description of input/output behavior, as well as any limitations an IANN might face to achieve your goal.
IANNs (in theory) could work for any classification problem! For example, IANNs could help a cell decide whether a newly produced protein is likely to be properly folded, or misfolded or whether an environment that cell is placed in is a healthy tissue, inflamed tissue, or damaged tissue. The inputs could include oxygen level, pH, glucose level, inflammatory signals, cell-density signals, stress-response markers etc.
Each input alone is not enough for a reliable decision such as low oxygen might happen normally in some tissues, and inflammation alone does not always mean damage. But the combination of several signals can give a better estimation. A limitation is that these signals are still context-dependent. The same oxygen, pH, or inflammation levels might mean different things in different tissues. So the IANN would need careful calibration, and it might work well in one environment but less reliably in another.
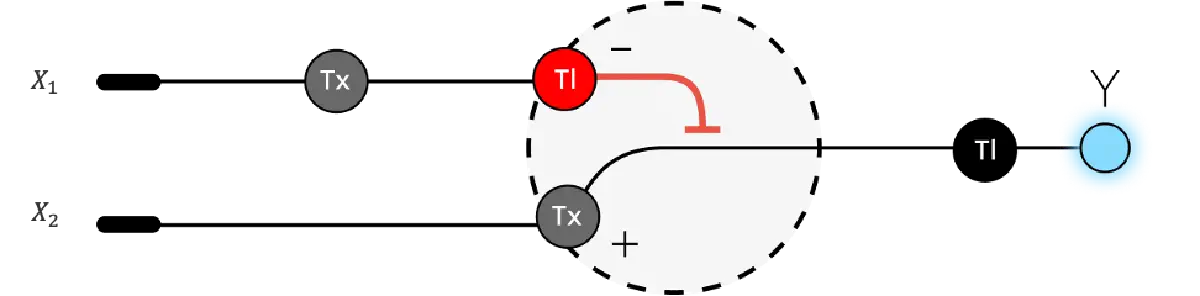
3. Below is a diagram depicting an intracellular single-layer perceptron where the X1 input is DNA encoding for the Csy4 endoribonuclease and the X2 input is DNA encoding for a fluorescent protein output whose mRNA is regulated by Csy4. Tx: transcription; Tl: translation.
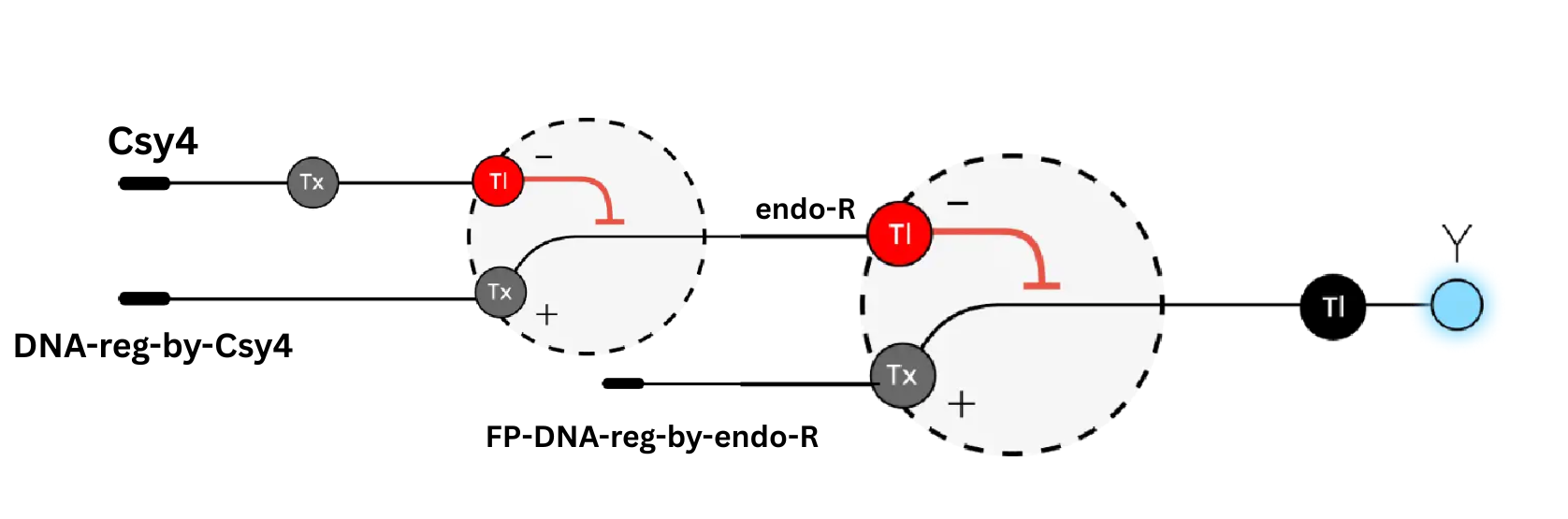
Draw a diagram for an intracellular multilayer perceptron where layer 1 outputs an endoribonuclease that regulates a fluorescent protein output in layer 2.
