Week 2 HW: DNA Read, Write and Edit
Part 1: Benchling & In-silico Gel Art
For this exercise, the full genome of Bacteriophage Lambda (GenBank accession J02459.1, 48,502 bp) was imported into Benchling from NCBI. A virtual restriction enzyme digestion was performed using seven enzymes: EcoRI, HindIII, BamHI, KpnI, EcoRV, SacI, and SalI. Each enzyme was applied individually to identify its recognition sites across the Lambda genome. The digest results were visualized using Benchling’s simulated gel electrophoresis tool. To get a nice visual, I tried different ladders and different lane orderings. The final output consisting of for each enzyme’s fragment pattern is below.

Experiments with DNA Gel Art Interface. My initials below figure B. K.

Note: As a possible extension, it could be cool to build an interactive version of Ronan’s website where you can pick genome, enzymes, drag lanes around, swap ladders, add timing, etc. Could be a nice tool for future HTGAA classes.
Note: As a possible extension, it could be cool to build an interactive version of Ronan’s website where you can pick genome, enzymes, drag lanes around, swap ladders, add timing, etc. Could be a nice tool for future HTGAA classes.
Part 3: DNA Design Challenge
3.1. Choose your protein
I chose PIGH (Phosphatidylinositol N-acetylglucosaminyltransferase subunit H), a human protein encoded by the PIGH gene on chromosome 14q24.1. PIGH is a 188-amino-acid subunit of the GPI-GnT complex, which catalyzes the first step of GPI-anchor biosynthesis — transferring N-acetylglucosamine to phosphatidylinositol on the cytoplasmic side of the endoplasmic reticulum. GPI anchors tether many important proteins to the cell surface.
I chose this protein because mutations in PIGH cause GPIBD17 (Glycosylphosphatidylinositol Biosynthesis Defect 17), a rare autosomal recessive disorder characterized by developmental delay, seizures, and autistic features. The gene was only linked to disease in 2018, so it is likely underdiagnosed. Its small size (188 aa) also makes it practical for the synthesis and expression exercises in this homework.
I obtained the FASTA sequence from UniProt.
3.2. Reverse Translate
Using the Sequence Manipulation Suite reverse translation tool from bioinformatics.org, I converted the 188-amino-acid protein sequence into a 564 bp DNA sequence using the most likely codons:
3.3. Codon Optimization
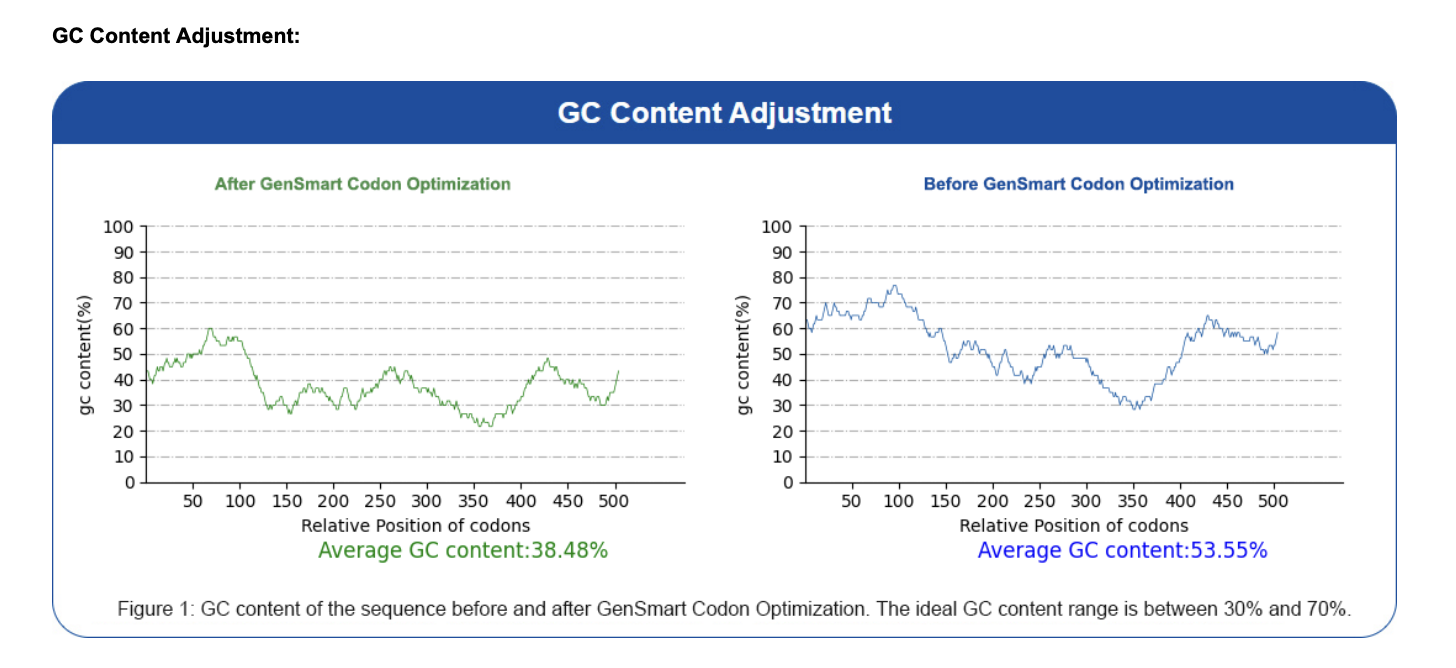
I used the GenSmart Codon Optimization Tool to optimize the PIGH coding sequence for expression in E. coli and Staphylococcus aureus. Codon optimization is necessary because different organisms have different preferences for which codons they use most efficiently related to tRNA availability. Each organism has a different pool of tRNA molecules, and some tRNAs are abundant while others are rare. When the ribosome hits a codon whose matching tRNA is scarce in that organism, translation slows down or stalls. A human gene expressed directly in E. coli may use rare codons that slow down or stall translation. Optimization replaces these with codons preferred by the host organism, improving expression levels without changing the protein sequence. Optimization report available here.
Optimized sequence:
3.4. You have a sequence! Now what?
To produce the PIGH protein, the codon-optimized DNA sequence is incorporated into an expression construct containing regulatory elements — a promoter (BBa_J23106), ribosome binding site (BBa_B0034), start codon, coding sequence, 7×His tag for purification, stop codon, and terminator (BBa_B0015). This can be ordered as a synthetic plasmid from Twist Bioscience in a backbone like pTwist Amp High Copy.
Protein production follows the central dogma in two stages. During transcription, RNA polymerase binds the promoter and synthesizes a complementary mRNA strand. During translation, ribosomes assemble at the RBS and read the mRNA in three-nucleotide codons, with tRNA molecules delivering amino acids from the start codon (AUG/methionine) until the UAA stop codon is reached, producing the folded 188-amino-acid PIGH polypeptide.
Cell-Dependent Production: The plasmid could be transformed into E. coli BL21 via heat shock or electroporation, with ampicillin selection to identify successful transformants. The host cell’s machinery and metabolic resources would then drive protein expression. Since PIGH is relatively small and does not appear to require complex eukaryotic modifications, E. coli may be a suitable host. The His-tagged protein could then be purified via Ni-NTA chromatography. This approach tends to be scalable but requires time for cell growth and purification.
Cell-Free Production: Alternatively, a TX-TL cell-free system (e.g., PURExpress) could be used, where the linear expression cassette is mixed directly with ribosomes, polymerase, tRNAs, and amino acids from lysed cells. This would likely produce protein within hours without living cultures, making it potentially ideal for rapid prototyping. PIGH’s small size (~564 bp) should be well within the efficient range for cell-free systems, though yields are generally lower and cost per reaction tends to be higher.
Part 4: Prepare a Twist DNA Synthesis Order
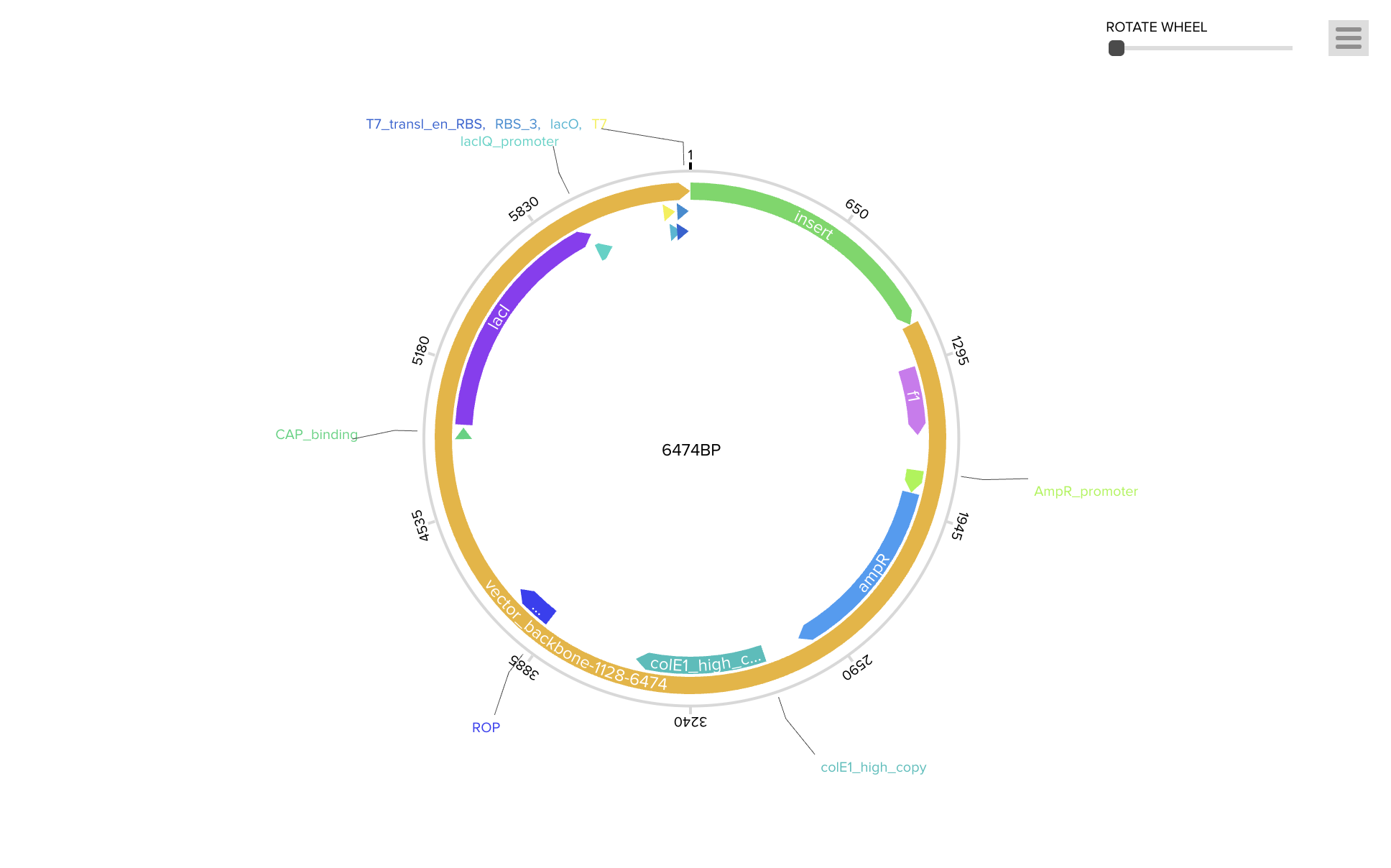
Part 5: DNA Read/Write/Edit
5.1 DNA Read
I would want to sequence the PIGH gene in patients presenting with developmental delay, seizures, and autistic features who lack a molecular diagnosis. Sequencing PIGH in such individuals could potentially help identify pathogenic variants and improve diagnostic rates for GPI-anchor biosynthesis disorders.