Assignment Part 2: Fungal Materials
What are some examples of existing fungal materials and what are they used for? What are their advantages and disadvantages over traditional counterparts?
Most current fungal materials are based on mycelium composites. Mycelium is the filament-like growth network of a fungus. In these materials, the fungus is grown through a plant-based substrate such as straw, corn stalks, or other agricultural waste. As it grows, the mycelium binds the loose material together. The final object is usually dried or heat-treated so the organism stops growing and the remaining material becomes a lightweight composite.
Use Cases:
Protective packaging. Mycelium can be grown inside molds to make custom shapes that protect products during shipping. This makes it a possible alternative to expanded polystyrene foam. Its advantages are that it can use agricultural waste, can be compostable, and requires less conventional manufacturing. The main limitations are slower production and lower mechanical strength compared with many petroleum-based foams.
Thermal and acoustic insulation. Because these materials are lightweight and porous, they can trap air and absorb sound. This makes them useful for interior panels, sound-absorbing tiles.
Architecture and construction. Mycelium has also been used mostly through experimental bricks, panels, and temporary structures. A well-known example is Hy-Fi, a temporary pavilion at MoMA PS1 built from mycelium bricks grown from agricultural waste. This project showed that mycelium materials can be used at architectural scale, but it also shows the current limits: these materials are better suited for lightweight, temporary, or insulating structures than for directly replacing concrete, steel, or structural timber.
A distinct product example is the Loop Living Cocoon, a mycelium-based coffin. This is different from packaging or construction because it uses mycelium for an end-of-life product where biodegradability is the main value. The coffin is grown from mycelium and plant fibers and is designed to decompose after burial.
Overall strengths and weaknesses
The main strengths of fungal materials are:
they can be grown from low-value biomass or waste
they are lightweight
they can be molded into specific shapes
they can be compostable or biodegradable
they can have useful insulation and acoustic properties
their texture and density can be tuned through growth conditions
The main weaknesses are:
lower strength than conventional structural materials
slower production compared with industrial foam or plastic manufacturing
contamination risk during growth
limited outdoor durability without coating or treatment
What might you want to genetically engineer fungi to do and why? What are the advantages of doing synthetic biology in fungi as opposed to bacteria?
Fungi are useful for material applications because they naturally form large, connected, three-dimensional networks. Bacteria are easier to engineer in many cases, especially common lab organisms like E. coli, but bacteria usually produce molecules, biofilms, or materials that need extra processing. Fungi directly grow into bulk structures through hyphal extension and entanglement.
Fungi also grow well on many cheap plant-based substrates. This is important because material production only becomes practical if the feedstock is inexpensive. Many mycelium-forming fungi can use sawdust, straw, coffee grounds, hemp, or agricultural residues.
Another advantage is that fungi are eukaryotes. This means they can sometimes process more complex proteins or biomolecules than bacteria. This matters if the material is expected to produce pigments, enzymes, structural proteins, or other functional additives.
The tradeoff is that fungi are harder to engineer than bacteria. Bacteria grow quickly and have standardized genetic tools. Many mushroom-forming fungi grow more slowly and have less developed engineering toolkits. Tools that work in yeast or common lab fungi do not always transfer easily to species such as oyster mushroom or reishi.
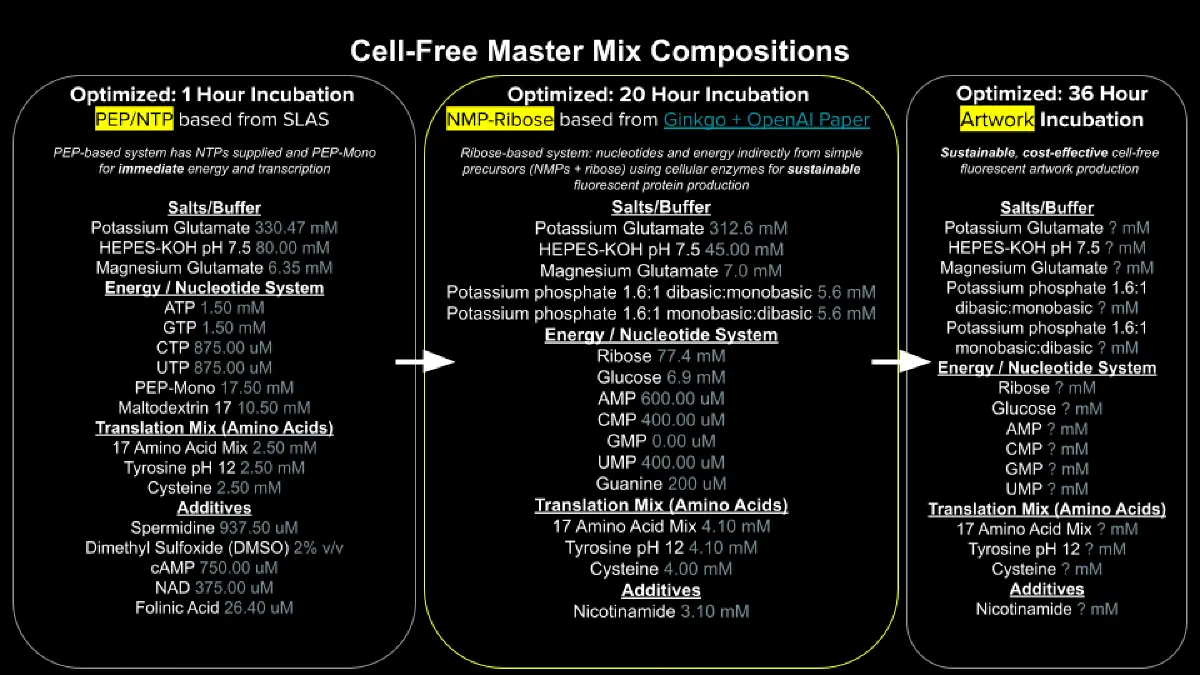
Week 9 HW: Cell-Free Systems
General Homework Questions
1. Explain the main advantages of cell-free protein synthesis over traditional in vivo methods, specifically in terms of flexibility and control over experimental variables. Name at least two cases where cell-free expression is more beneficial than cell production.
Cell-free protein synthesis (CFPS) skips the cell membrane barrier, so the user has direct access to the reaction mixture. This means parameters like pH, redox state, magnesium and potassium concentrations, temperature, and amino acid pools can all be tuned freely without worrying about cell viability. There is also no need for cloning, transformation, or growing cultures, so the time from DNA template to protein is hours instead of days.
Two cases where CFPS is clearly better than in vivo production:
Toxic proteins: Antimicrobial peptides or cytotoxic enzymes would kill an E. coli host, but in a cell-free reaction there is no host to harm.
Non-canonical amino acids: Incorporating unnatural amino acids or isotopic labels is straightforward because the amino acid pool can be depleted and replaced directly in the reaction tube.
2. Describe the main components of a cell-free expression system and explain the role of each component.
Cell extract (lysate): Crude cytoplasm from E. coli, wheat germ, rabbit reticulocyte, or CHO cells. Provides ribosomes, tRNAs, aminoacyl-tRNA synthetases, and translation factors.
DNA template: Plasmid or linear PCR product encoding the target gene under a strong promoter (commonly T7). Carries the genetic information for the protein.
Energy mix and substrates: NTPs, dNTPs, amino acids, and salts (Mg²⁺, K⁺) that fuel transcription and translation.
Energy regeneration system: A secondary high-energy phosphate donor (e.g., phosphoenolpyruvate, creatine phosphate) plus its kinase, which keeps ATP and GTP replenished throughout the reaction.
3. Why is energy provision regeneration critical in cell-free systems? Describe a method you could use to ensure continuous ATP supply in your cell-free experiment.
Protein synthesis consumes multiple high-energy phosphate bonds per peptide bond formed. Without a living metabolism in the tube, free ATP runs out within minutes and inorganic phosphate accumulates, inhibiting further translation. Regeneration is what determines how long the reaction lasts and how much protein it produces.
A common method is the PEP / pyruvate kinase system: phosphoenolpyruvate acts as a high-energy phosphate donor, and pyruvate kinase transfers that phosphate onto ADP to regenerate ATP. Creatine phosphate with creatine kinase works the same way. For longer reactions, a continuous-exchange setup (CECF) with a semipermeable membrane can also be used to feed in fresh substrates and remove inhibitory byproducts.
4. Compare prokaryotic versus eukaryotic cell-free expression systems. Choose a protein to produce in each system and explain why.
Prokaryotic systems (typically E. coli-based) are fast, cheap, and high-yield, often reaching mg/mL concentrations within a few hours. They lack post-translational modifications such as glycosylation and proper disulfide bond formation. Eukaryotic systems (wheat germ, rabbit reticulocyte, HeLa, CHO) are slower and produce less protein, but they carry the chaperones, ER/microsomal membranes, and modification enzymes needed for complex folding.
Prokaryotic choice — GFP: GFP folds correctly on its own and needs no glycosylation. E. coli lysate produces large amounts cheaply, which is ideal for reporters and screening.
Eukaryotic choice — Erythropoietin (EPO): EPO requires sialic acid glycosylation to be biologically active. A mammalian lysate (CHO or HEK293) provides the modification machinery prokaryotes lack.
5. How would you design a cell-free experiment to optimize the expression of a membrane protein? Discuss the challenges and how you would address them in your setup.