Since the PIGH coding region is relatively short (~564 bp), Sanger sequencing would likely be a suitable approach. Sanger is generally considered a first-generation method, reading one sequence at a time rather than in parallel like next-gen methods. I would extract genomic DNA from a patient sample, PCR-amplify the PIGH region using forward and reverse primers, and use the purified product as input for the Sanger reaction. A forward and reverse read should be sufficient to cover the full sequence with overlap.
The Sanger reaction mix contains normal dNTPs along with fluorescently labeled ddNTPs. During extension, the polymerase occasionally incorporates a ddNTP, terminating the chain at that position. This produces fragments of various lengths, each capped with a color for A, T, G, or C. Capillary electrophoresis separates them by size, and a detector reads the color at each position. The output is a chromatogram with color-coded peaks and a derived nucleotide sequence that could be compared against the PIGH reference to look for mutations.
5.2 DNA Write
I would want to synthesize the codon-optimized PIGH expression cassette from Parts 3–4. It could also be useful to order variant versions carrying known disease-associated mutations alongside wild-type to compare their effects on protein function. I would likely use phosphoramidite oligonucleotide synthesis as offered by companies like Twist Bioscience. Short oligos are built base-by-base on a solid support through repeated cycles of de-protection, coupling, capping, and oxidation, then assembled into the full-length gene and sequence-verified. Error rate tends to increase with length, so longer constructs may need to be built from smaller fragments. For PIGH (~564 bp), this should be well within a comfortable range.
5.3 DNA Edit
I would want to correct the M1L mutation (c.1A>T) in the PIGH gene. This change appears to disrupt the start codon and may prevent normal protein production. An adenine base editor (ABE) could be a reasonable choice for this kind of single-base correction. ABE generally uses a nickase Cas9 fused to an adenosine deaminase. A guide RNA directs the complex to the target site, the deaminase converts the target base without creating a double-strand break, and the cell’s repair machinery completes the correction. Inputs would include the ABE protein or mRNA, a guide RNA targeting the mutation region, and cells carrying the variant. Some limitations include the narrow editing window of the deaminase, dependence on a nearby PAM sequence, possible bystander edits on other adenines in the window, and variable delivery efficiency across cells.