The main challenge is hydrophobicity. Membrane proteins have transmembrane domains that misfold and aggregate when translated in aqueous extract without a lipid environment.
The setup would add a hydrophobic phase to the reaction so the protein has somewhere to insert as it leaves the ribosome:
Nanodiscs: Pre-formed lipid bilayer discs stabilized by membrane scaffold proteins. The nascent protein inserts co-translationally and ends up in a native-like bilayer.
Detergent micelles: Mild detergents like DDM, Brij-35, or Triton X-100 can solubilize the protein during synthesis. Cheaper than nanodiscs but may interfere with downstream assays.
Liposomes or SUVs: Synthetic vesicles can also serve as insertion targets.
Additional optimizations: tune Mg²⁺ concentration, add chaperones (DnaK/DnaJ/GrpE), and use a slower expression strain or lower temperature to avoid aggregation. Activity can be confirmed with a functional assay (binding, transport, or ligand response) rather than just SDS-PAGE.
6. Imagine you observe a low yield of your target protein in a cell-free system. Describe three possible reasons for this and suggest a troubleshooting strategy for each.
Energy depletion: ATP/GTP runs out before translation finishes. Fix: Add a stronger regeneration system (creatine phosphate + creatine kinase) or switch to a continuous-exchange format.
Template degradation by nucleases or protein degradation by proteases: Endogenous RNase E, Lon, or OmpT can chew up mRNA or the product. Fix: Add RNase inhibitors, use a circular plasmid instead of linear template, or switch to extracts from knockout strains (Δlon, ΔompT).
Inefficient codon usage: Rare codons stall ribosomes. Fix: Codon-optimize the gene for the extract organism, or supplement with rare-codon tRNAs.
Homework Question from Kate Adamala
Design a synthetic minimal cell: a pesticide-sensing SMC that signals to reporter bacteria
1. Function and system logic
a. What does the synthetic cell do? Input and output?
The SMC detects the agricultural pollutant atrazine in the environment and converts that signal into release of IPTG, which then activates GFP expression in nearby reporter E. coli. Input: atrazine (diffuses across the membrane). Output: IPTG released into the surroundings.
b. Could this work as cell-free TX/TL alone, without encapsulation?
No. Without a membrane, the IPTG would immediately reach the reporter bacteria regardless of atrazine, so there would be no conditional sensing. Encapsulation is what makes the sensor gating possible.
c. Could this work in a genetically modified natural cell?
In principle yes — an atrazine-responsive riboswitch could be engineered into live bacteria. But using an SMC avoids genetic drift, biocontainment issues, and host fitness costs. The same SMC architecture can also be re-targeted to other small molecules by swapping the riboswitch, which is harder to do in living cells.
d. Desired outcome:
In the presence of atrazine, the SMC synthesizes a membrane pore, IPTG escapes, and reporter E. coli glow green. Without atrazine, no pore forms and the bacteria stay dark.
2. Component design
a. Membrane: POPC (palmitoyl-oleoyl-phosphatidylcholine) with cholesterol to control fluidity and reduce leakage.
b. Encapsulated inside:E. coli TX/TL extract, a pool of free IPTG, and a plasmid encoding α-hemolysin (aHL) under control of an atrazine-binding RNA riboswitch.
c. TX/TL origin: Bacterial (E. coli), because the riboswitch is bacterial and integrates cleanly with prokaryotic translation.
d. Communication with the environment: Atrazine is small and lipophilic enough to cross the bilayer passively. IPTG is membrane-impermeable and stays inside until aHL is expressed and forms a pore.
3. Experimental details
a. Lipids and genes:
Lipids: POPC, cholesterol.
Gene: α-hemolysin (aHL) downstream of an engineered atrazine aptamer/riboswitch.
Reporter cells:E. coli transformed with GFP under a T7 promoter with a lac operator.
b. Measurement:
Co-incubate SMCs with reporter E. coli and titrate atrazine across a concentration range. Read bulk GFP fluorescence over time on a plate reader (or use flow cytometry for single-cell distributions). A no-atrazine control and a free-IPTG positive control define the dynamic range.
Homework Question from Peter Nguyen
Application field: Textiles / Fashion
a. One-sentence pitch
A field-worker jacket woven with freeze-dried cell-free biosensor patches that change color when exposed to airborne organophosphate pesticides.
b. How it works (more detail)
Disposable paper-matrix patches embedded into the jacket’s sleeves and shoulders contain freeze-dried E. coli extract, a constitutive expression circuit, and a butyrylcholinesterase-based reporter. When sweat or ambient humidity rehydrates the patch, the cell-free reaction starts producing a colored enzymatic output as a baseline. Organophosphate pesticides in the air inhibit the cholinesterase, stopping the reaction and triggering a visible color change on the fabric. The worker sees the warning directly on their sleeve without any electronics.
c. Societal challenge / market need
Agricultural laborers are exposed to pesticide overspray daily, and acute and chronic organophosphate poisoning are major causes of occupational illness, especially in low-income farming regions. Existing electronic monitors are expensive and rare in the field. A passive textile sensor is cheap, requires no power, and gives immediate visual feedback.
d. Addressing cell-free limitations
Activation by water: The patch stays dry-stable and is activated by sweat or humidity during use. To prevent unwanted activation, patches can sit behind a thin water-permeable membrane.
Stability: Freeze-drying preserves the lysate for months at ambient temperature; patches are sealed in foil until first use.
One-time use: Patches are modular and clipped/stitched into the jacket so they can be unclipped and swapped after a positive event or after the day’s shift, without replacing the whole garment.
Homework Question from Ally Huang
Mock Genes in Space proposal — Monitoring radiation-induced DNA damage in astronauts using BioBits®
1. Background
Astronauts on long-duration missions face chronic cosmic radiation that causes DNA double-strand breaks and oxidative stress, accumulating cancer and tissue-damage risk over time. Current diagnostic tools for tracking this damage rely on bulky lab equipment that is impractical aboard a spacecraft. A lightweight, power-efficient, room-temperature-stable biological assay would let crews monitor their own radiation exposure in real time and test the effectiveness of shielding materials. Freeze-dried cell-free systems are an ideal fit because they require no live cells, no cold storage, and minimal hardware — exactly the constraints of deep-space missions.
2. Molecular target
The human p53 transcript and protein, a master regulator of the DNA damage response that is upregulated in proportion to genomic stress.
3. Relation of the target to the challenge
p53 is the central node of the DNA damage response: when double-strand breaks occur, ATM/ATR signaling stabilizes and activates p53, which then drives transcription of repair and apoptosis genes. The level of p53 expression therefore tracks the intensity of recent radiation exposure. By using p53-responsive reporters in a freeze-dried cell-free system, we can quantify radiation-induced damage in real time aboard the ISS without complex equipment, and compare exposures across different shielding configurations or mission phases.
4. Hypothesis / research goal
Hypothesis: A freeze-dried BioBits® cell-free reaction containing a p53-responsive fluorescent aptamer reporter can be rehydrated aboard the ISS and used to quantify DNA damage in astronaut-derived samples, producing fluorescence proportional to radiation dose.
The reasoning rests on three points. First, cell-free reactions remain stable in freeze-dried form for long periods and do not need live cell culture, which fits the operational constraints of a spaceflight environment. Second, p53 activation is a well-validated, dose-dependent biomarker of DNA damage, with extensive ground-based calibration data already available. Third, fluorescent aptamer reporters provide a directly visible readout that can be quantified with the P51 Molecular Fluorescence Viewer, requiring no benchtop fluorimeter. If validated, this platform becomes a general template for in-flight molecular diagnostics.
5. Experimental plan
Astronauts collect small blood or cheek-swab samples at scheduled intervals. Each sample is mixed with rehydrated BioBits® extract carrying a p53-responsive fluorescent aptamer plasmid. Reactions are incubated in the miniPCR® thermal cycler at 37 °C, and fluorescence is read with the P51 viewer. Controls include: (i) a ground-prepared positive control with known radiation-damaged DNA, (ii) a shielded ground sample to establish baseline, and (iii) a no-template negative control. Data collected: fluorescence intensity over time and across sampling sessions, correlated with onboard dosimeter readings to validate the assay against physical radiation measurements.
References
Lentini, R. et al. (2014). Integrating artificial with natural cells to translate chemical messages that direct E. coli behaviour. Nature Communications, 5, 4012.
Martini, L. & Mansy, S. S. (2011). Cell-like systems with riboswitch controlled gene expression. Chemical Communications, 47(38), 10734.
Pardee, K. et al. (2016). Portable, on-demand biomolecular manufacturing. Cell, 167(1), 248–259.
Stark, J. C. et al. (2018). BioBits™ Bright: A fluorescent synthetic biology education kit. Science Advances, 4(8), eaat5107.
Week 10 HW: Imaging and Measurement
Homework: Waters Part I — Molecular Weight
An eGFP standard was analyzed on a Waters Xevo G3 QTof MS system to determine the molecular weight of intact eGFP and observe its charge state distribution in the native and denatured (unfolded) states.
Values obtained from Expasy’s ProtParam tool for the eGFP sequence containing the LE linker and His-purification tag (HHHHHH):
Property
Result
Number of amino acids
247
Average molecular weight
28006.60 Da
Monoisotopic molecular weight
27988.96 Da
Theoretical pI
5.90
I will use the average MW = 28006.60 Da.
However, the mature eGFP chromophore forms by autocatalytic cyclization, which causes a nominal mass loss of approximately H₂O + H₂ ≈ 20.03 Da (dehydration and loss of two hydrogen atoms).
MW_theory = 28006.60 − 20.03
MW_theory = 27986.57 Da
2. Calculate the molecular weight using the adjacent charge state approach
I selected the following adjacent charge state peaks from Figure 1:
(m/z)ₙ = 1000.4302
(m/z)ₙ₊₁ = 965.9684
2.1 Determine z for the adjacent pair (n, n+1):
The formula is: z = (m/z)ₙ₊₁ / [ (m/z)ₙ − (m/z)ₙ₊₁ ]
z = 965.9684 / (1000.4302 − 965.9684)
z = 965.9684 / 34.4618
z = 28.03 ≈ 28
2.2 Determine the MW of the protein:
Using the relationship (m/z)ₙ = (MW + n·H) / n, where H ≈ 1.0073 Da (mass of a proton), solving for MW gives:
MW = n · (m/z)ₙ − n · H
Using n = 28 and (m/z)ₙ = 1000.4302:
MW = 28 × (1000.4302) − 28 × (1.0073)
MW = 28011.05 − 28.20
MW ≈ 27983.84 Da
As a check, applying the same formula to the adjacent peak with n+1 = 29 and (m/z)ₙ₊₁ = 965.9684:
MW = 29 × (965.9684) − 29 × (1.0073)
MW ≈ 27983.87 Da
Average experimental MW:
MW_exp = (27983.84 + 27983.87) / 2
MW_exp ≈ 27983.86 Da
2.3 Calculate the accuracy of the measurement:
The formula is: Accuracy = | MW_exp − MW_theory | / MW_theory
Accuracy = | 27983.86 − 27986.57 | / 27986.57
Accuracy = 2.71 / 27986.57
Accuracy = 9.68 × 10⁻⁵
Converting to ppm:
ppm error = 9.68 × 10⁻⁵ × 10⁶
ppm error ≈ 97 ppm
3. Can you observe the charge state for the zoomed-in peak in the mass spectrum for the intact eGFP?
Yes. In the zoomed-in inset around m/z ≈ 1473, the isotopic peaks within the envelope are separated by approximately Δ(m/z) ≈ 0.052. Since the isotopic spacing equals 1/z:
z = 1 / 0.052
z ≈ 19
So the charge state of the zoomed-in peak is +19.
Homework: Waters Part II — Secondary / Tertiary structure
1. Difference between native and denatured protein conformations
A native protein is folded into its biologically active 3D structure: secondary structure (α-helices, β-sheets) is organized into a tertiary fold, and the hydrophobic residues are buried in the core. When a protein denatures, the non-covalent interactions (hydrogen bonds, hydrophobic packing, salt bridges) break down. The chain opens up and many previously buried basic residues become exposed to the solvent.
On a mass spectrometer this difference shows up as a change in the charge state distribution, not the mass itself. In native MS (gentle solvents, neutral pH), the protein keeps its compact fold and only a few exposed sites can pick up protons — so the spectrum shows a small number of charge states clustered at high m/z (e.g. around 2500–3000). In denaturing conditions (organic solvent, low pH), all basic residues are exposed, so the protein picks up many more protons — the spectrum shifts to a broader distribution of charge states at much lower m/z (e.g. 700–1500).
In Figure 2, the denatured eGFP (top) shows a wide envelope of many closely spaced peaks at low m/z, while the native eGFP (bottom) shows only a few peaks at higher m/z. The total deconvoluted mass is the same — only the protonation pattern differs.
2. Charge state of the peak at ~2800 m/z in the native spectrum
In Figure 3, the zoomed-in inset around m/z ≈ 2800 shows isotopic peaks separated by approximately Δ(m/z) ≈ 0.1. Since the isotopic spacing equals 1/z:
z = 1 / 0.1
z = 10
So the charge state is approximately +10. This low charge state is consistent with a folded, native conformation in which only a small number of basic residues are exposed and available for protonation.
Homework: Waters Part III — Peptide Mapping (primary structure)
1. How many Lysines (K) and Arginines (R) are in eGFP?
2. How many peptides will be generated from tryptic digestion of eGFP?
Trypsin cleaves after K and R (except when followed by P). Using the Expasy PeptideMass tool with the parameters shown in Figure 4, the digest yields 19 peptides (some short cleavage products are filtered below the default mass cutoff, and a few KP/RP sites are not cleaved).
5. Identify the m/z, charge, and singly-charged mass of the peptide in Figure 5b
The most abundant peak is at m/z = 525.76.
The isotope peaks in the inset are separated by approximately Δ(m/z) ≈ 0.5, so the charge state is:
z = 1 / 0.5
z = 2
Calculating the singly-charged form [M+H]⁺ from m/z = (M + n·H) / n:
525.76 = (M + 2 × 1.00727) / 2
M = 2 × 525.76 − 2 × 1.00727
M = 1051.52 − 2.01
M ≈ 1049.51 Da
Therefore:
[M+H]⁺ = M + 1.00727
[M+H]⁺ ≈ 1050.51 Da
6. Identify the peptide and calculate mass accuracy
Comparing the observed [M+H]⁺ = 1050.51 Da against the predicted peptide list from PeptideMass, the closest match is the tryptic peptide FEGDTLVNR (theoretical [M+H]⁺ = 1050.5214 Da).
7. What is the percentage of the sequence confirmed by peptide mapping?
Based on the amino acid coverage map in Figure 6, approximately 88% of the eGFP sequence is confirmed by identified peptides.
Bonus — Does the peptide map data make sense?
Yes. The peptide FEGDTLVNR matches an expected tryptic peptide of eGFP within ~11 ppm, and ~88% of the full sequence is covered by identified peptides with correct masses and fragmentation patterns. Combined with the intact-mass measurement from Part I (which matched the expected MW within ~100 ppm), this strongly supports the conclusion that the protein analyzed is indeed the eGFP standard.
Homework: Waters Part IV — Oligomers
Using the known KLH subunit masses to assign oligomeric species on the CDMS spectrum:
Oligomeric species
Calculation
Mass
7FU Decamer
340 kDa × 10
3.4 MDa
8FU Didecamer
400 kDa × 20
8.0 MDa
8FU 3-Decamer
400 kDa × 30
12 MDa
8FU 4-Decamer
400 kDa × 40
16 MDa
Each of these masses corresponds to a distinct peak in the CDMS spectrum of Figure 7, showing that KLH exists in solution as a mixture of these higher-order oligomeric states.
Homework: Waters Part V — Did I make GFP?
Theoretical
Observed (Intact LC-MS)
PPM Mass Error
Molecular weight of eGFP (Da)
27986.57
27983.86
~97 ppm
Peptide FEGDTLVNR (Da, [M+H]⁺)
1050.5214
1050.51
~10.85 ppm
The intact-mass and peptide-map measurements both agree with the predicted values within typical LC-MS mass accuracy, confirming that the analyzed protein is the eGFP standard.
Heck, A. J. R. (2008). Native mass spectrometry: a bridge between interactomics and structural biology. Nature Methods, 5(11), 927–933.
Week 11 HW: Building Genomes
Part A: The 1,536 Pixel Artwork Canvas | Collective Artwork
Unfortunately, I missed the deadline, I guess I have to become a TA next year (hurray)! ¯_(ツ)_/¯
I thought the sense of community behind the project was really well designed, and I appericated the idea of many people contributing small pieces to one shared bioartwork.
This also made me think about my own project on protein steganography. It could be interesting to create a message-encoding project where people enter their names, messages, or short memories, and we encode them into DNA or protein sequences. Although this would not be as immediately visual as the pixel canvas, it could still be very aligned with HTGAA. One possible version could even involve producing the encoded DNA in bacteria and sharing it with committed listeners. Adrian from the Ottawa node also suggested that it might be beautiful to encode the information into a plant seed or something similar—a living memorial for HTGAA. :')
Storyboard generated with GPT Image 2.0
Update on artwork
That’s me, the blue one in the middle!
I took my place in the second version. I’m so happy!
Part B: Cell-Free Protein Synthesis | Cell-Free Reagents
Referencing the cell-free protein synthesis reaction composition (the middle box outlined in yellow on the image above, also listed below), provide a 1-2 sentence description of what each component’s role is in the cell-free reaction.
Category
Component
Role
Lysate
BL21(DE3) Star lysate (with T7 RNAP)
Provides the actual machinery doing the work — ribosomes, tRNAs, T7 RNA polymerase, and the metabolic enzymes that recycle ATP and build NTPs.
Salts / Buffer
Potassium glutamate
Main potassium source. Glutamate is used instead of chloride because it mimics the inside of E. coli and supports ribosome activity.
HEPES-KOH pH 7.5
Holds the pH near 7.5 as acidic byproducts build up during the reaction.
Magnesium glutamate
Mg²⁺ is essential for RNA polymerase, ribosomes, and almost every step that touches a nucleotide. The level is tuned per lysate batch.
Potassium phosphate (mono- and dibasic)
Supplies the phosphate needed to turn NMPs into NTPs and ADP back into ATP. The mix of mono- and dibasic forms also helps set the starting pH.
Energy / Nucleotide
Ribose
Feeds into PRPP (an activated sugar) used by the salvage pathway to build nucleotides from free bases.
Glucose
Cheap energy source. The lysate runs glycolysis on it to regenerate ATP, replacing expensive PEP in long reactions.
AMP, CMP, GMP, UMP
Cheaper monophosphate forms of the nucleotides. Lysate kinases phosphorylate them up to NTPs for transcription.
Guanine
Free purine base that enters the GTP pool through the salvage pathway (see Q3). Cheaper than GMP or GTP.
Translation Mix
17 AA mix
The 20 standard amino acids minus tyrosine and cysteine, which need separate handling. These are the building blocks for the protein.
Tyrosine
Added separately because it doesn’t dissolve well, and because it’s part of the chromophore in most fluorescent proteins — often a limiting reagent for FP yield.
Cysteine
Added separately because it oxidizes easily and forms disulfide bonds, so it’s sensitive to the reaction’s redox state.
Additives
Nicotinamide
Precursor for NAD⁺/NADH. Glycolysis needs NAD⁺ to keep running, which matters when glucose is the energy source.
Backfill
Nuclease-free water
Brings everything to the right volume; nuclease-free so the mRNA and DNA template don’t get chewed up.
Describe the main differences between the 1-hour optimized PEP-NTP master mix and the 20-hour NMP-Ribose-Glucose master mix shown in the Google Slide above.
The PEP-NTP system hands the reaction everything pre-made: NTPs go straight into transcription, and PEP rapidly regenerates ATP from ADP. This produces protein quickly but burns out within an hour as PEP runs out and pyruvate and phosphate build up and acidify the mix — and it’s expensive. The NMP-Ribose-Glucose system instead supplies cheap precursors (NMPs and glucose) and lets the lysate’s own metabolism phosphorylate them up to NTPs and recycle ATP through glycolysis. It starts slower but keeps producing protein for 20+ hours at a fraction of the cost.
How can transcription occur if GMP is not included but Guanine is?
The lysate still has E. coli’s purine salvage pathway intact. An enzyme called XGPRT (Gpt) combines free guanine with PRPP — an activated sugar made from the ribose in the mix — to form GMP. From there, lysate kinases phosphorylate GMP to GDP and then to GTP, which T7 RNA polymerase uses for transcription. So guanine works as a much cheaper drop-in for GMP, with the tradeoff that the conversion is slower — which is fine, even useful, in a long reaction since it avoids burning through the GTP pool too fast.
Part C: Planning the Global Experiment | Cell-Free Master Mix Design
Given the 6 fluorescent proteins we used for our collaborative painting, identify and explain at least one biophysical or functional property of each protein that affects expression or readout in cell-free systems. (Hint: options include maturation time, acid sensitivity, folding, oxygen dependence, etc) (1-2 sentences each)
Protein
Color
Property affecting CFPS readout
sfGFP
Green (~510 nm)
Folds fast and matures fast, which is why it’s the standard CFPS reporter. The final maturation step still needs molecular O₂, so sealed wells may become oxygen-limited late in a long run.
mRFP1
Red (~607 nm)
Slow-maturing monomer derived from DsRed; matures >10× faster than DsRed but is still much slower than GFPs, with lower brightness per molecule. A long incubation should help, but a large fraction of protein will still be immature at any given timepoint.
mKO2
Orange (~559 nm)
A faster-maturing version of mKO1, with unusually strong pH stability — meaning it should tolerate the gradual acidification of a long CFPS reaction well. The chromophore contains a cysteine, so it could potentially be sensitive to redox conditions.
mTurquoise2
Cyan (~474 nm)
The brightest cyan FP available (quantum yield ~0.93), but maturation is multi-step and the oxygen-dependent step is rate-limiting. Likely benefits from sustained O₂ supply and stable pH over the 36-hour run.
mScarlet-I
Red (~594 nm)
A faster-maturing variant of mScarlet (half-time ~36 min), trading a bit of brightness (QY 0.54) for speed. Practical RFP choice for CFPS since it matures much faster than mRFP1.
Electra2
Blue (ex ~405 / em ~456 nm)
A newer (2022) BFP derived from anemone eqFP611, with brightness similar to mTagBFP2 in mammalian cells. Behavior in E. coli CFPS is less characterized — aggregation has been reported in some contexts, so its folding over 36 h is somewhat uncertain.
Create a hypothesis for how adjusting one or more reagents in the cell-free mastermix could improve a specific biophysical or functional property you identified above, in order to maximize fluorescence over a 36-hour incubation. Clearly state the protein, the reagent(s), and the expected effect.
Protein: mRFP1 — slow maturation, two oxidative steps, dimmer per molecule than the other RFPs.
Reagent change: Within the 2 µL adjustable volume, add ~20 mM extra HEPES-KOH (pH 7.5) and ~0.5–1 mM extra tyrosine.
Reasoning and expected effect: Over 36 hours, phosphate, pyruvate, and lactate will accumulate and slowly acidify the reaction. The translation and folding machinery is more pH-sensitive than the mRFP1 chromophore itself, so stronger buffering should keep the ribosomes working productively for longer. Tyrosine is one of the three residues that form the chromophore and is supplied separately because of its low solubility — topping it up directly should reduce the chance it becomes rate-limiting in a long run. The expected outcome is brighter red fluorescence at 36 h compared to the baseline composition.